
Abstract
Profilin is a ubiquitously expressed protein well known as a key regulator of actin polymerisation. The actin cytoskeleton is involved in almost all cellular processes including motility, endocytosis, metabolism, signal transduction and gene transcription. Hence, profilin’s role in the cell goes beyond its direct and essential function in regulating actin dynamics. This review will focus on the interactions of Profilin 1 and its ligands at the plasma membrane, in the cytoplasm and the nucleus of the cells and the regulation of profilin activity within those cell compartments. We will discuss the interactions of profilin in cell signalling pathways and highlight the importance of the cell context in the multiple functions that this small essential protein has in conjunction with its role in cytoskeletal organisation and dynamics. We will review some of the mechanisms that control profilin expression and the implications of changed expression of profilin in the light of cancer biology and other pathologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Profilin
Profilin is a key regulator of actin polymerisation and is critically important to cellular function (Ding et al. 2012). The actin cytoskeleton is involved in almost all cellular processes including motility, endocytosis, metabolism, signal transduction and gene transcription (Olson and Nordheim 2010); however, profilin’s role in the cell goes beyond its direct and essential function in actin dynamics. This review will focus on the interactions of actin, profilin and its ligands in the plasma membrane, and the regulation of profilin activity within the cell. The diverse nature of the pathologies that can arise from disruptions to profilin demonstrate the breadth of roles profilin fulfils in a cell. The diverse pathologies include neurological conditions such as spinal muscular atrophy (Nolle et al. 2011), diabetes and glucose metabolism (Pae and Romeo 2014; Romeo and Kazlauskas 2008), vascular hypertrophy and hypertension (Jin et al. 2012; Moustafa-Bayoumi et al. 2007) and tumour progression in cancer (Mouneimne et al. 2012; Yap et al. 2012). Changes in levels of profilin expression can up- or downregulate the level of expression of proteins involved in motility, proliferation, apoptosis (Coumans et al. 2014) and oncogenesis (Frantzi et al. 2016) giving profilin a key role in multiple pathways to cell fate beyond its role as a resource for actin polymerisation.
Profilins are a family of small, 15-kD multi-ligand proteins structurally conserved among eukaryotes (Pandey and Chaudhary 2017; Schluter et al. 1997). The four mammalian isoforms show tissue specificity. Profilin 1 is ubiquitously expressed while Profilin 2 is associated with nervous tissue (Birbach 2008). Profilin 3 and 4 are expressed in the testis and Profilin 3 in kidney (Hu et al. 2001). The different mammalian profilins have non-redundant functions that have critical roles developmentally (Birbach 2008; Khadka et al. 2009) and neurologically (Michaelsen et al. 2010). This review focuses on Profilin 1 but one should note that changes in expression of Profilin 2 alters the progression and characteristics of various cancers (Ma et al. 2011; Mouneimne et al. 2012; Zhang et al. 2018) and neurological disorders (Chakraborty et al. 2014). For simplicity, Profilin 1 will be referred to as “profilin” from this point onward.
The complexity of its interactions has long been investigated (Sohn and Goldschmidt-Clermont 1994). Profilin 1 was first identified as a 15-kDa actin binding protein (Carlsson et al. 1977) and is recognised as one of the essential proteins for cell survival (Witke et al. 2001) by regulating the dynamic actin cytoskeleton (Davidson and Wood 2016). Through its interactions with various actin binding proteins (ABP) containing proline repeat domains (PRD), profilin facilitates membrane protrusion and cell motility (Ding et al. 2009; Witke 2004). Major proteins that connect profilin to the plasma membrane and modulate actin polymerisation through PRDs are Neuronal Wiskott-Aldrich syndrome protein/WASP-family verprolin-homologous protein (N-WASP/WAVE), enabled/mammalian enabled/vasodilator stimulated phosphoprotein (Ena//Mena/VASP), formins and the actin-related protein 2/3 complex (ARP2/3) (Jockusch et al. 2007). Profilin has other links to membrane-bound proteins such as phosphatase and tensin homologue (PTEN) (Zaidi and Manna 2016), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) (Bhargavi et al. 1998) and vascular endothelial growth factor receptor (VEGFR2) (Fan et al. 2012; Simons and Schwartz 2012). Some interactions between profilin and membrane proteins may be indirect, such as the recruitment and activation of formins by the receptor for advanced glycation end products (RAGE) (Sorci et al. 2013) that brings profilin into oxidative stress pathways (Lu et al. 2015). Finally, the other major membrane ligand class of profilin are the phosphoinositol lipids (PPI), and through these interactions, profilin is connected to a vast array of cellular functions (Lu et al. 1996; Sun et al. 2013).
Profilin: a key player in cytoskeleton organisation
For cytoskeletal reorganisation, the direct interaction of profilin with cortical actin, PRD and PPI will determine how profilin favours the development and persistence of one type of structure over another (e.g. lamellipodia, filopodia and invadopodia) to regulate cell motility (Lorente et al. 2014; Skruber et al. 2020). The results of this can be seen in a number of cancer cells including breast, hepatic and pancreatic tumour cells where overexpression of profilin results in a reduction in motility and migration. For these cell types, reduction in profilin levels results in an increase in metastatic ability (Janke et al. 2000; Roy and Jacobson 2004; Schoppmeyer et al. 2017; Wang et al. 2004). Reduction of profilin expression in human mammary epithelial cells (HMECs) also results in enhanced motility (Zou et al. 2007) which is in contrast to other cell types such as human umbilical vein epithelial cells (HUVECs) (Ding et al. 2006) and in other types of cancers such as renal cell carcinoma (Minamida et al. 2011) and gastric cancer where metastatic disease is associated with high profilin expression (Cheng et al. 2015).
The orientation of the various binding sites of profilin is important in understanding the interplay of its ligands. The profilin poly-L-proline binding site is in a hydrophobic pocket that involves the N and C terminal α-helices (Bjorkegren et al. 1993; Haarer et al. 1993; Metzler et al. 1994). This is on the orthogonal surface to the actin binding site allowing for ternary complexes to be formed between profilin, actin and proteins with PRD containing proteins. Profilin-specific PRDs containing ligands can bind in two orientations and these different spatial registers also allow for further regulation (Mahoney et al. 1999). Profilin’s affinity for both actin and PRDs is affected by the electrostatic environment. Local changes that lead to a decrease in pH will lead to a partial unfolding of profilin and decrease in binding to both ligands, increasing the local free G-actin population (McLachlan et al. 2007). Additionally, two binding regions for PPIs have been identified. They overlap the actin and PRD binding domains (Lambrechts et al. 2002) and form part of a broad surface of exposed hydrophobic residues (for reviews see (Coumans et al. 2018; Krishnan and Moens 2009); Witke (2004)). Binding of either actin or PRD can release profilin from the lipids which allows it to move from the membrane to the cytoskeleton (Krishnan and Moens 2009). As PPIs compete with actin (Lassing and Lindberg 1985) and PRD containing proteins for binding to profilin (Lambrechts et al. 2002), they fine tune the availability of profilin for its role as a regulator of cell signalling pathways (Ding et al. 2012).
The actin and microtubule cytoskeletons exist in conjunction with each other and there is growing evidence that profilin has a role in regulating the dynamics of microtubules as well as actin filaments (Pinto-Costa and Sousa 2020). Partial co-localisation of profilin and microtubules were observed in fibroblasts (Grenklo et al. 2004). The formin mDia was suggested as a link between actin and microtubules as it is involved in actin dynamics and the stabilisation of the (+) ends of microtubules (Bartolini et al. 2012), and profilin appears to be the pivot that allows mDia to regulate the behaviour of actin and microtubule (Nejedla et al. 2016). Profilin may bind directly to microtubules in addition to its interaction via formins, through residues not involved with the PRD domain (Pinto-Costa and Sousa 2020). These residues are in the region of profilin that are mutated in some cases of the neurological condition amyotrophic lateral sclerosis (ALS) and perturb the dynamics of microtubules or the cross-talk between the actin and microtubule cytoskeletons (Henty-Ridilla et al. 2017). Further discussion of profilin’s role in regulating microtubule dynamics, and pathologies arising from this misregulation is not in the scope of this review. Our aim in this section was only to highlight the central role of profilin in regulating the cytoskeleton and to point readers to some of the recent developments in this topic.
Profilin as a regulator of actin dynamics
Profilin’s role as a cell signalling molecule and regulator of actin dynamics overlap (Bae et al. 2010) so a brief description of the role profilin plays in actin dynamics provides a context for the main focus on the role of profilin in cell signalling. The recruitment of profilin-ATP-actin monomers is central to the activity of proteins involved in the elongation of actin filaments and thus is essential for the cellular processes that rely on the fine-tuning of the actin cytoskeleton (Jockusch et al. 2007; Skruber et al. 2018). Nucleation promoting factors (NPFs) such as formins and elongation factors such as Ena/VASP and ARP2/3 specifically recruit and maintain high concentrations of profilin-ATP-actin monomers to sites of active actin remodelling through multiple polyproline domains (Campellone and Welch 2010; Dominguez 2010; Skruber et al. 2020). At high concentrations, profilin paradoxically accelerates faster turnover of actin filaments by competing for barbed ends and increasing the off-rate of actin monomers (Ding et al. 2012; Pernier et al. 2016). Two major classes of profilin-PRD partners involved in actin filament elongation are formins and members of the Ena/Mena/VASP family referred to as Ena/VASP. Profilin accelerates both formin (Romero et al. 2004) and Ena/VASP (Hansen and Mullins 2010)-mediated actin filament elongation. Formins and Ena/VASP are enriched at the tips of filopodia and lamellipodia and result in long unbranched actin filaments and have specific, non-redundant functions but both actively recruit profilin-actin complexes (Blanchoin et al. 2014; Yang et al. 2007). Formins can both nucleate and elongate linear actin structures while Ena/VASP proteins are elongation factors only. Changes in profilin concentration determine whether filaments nucleated by Ena/VASP or dendritic networks formed by ARP2/3 develop at the leading edge (Skruber et al. 2020). The multiple domains on formins, Ena/VASP, various NPFs and ARP2/3 allow spatially defined interactions with other proteins and lipids that serve as molecular scaffolds to link the actin cytoskeleton to the cell membrane (recently reviewed in (Boczkowska et al. 2014; Dominguez 2016; Saarikangas et al. 2010; Siton-Mendelson and Bernheim-Groswasser 2017). Profilin has recently been discovered to participate in the post-translational N-terminal acetylation of actin, critical for its interaction with actin binding proteins, by providing the binding site of the Nt-acetyltransferase NAA80 on profilin-actin complex (Rebowski et al. 2020). This is another example of the expanding repertoire of known profilin functions.
Profilin, lipids and signalling
Lipid ligands of Profilin
The importance of profilin: PIP2 interactions were noted decades ago firstly with observations that the actin polymerisation activity of profilin is regulated by binding to PIP2 (Lassing and Lindberg 1985; Lassing and Lindberg 1988). Profilin has a role as a regulator of cell metabolism through its interactions with membrane PPI (Bae et al. 2010). The PPIs have specific and fundamental roles in all aspects of cell physiology as second messengers and through interactions with cytoskeletal and regulatory proteins (Balla 2013; Viaud et al. 2016). An important membrane PPI, PI(4,5)P2 (PIP2) and two of its derivatives PI(3,4,5)P3 (PIP3) and PI(3,4)P2 are profilin ligands (Lu et al. 1996). PIP2 is the predominant PPI in plasma membrane even if it only makes up approximately 1% of the total lipid content. PIP3 and PI(3,4)P2 are present in much lower concentrations (Balla et al. 2009; Di Paolo and De Camilli 2006). As a second messenger, precursor to second messengers and a regulator of membrane bound proteins, PIP2 is intimately involved in cytoskeletal organisation and a plethora of signalling cascades (Delage et al. 2013).
Profilin’s affinity for PIP2 changes with the size of PIP2 clusters and has much higher affinity for self-associated PIP2 clusters than for single PIP2 molecules. This has implications for its functions (Moens and Bagatolli 2007). In the plasma membrane, PIP2 exists as freely diffusing single molecules with transient small clusters and larger stable aggregates with independent pools serving specific cellular functions (Delage et al. 2013; Kwiatkowska 2010; Levental et al. 2009; Wang and Richards 2012). Profilin can potentially bind up to five inositol headgroups of PIP2 (Ostrander et al. 1995; Richer et al. 2008). In vitro profilin has a higher affinity for micellar PI(3,4)P2 (Kd 1.1 μM) and PIP3 (Kd 5.7 μM) than for PIP2 (Kd 11 μM) (Lu et al. 1996). The relative abundance of PIP2 is the reason it has been proposed as the predominant membrane ligand of profilin despite its higher affinity for the rarer PPIs (Ding et al. 2012). Profilins increased, but still, its relatively low affinity for higher PIP2 densities allows it to be concentrated at sites of high actin polymerisation in a way that allows for rapid dynamics (Senju et al. 2017). Separate pools of PPIs are maintained by synthesis by inositide kinases and degradation by phosphatases which allow rapid but localised changes of PIP2 concentration (Hammond and Burke 2020; Krauss and Haucke 2007; Sun et al. 2013) that in turn will alter profilin dynamics.
Electrostatic attraction is important for the organisation of PIP2 in the membrane and is also the mechanism by which profilin and other ABP are recruited (McLaughlin and Murray 2005). The different affinities of the various proteins recruited to the plasma membrane by electrostatic attraction allow for different dynamics. Profilin has a lower affinity for PIP2 than n-WASP and the formin Dia2 and thus is more dynamic but the scaffolding proteins ezrin and moesin have even greater affinity and slower dynamics and are less sensitive to changes in PIP2 concentration (Senju et al. 2017). Consistent with earlier work demonstrating that the affinity of profilin to both PIP2 and PIP3 increased as lipid concentration increased from monodispersed to micellar (Moens and Bagatolli 2007), the percentage of profilin bound to the artificial membranes increased in line with PIP2 concentration (Senju et al. 2017). Profilin will therefore be selectively recruited to areas of the cell with locally high PIP2 concentrations and these areas typically have higher rates of actin turnover (Janmey and Lindberg 2004).
Profilin, PIP2 and PLC
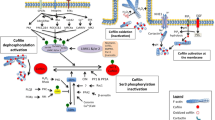
The participation of profilin in PIP2-mediated cell signalling events was also recognised decades ago with the observation that profilin inhibits phospholipase C-gamma (PLCγ) hydrolysis of PIP2 until PLCγ is phosphorylated and activated by receptor protein-tyrosine kinases (RTK) (Goldschmidt-Clermont et al. 1991). Alterations of membrane phosphoinositide levels by activation of PLC have profound effects on the activities of membrane and cytoskeletal proteins (McLaughlin et al. 2002). Hydrolysis of PIP2 by PLC converts it into the second messengers’ inositol 1,4,5-triphosphate (IP3) and diacetyl glycerol (DAG). DAG activates protein kinase C (PKC) cascades and IP3 induces Ca2+ intracellular fluxes (Fig. 1) (Janmey and Lindberg 2004). Intracellular calcium stores and overall PIP2/PIP3 balance are maintained by PLC enzymes. Alterations in the balance of PIP2 and PIP3 has a direct impact on cell fate as a high concentration of PIP2 tends to induce a bias towards differentiation while a high PIP3 concentration tends to promote cell proliferation and survival (Fukami et al. 2010).

Profilin interactions in the membrane. a Activation of the epidermal growth factor receptor (EGFR) by EGF phosphorylates phospholipase C (PLC) allowing the hydrolysis of profilin (Pfn)-bound PIP2 into diacetyl glycerol (DAG) and inositol 1,4,5-triphosphate (IP3) profilin. Both DAG and the Ca2+ fluctuations downstream of IP3 release activate phosphokinase C (PKC). b Activation of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) by growth factor (GF) activation of receptor tyrosine kinases (RTK), other agonists, and even extracellular profilin through an unknown receptor signal down the PI3K/Akt pathway, extracellular signal-related kinase (ERK) and mitogen-activated protein kinase (MEK) pathways, and also activate PKC. The activation of PKC phosphorylates profilin at S137. S137 phosphorylated profilin may interact with the p85 subunit of PI3K to downregulate signalling through the Akt-mTOR pathway. This results in the downregulation of the nuclear transcription factor NF-κβ and genes involved in cytoskeletal reorganisation. c The phosphatase and tensin homologue (PTEN) converts PI(3,4,5)P2 to PIP2, and the interaction with profilin prevents the ubiquitination and degradation of this enzyme, thus regulating the balance of PI(3,4,5)P2 and PIP2. PTEN is pro-apoptotic and the interaction with profilin promotes apoptotic pathways. The dashed line indicates that this may be an indirect effect
Profilin, PIP2 and PI3K
Phosphorylation of PIP2 by PI3K class 1 enzymes produces PIP3 that in turn is the source of PI(3,4)P2. These PPIs control different branches of PI3K signalling as well as interacting with profilin and other regulatory proteins (Li and Marshall 2015; Viaud et al. 2016). A high cellular concentration of profilin and its affinity for PIP2 was shown to be sufficient to account for its effect in protecting PIP2 from hydrolysis by PLC (Machesky et al. 1990). In resting human platelets 13.8% of cellular profilin is associated with plasma membrane PIP2 and this transiently increased to 26.6% after activation by thrombin (Hartwig et al. 1989). Changing the turnover and distribution of PIP2 by altering the expression of profilin will therefore have profound effects on virtually all cell signalling pathways with significant consequences for cancer and other disease states (Bae et al. 2009; Frantzi et al. 2016; Zoidakis et al. 2012; Zou et al. 2010).
PIP2 interactions at the plasma membrane
PIP2 recruits multiple ABP including profilin to the membrane creating a dynamic and complex milieu (Saarikangas et al. 2010; Senju et al. 2017). Some of these proteins are structural, such as the membrane deforming Bin, Amphiphysin Rvs proteins (BAR) proteins (Takenawa 2010) and tubulin (Popova et al. 2002). Similarly, other actin binding proteins such as cofilin and regulatory proteins such as Ak strain thymoma (Akt), 3-phosphoinositide-dependent protein kinase-1, (PDK1), PLC and guanosine exchange factors (GEF) such as Ras-related C3 botulinum toxin substrate (Rac) that have roles in the same signalling pathways as profilin are recruited and/or activated at the plasma membrane via PIP2 binding (Bezanilla et al. 2015; Senju et al. 2017; Takenawa 2010). Perturbations of PIP2 concentration will therefore have indirect effects on profilin (Fig. 1) This complexity illustrated by the interactions of profilin in signalling pathways, with PI(3,4)P2 and PTEN in PI3K pathways, and with members the non-canonical wingless/integration-1 (Wnt)/calcium pathway has made it difficult to isolate the role of profilin in regulating PPI interactions and cell fate in live cells.
Profilin modulates PI3K activity through interaction with PI(3,4)P2
The first in vivo example confirming the role of profilin in regulating other proteins at the cell membrane through PPIs was observed in MDA-MB-231 cells (Bae et al. 2010). These cells express low levels of profilin but have hypermotile phenotype in contrast to other cells where low profilin limits motility. In MDA-MB-231 cells, the depletion of profilin allows an increased accumulation of PI(3,4)P2 at the leading edge which is rescued by profilin overexpression (Bae et al. 2010). While PI(3,4)P2 is a very minor component of the plasma membrane, one important function is to serve as a membrane anchor for lamellipodin that in turn recruits Ena/VASP to promote actin filament assembly (Yoshinaga et al. 2012). Two explanations for an increased PI(3,4)P2 concentration due to profilin depletion are possible, and not mutually exclusive. The first is that profilin-PIP2 interaction dampens PI3K signalling by limiting access to PIP2. The second possibility is that lamellipodin competes for PI(3,4)P2 binding with profilin (Bae et al. 2010).
The PI3K pathway exerts its influence through the activation of the serine/threonine kinase Akt by PIP3. The downstream targets phosphorylated by Akt including extracellular signal-related kinase (ERK) and Mitogen-activated protein kinase (MEK) (Fig. 1) are involved in transcription, translation, proliferation, cell survival, cytoskeletal organisation and motility (Osaki et al. 2004; Wu et al. 2014). In certain disease states such as glomerular nephritis, diabetes and vascular disease, extracellular profilin is an agonist of the PI3K/Akt and Ras/MEK/ERK pathways (Fig. 1) (Caglayan et al. 2010; Jin et al. 2012; Tamura et al. 2000).
Stimulation of receptor tyrosine kinases (RTKs) or G-protein coupled receptors (GPCRs) by growth factors such as epithelial growth factor (EGF), platelet-derived growth factor (PDGF) or insulin activate both PI3K and PLC pathways and both have a role in motility (Wu et al. 2014). The direct involvement of profilin in the PI3K pathway was demonstrated in breast cancer cells as the depletion of profilin increased EGF-induced Akt phosphorylation downstream of PI3K and so enhanced membrane protrusion (Ding et al. 2014). For directional motility, the propagation of PI3K signals need to persist long enough to form stable protrusions and affect movement (Welf et al. 2012) so the effect of profilin on the timing of signal propagation is significant. Profilin depletion in MDA-MB-231 cells also correlates with an increase in the gelatinase, matrix metalloproteinase (MMP 9), a downstream transcription target of PI3K important in extracellular matrix degradation, and increased the secretion of pro-metastatic factors including VEGF (Ding et al. 2014). Overexpression of profilin in these cells reduced MMP9 transcription pointing to a possible role for profilin role in the nucleus. The hyperinvasive properties of MDA-MB-231 profilin knock down cells were diminished when PI(3.4)P2 levels were reduced. This demonstrates that the invasive phenotype of these cells due to the loss of profilin involves the PI3K/ PI(3.4)P2 signalling pathway (Ding et al. 2014).
PI3K signalling, PTEN and profilin
The plasma membrane spatial organisation allows signals to be restricted or propagated to generate signals downstream of receptor activation that are context-dependent (Grecco et al. 2011). This is seen in PI3K/Akt signalling where the role of different types of microdomains within the plasma membrane coordinate and regulate PI3K/Akt activity (Gao et al. 2011). How profilin alters PI3K signalling remains an open question. Recent in vivo experiments show that profilin affects events downstream of PI3K signalling and at the same time profilin itself is regulated by that pathway (Ding et al. 2014; Rizwani et al. 2014). When profilin is overexpressed in MDA-MB-231 cells, the PI3K/Akt pathway is downregulated (Jiang et al. 2015). One way this is achieved is through the upregulation of the PI3K/Akt antagonist PTEN (Das et al. 2009). By converting PIP3 to PIP2, PTEN acts as a tumour suppressor agent (Fig. 1) (Gericke et al. 2013). The study by Das et al. (2009) reported that increase in PTEN levels due to the upregulation of profilin was not related to an increase in PTEN transcription and speculated that profilin overexpression had a role in altering the stability of PTEN. This speculation has been validated in recent work that has shown that profilin protects PTEN from degradation by ubiquitination through binding to a PRD on PTEN (Zaidi and Manna 2016). Overexpression of profilin in MDA-MB-231 cells downregulates the PIP3/PI(3,4)P2 signalling axis by limiting the recruitment of pro-migratory proteins such as lamellipodin (Bae et al. 2010) and the scaffolding adaptor Tks5 that stabilises invadopodia (Valenzuela-Iglesias et al. 2015). While direct interactions of profilin and phosphoinositides are reported, indirect interactions are also possible. Intriguingly, a pool of immobile G-actin monomers bound to PIP3 is associated with profilin in dendritic spines in neurones. As actin competes with phosphoinositide binding on profilin, the link between profilin and PIP3 is possibly through a yet unidentified intermediary protein (Schlett 2017).
Profilin, PLC and Ca2+ signalling
The Wnt signalling pathways are involved in a variety of cellular process that include motility, polarity and organogenesis. One of the three main pathways of Wnt signalling, the non-canonical Wnt/calcium pathway acts downstream of PLC after co-stimulation of Dishevelled (Dsh) and a G-protein by Frizzled (Fz) to stimulate the release of calcium from the endoplasmic reticulum (Komiya and Habas 2008). The role of profilin in modulating calcium-dependent cell processes downstream of the hydrolysis of PIP2 by PLC pathways is demonstrated in bladder cancer where a reduction in profilin expression leads to a reduction of PLCβ4 levels and a downregulation of the non-canonical Wnt/Ca2+ pathway (Frantzi et al. 2016).
Bladder cancers that invade into the muscle tissue are inversely associated with profilin expressions levels (Frantzi et al. 2016; Zoidakis et al. 2012). In vitro experiments with the metastatic bladder cancer cell line T24M suggest that profilin is essential in linking integrin signalling to the non-canonical Wnt/Ca2+ pathway through PIP2 (Frantzi et al. 2016). Silencing profilin in these cells led to a 0.19 and 0.51-fold decrease in PLCβ4 and PLCβ2 expression, respectively. Other molecules involved in calcium-driven processes of the cell including members of the non-canonical Wnt/Ca2+ pathway were also downregulated. Silencing profilin expression in the T4M cell line led to cells that were hypomotile, had reduced levels of actin polymerisation and reduced adhesion to fibronectin. Explants of profilin silenced T24M cell lines in non-obese diabetic severe combined immunodeficient (NOD/SCID) mice formed smaller tumours than controls, and there was no evidence of metastasis from the profilin silenced tumours (Frantzi et al. 2016). The loss of motility in profilin-silenced T24M cells is a contrast to the hypermotile phenotype of MDA-MB-231 breast cancer cells when profilin expression is downregulated (Bae et al. 2009). The involvement of profilin in regulating cell motility by the non-canonical Wnt/Ca2+ pathway adds to the complexity of our understanding of profilin. It reinforces the importance of the context of profilin in each cell type in promoting or inhibiting tumour progression (Ding et al. 2012).
Protein ligands of profilin
When stimulated, growth factor receptors, cytokine receptors, integrins and cadherins all activate guanine exchange factors that in turn activate specific guanosine tri-phosphatases (GTPases) to mediate cellular responses that includes actin polymerisation (Parri and Chiarugi 2010). Profilin is a direct phosphorylation target of Rho-activated kinase (ROCK) (Fig. 2) (Shao et al. 2008) but is also linked to many of the GTPases of the plasma membrane through intermediate proteins. For the GTPase Ras-associated protein-1 (Rap1), the intermediate proteins that are profilin ligands include ARF-6 and RIAM (Kim et al. 2012). AF-6 mediates actin polymerisation in cell-cell junctions (Boettner et al. 2000) and RIAM is a ligand of both profilin and Ena/VASP and links integrin activation to actin polymerisation downstream of Rap1 (Lafuente et al. 2004). The Rho GTPase RhoA interacts with profilin, the formin mDia and also ROCK (Heasman and Ridley 2008; Lu et al. 2015; Shao et al. 2008; Watanabe et al. 1997). The Rho GTPase CDC42 also interacts with mDia (Heasman and Ridley 2008) and WASP (Sit and Manser 2011) while another Rho GTPase Rac1 activates the ARP2/3 complex downstream of WAVE2 and formin Dia2 (Sit and Manser 2011). Therefore, multiple signalling pathways that activate GTPases and are involved in the modulation of the actin cytoskeleton are able to recruit profilin to the membrane. The protein ligands of profilin mentioned herein is not exhaustive and does not consider the complexity of the signalling networks resulting from the cross-talk between different signalling pathways. However, what stands out from that brief overview is that profilin is intimately associated with the plasma membrane through a large number of ligands.

Profilin can be phosphorylated at S137 by Rho associated kinase (ROCK) downstream of vascular endothelial growth factor (VEGF) signalling, reducing actin polymerisation rates. The phosphorylation of profilin at S137 prevents the binding of proteins through proline repeat domains (PRD). Many of these proteins regulate actin polymerisation resulting in decreased actin polymerisation, cell survival and proliferation. The protein phosphatase 1 (PP1) enzyme regulates the balance between phosphorylated and de-phosphorylated profilin. ROCK may also have a role in regulating the interaction between profilin and the formin mDia VEGFR and the downstream kinase Src directly phosphorylate profilin at Y129, with opposite effect to the Rho/ROCK pathway. Y129 phosphorylated profilin has a higher affinity for actin, and accelerates actin polymerisation rates, leading to angiogenesis and vascular hypertrophy
Profilin has been described as having pro-apoptotic functions (Das et al. 2009; Yao et al. 2013; Zaidi and Manna 2016) and these pro-apoptotic functions of profilin seem to be linked to its binding to PRD proteins (Pecar Fonovic et al. 2013). The C-terminal Try139 residue is part of the PRD binding site (Bjorkegren-Sjogren et al. 1997). Truncating profilin at this site by cathepsin X carboxypeptidase cleavage reduces the anti-tumour activity of profilin by reducing the affinity of profilin for clathrin, decreasing clathrin-mediated endocytosis and increasing cell migration and invasion (Pecar Fonovic et al. 2013; Pecar Fonovic and Kos 2015). Profilin binds to PTEN through a PRD domain (Zaidi and Manna 2016) so cleavage of profilin by cathepsin X could also disrupt this interaction that promotes apoptosis by suppressing PI3K/Akt signalling.
Pathologies linked to the phosphorylation of Profilin
Profilin was first described as a phosphorylation target of protein kinase C (PKC) in 1988 (Hansson et al. 1988). The first identified phosphorylation site in profilin, S137 residue, is located near the proline repeat recognition domain at the C-terminal α-helix (Singh et al. 1996b). Phosphorylation of profilin is possible by serine-threonine or tyrosine kinases and databases reveal many theoretical or experimentally validated sites (Hornbeck et al. 2012). While some of these residues are phosphorylated by specific pathways, others such as S91, Y128 and S137 are the targets of multiple kinases at least theoretically. These are summarised in the supplemental material in Gau et al. (2016). One of these alternate pathways of S137 phosphorylation involves the activation of Rho-associated kinase (ROCK) downstream of Rho-GTPase (Shao et al. 2008). Profilin S137 phosphorylation alters the binding to PRD ligands and this is biologically relevant in cancer (Diamond et al. 2015) and Huntington disease (Shao et al. 2008). In the nucleus, S137 phosphorylation of profilin blocks apoptosis and promotes tumour progression (Fig. 3) (Diamond et al. 2015). WT profilin inhibits the formation of aggregates of the Huntington protein (Htt) but S137 phosphorylated profilin does not (Shao et al. 2008). Phosphorylation of other residues also alters profilin interactions and Y129 is important in endothelial cell migration and angiogenesis (Fan et al. 2012) and there is a potential link in apoptosis for the phosphorylation of Y139 (Pecar Fonovic and Kos 2015). Phosphorylation of other residues such as T89 that have been shown to occur in vivo may also have functional significance (Gau et al. 2016).

Interactions of profilin (purple oval) and actin (red oval) in the nucleus. a Profilin (Pfn) is small enough to pass freely through the nuclear pore (green polygons). b Profilin-actin is specifically transported from the nucleus by Exportin-6. c The interaction of profilin and undefined proteins containing PRDs is blocked by S137 phosphorylated profilin, leading to the inhibition of apoptosis and an increase in proliferation. d The phosphorylation of profilin at Y129 by vascular endothelial growth factor receptor (VEGFR) in response to ligand activation blocks the degradation of Hypoxia induced factor 1α (HIF-1α) by von Hippel-Lindau protein. The accumulation of HIF-1a in the nucleus upregulates angiocrine factors including VEGF that in turn promotes profilin phosphorylation, increased actin polymerisation and vascular hypertrophy. e When the transcription factors such as MKL and NF-κβ translocate from the cytoplasm to the nucleus they upregulate a number of genes involved in proliferation including profilin. This is an oversimplification of the relationship between profilin and MKL as there are indirect interactions with other factors (not shown) that regulate the relationship. f Profilin acts a corepressor or oestrogen receptor α (ERα) even in the presence of oestrogen (E2), preventing ERα from activating oestrogen response elements with the result that proliferative pathways are downregulated and apoptosis increases
The lipid and isoenzyme environment at the plasma membrane is important in PKC phosphorylation of S137 at the C terminal as in vivo and in vitro experiments established that the pathways involved in S137 phosphorylation were downstream of PI3K (Sathish et al. 2004; Singh et al. 1996a). Profilin phosphorylation was found maximal with PKCζ in the presence of PIP2 even in the absence of conventional agonists such as DAG. The stoichiometry suggested that only one residue of profilin was phosphorylated by PKC. The proposed mechanism of PI3K in this pathway is that upon ligand stimulation, profilin-bound PIP2 is phosphorylated to PIP3, releasing profilin, activating PKCζ that in turn phosphorylates profilin (Vemuri and Singh 2001). A feedback loop may exist where profilin interaction with the regulatory p85α subunit of PI3K via a PRD, increases PI3Kinase activity, activating PKC that in turn phosphorylates profilin, leading to a downregulation of PI3K activity (Fig. 1) (Rizwani et al. 2014; Vemuri and Singh 2001). The other effect of profilin S137 phosphorylation is the inactivation of profilin functions that depend on PRD binding (Diamond et al. 2015).
Mechanisms to de-phosphorylate profilin at S137 are significant as the switch between phosphorylated and de-phosphorylated profilin is important in determining cell fate (Diamond et al. 2015) and protein phosphatase-1 (PP1) specifically targets phosphorylated profilin S137 (Shao and Diamond 2012). The relevance of this is seen in a breast cancer model using MDA-MB-231 explants in mice. Tumours from cells with the phosphomimetic profilin S137D mutation were larger than controls, or tumours overexpressing wild-type profilin or the S137A phospho-resistant mutation (Diamond et al. 2015). When tagged with a nuclear location sequence, S137D mutant cell lines formed more colonies than cells overexpressing profilin or the S137A mutation demonstrating that the phosphorylation status of nuclear profilin determines if it acts in inhibiting or promoting tumours. Profilin is known to induce cell-cycle arrest and have pro-apoptotic functions (Janke et al. 2000; Wittenmayer et al. 2004; Zou et al. 2007, 2010). The subcellular location of profilin is important as the nuclear but not cytoplasmic S137D profilin in MDA-MB-231 cells blocked the pro-apoptotic pathways and also interfered with cell cycle arrest (Diamond et al. 2015). Further experiments showed in the cytoplasm that profilin S137A was more effective at rescuing profilin null chondrocytes than WT or S137D and suggests that it is the switch toggling between phosphorylated and de-phosphorylated states that regulates proliferation and survival through interactions with proteins with PRDs (Diamond et al. 2015).
This is similar to results obtained in MCF-7 cells where cells treated with the PI3K agonist phorbol 12,13 dibutyrate (PDBu) to stimulate PKCζ activity phosphorylate wild-type profilin increased motility and the invasive phenotype activity (Rizwani et al. 2014). These cells showed upregulation of MMP2 and MMP9. The paper by Rizwani et al. (2014) showed that phosphorylated wild-type profilin, and profilin mutations inhibiting binding to actin (R74E) or PRD mutation (H133S), translocates to the nucleus. Unfortunately, this paper assigned a phosphomimetic function to S137A mutation rather than a phospho-resistant one that places a query over the accuracy of the conclusions they draw from their results with this mutation. Their results however showed that MCF-7 S137A cells had reduced migration, formed fewer colonies in soft agar, were less invasive, and that increased PKCζ activity did not induce profilin translocation into the nucleus. This is consistent with the results from Diamond et al. (2015) who demonstrated that phospho-resistant profilin S137A mutation inhibited cell cycle arrest and sensitised cells to apoptosis.
The role of phosphorylation in regulating profilin activity through altering its binding to proteins with PRDs in the nucleus is largely unknown. Profilin:actin complexes are removed from the nucleus by exportin 6 (Stuven et al. 2003) and profilin with reduced actin binding with a R74E mutation is predominantly nuclear in distribution (Rizwani et al. 2014). Profilin with a H133S mutation that disrupts PRD binding has a similar cellular distribution to wild-type profilin, but phosphorylation increases nuclear concentration in R74E, H133S and wild-type profilin (Rizwani et al. 2014). Therefore, the change in phosphorylated profilin distribution from the cytoplasm to the nucleus seems to be specific to phosphorylation rather than the inactivation of PRD binding as such and is independent of exportin 6.
Profilin dimers, but not tetramers, are resistant to phosphorylation by PKC at S137 (Korupolu et al. 2009). In the dimers, PRD binding is abolished, and the S137 residue is hidden as the monomers interface through the carboxy terminals. Tetramers have reduced PRD binding but are preferentially phosphorylated over monomers (Korupolu et al. 2009). Tetramers and dimers have been reported in cell extracts (Babich et al. 1996; Skare et al. 2003) and tetramers in solution (Rennella et al. 2017) but the function of oligomerization remained elusive. The studies to determine if oligomerization is biologically relevant in altering S137 phosphorylation are still to be carried out, but if validated, will add another layer to the regulation of profilin. Profilin oligomerization is promoted under oxidizing conditions (Mittermann et al. 1998), and as profilin is linked to the oxidative metabolism of whole organisms (Pae and Romeo 2014) and individual cells (Li et al. 2013; Yao et al. 2014), it will be fascinating to see further developments in the physiological effect of oligomerization of profilin. Profilin is upregulated by PKC/NF-κB in response to ROS (Li et al. 2013) and as it is a phosphorylation target of PKC, it seems possible that oligomerization may be an additional factor to consider in profilin mediated pathologies.
The biological significance of profilin S137 phosphorylation is also seen in conditions such as Huntington disease (HD) and spinobulbar muscular dystrophy (SBMA) (Shao et al. 2008) and in breast cancer (Diamond et al. 2015; Rizwani et al. 2014). The conditions HD and SBMA are the result of agglutination of Huntington protein (Htt) and androgen receptor (AR) respectively. In HEK293 cells, both wild-type profilin and phospho-resistant S137A profilin inhibited agglutination of AR and Htt. Similarly, chemical inhibition of ROCK phosphorylation of profilin prevented aggregates forming. In contrast the phosphomimetic S137D mutation led to aggregate formation of both AR and Htt. The S137D mutant had a moderately lower affinity for G-actin but did not bind to the PRD of Htt. The reduction in G-actin binding alone was sufficient to promote aggregation of AR, but Htt aggregation was primarily regulated by PRD, and the reduction in actin binding was an additional mechanism in promoting pathogenic changes (Shao et al. 2008).
Profilin activity is also regulated by tyrosine phosphorylation at the Y129 residue. This forms part of the actin binding site, and phosphorylation increased the affinity for actin but more importantly increased the rate of nucleotide exchange, accelerating actin polymerisation rates (Fan et al. 2012). At the leading edge of the cytoplasm of endothelial cells, Y129 phosphorylation occurs after stimulation of VEGFR2 by the growth factor VEGF-A (Fig. 2). Both VEGFR2 and its downstream target the kinase Src phosphorylate profilin at Y129 in response to VEGF-A. The VEGFR2-Src pathway is particularly relevant to angiogenesis and endothelial chemotaxis and cell migration in adults, with phosphorylated profilin promoting tissue repair after injury ischemia or wounding. This pathway is not relevant developmentally as VEGF stimulation of PI3K-Akt pathways contributed to endothelial cell chemotaxis in angiogenesis through PI3K/Akt pathways and was found to be responsible for developmental angiogenesis. Thus, the upregulation of profilin activity in angiogenesis through Y129 phosphorylation in adults is an example of specific regulatory mechanism of profilin (Fan et al. 2012).
The Y129 phosphorylation has metabolic implications not directly related to actin dynamics. In the brain tumour glioblastoma multiforme (GBM), profilin Y129 phosphorylation is part of a feed-forward mechanism in which endothelial cells release angiocrines in response to mediators released by the tumour to drive aberrant vascularization through the induction of hypoxia-inducible factor (HIF) even under normoxia (Fan et al. 2014). VEGF/Src phosphorylation of the Y129 residue allows profilin to form a complex with von Hippel-Lindau protein (VHL), preventing VHL from degrading HIF-1α. Accumulation of the transcription factor HIF-1α induces upregulation of angiogenic factors including VEGF-A, leading to further profilin phosphorylation (Fig. 3). When glioma cells were injected into mice with a conditional knock in phosphorylation-resistant mutation Y129F, the vasculature of the resulting tumours was significantly less dense than tumours from wild-type profilin. The results suggest that hyperplasia of GBM tumour microvascular system is driven by and dependent on profilin phosphorylation in response to angiocrine factors secreted by the tumour (Fan et al. 2014).
The distribution of tyrosine-phosphorylated profilin also changes under different stimuli. Tyrosine-phosphorylated profilin is present only in the cytoplasm of promyelocytic leukaemia cell line HL-60 under basal conditions, but treatment with etoposide to induce apoptosis caused it to appear in the nucleus as well. It was absent in both compartments when the cells were treated with retinoic acid to induce differentiation (Navakauskiene et al. 2004).
Profilin is involved in other pathways linked to apoptosis. In pancreatic adenocarcinoma, profilin functions as a tumour suppressor by inducing apoptosis through a mitochondrial pathway. A major deacetylase in the mitochondria, sirtuin 3 (SIRT3), is upregulated when profilin is overexpressed (Yao et al. 2014). SIRT3 functions to reduce oxidative stress and promote the tricarboxylic acid cycle. Loss of SIRT3 leads to reprogramming metabolic pathways towards glycolysis leading to cell proliferation and tumour progression by its interaction with HIF-1a. As in relation to GBM, profilin Y129 phosphorylation was associated with the progression of GBM by interfering with HIF-1α clearance and was anti-apoptotic (Fan et al. 2014). Therefore, the downregulation of profilin expression or inactivation by Y129 phosphorylation are anti-apoptotic.
Profilin expression
Control of profilin expression
Profilin acts as a pivot point to integrate signalling inputs and determines actin dynamic outputs (Davidson and Wood 2016). Therefore, changes in profilin expression levels will have significant effects on cell fate. This can be seen on a proteomic level where changing expression levels of profilin changes the expression of a host of other proteins (Coumans et al. 2014; Frantzi et al. 2016). Synthesis of profilin is itself sensitive to perturbations of actin networks. Disruption of cell to cell contacts and the underlying actin networks in an epithelial cell line activated a protein kinase cascade to phosphorylate the 40S ribosomal protein S6 which regulates many of the genes involved in cell survival and proliferation including profilin, VASP and vinculin (Quinlan 2004). This is interesting since the PI3K signalling pathway is seen as the major activator of S6 in oncogenesis (Bader et al. 2005). Disruption of actin cytoskeleton provides a route for S6 activation independently of PI3K/Akt to regulate profilin expression (Quinlan 2004). The paradox of profilin, an essential protein in the cell promoting motility and cell cycle progression yet being pro-apoptotic has been well noted (Diamond et al. 2015; Ding et al. 2012; Ding et al. 2014). As with every other aspect of profilin, the context of subcellular location, level of expression and modification is critical in determining how profilin can be involved in apoptosis. While several of these aspects of profilin have already been discussed in relation to PTEN suppression of the Akt pathways (Das et al. 2009), the role of microRNAs (miRNA) in regulating profilin expression and the activation of metabolic pathways is also an emerging area of interest.
The regulation of profilin expression is complex and the list of miRNA and transcription factors involved is growing. One report suggested that at least twenty miRNA and 27 transcription factors are involved in profilin regulation (Schoppmeyer et al. 2017). The database http://www.mirdb.org predicts that profilin could be the target of forty five miRNAs (Chen and Wang 2020; Liu and Wang 2019) while http://mirtarbase.cuhk.edu.cn (Chen and Wang 2020; Chou et al. 2018) list experimental evidence of over fifty interactions between profilin and miRNAs. Of the miRNAs that regulate profilin expression identified so far, miR-182 and miR-96 are associated with pancreatic (Yu et al. 2012) and breast cancers (Guttilla and White 2009). Downregulation of profilin in MDA-MB-231 cells has been reported as a result of aberrantly expressed miR-182 (Liu et al. 2013). Silencing miR-182 and miR-96 increased the expression of the transcription factor forkhead box 01 (FOX01) which is downregulated in breast cancer tissue (Guttilla and White 2009). Profilin is a target of FOX01 (Lin et al. 2012), so overexpression of miR-182 and miR-96 downstream of PI3K/Akt signalling results in the suppression of profilin and increase in proliferation (O'Bryan et al. 2017). In addition, miR-96 suppresses FOX03a, a transcription factor of the cyclin-dependent kinase inhibitor p27kip1 important in regulating the cell cycle G1/S transition (Lin et al. 2010). The links between profilin and p27 are complex as profilin overexpression in MDA-MB-231 cells increased the stability of p27kip1 inducing cell cycle arrest at G1 and increased apoptosis (Zou et al. 2010). The Akt pathway suppresses p27, and as profilin overexpression upregulates PTEN, inhibiting Akt (Das et al. 2009), it is a potential mechanism by which profilin acts as tumour suppressor in these cells (Zou et al. 2010). In conditions of metabolic stress such as serum starvation when the AMP:ATP ratio increases, AMP-activated protein kinase (AMPK) is stabilising p27 by T198 phosphorylation. In MDA-MB-231 cells, overexpression of profilin stimulates AMPK activation, promotes the reversion of the mesenchymal phenotype to an epithelial type and leads to cell-cycle arrest (Jiang et al. 2015).
In the lung cancer cell line A549, the cytoplasmic or nuclear location of profilin, cofilin and VASP can be altered by changing the expression of miR-17-92 by docosahexaenoic acid (DHA) (Ali et al. 2016). DHA treatment inhibited the phosphorylation of VASP by PKC at S157 downstream of cAMP signalling but promoted its phosphorylation at S239 by PKG. VASPS157 was associated with profilin and is pro-metastatic while VASPS239 was associated with cofilin and was pro-apoptotic. Attenuating miR-17-92 by DHA reduced VASPS157, altered the location of profilin from the cytoplasm to the nucleus, reduced F-actin content in the cell and increased apoptosis (Ali et al. 2016).
In hepatocellular carcinoma (HCC), miR-19a-3p is implicated as an inhibitor of profilin expression (Wang et al. 2019). The study by Wang et al. (2019) showed that there was an inverse relationship between levels of miR-19a-3p and profilin in clinical samples, and that low levels of profilin expression in tumours compared with surrounding tissues were associated with a poor prognosis. They also demonstrated that profilin is a direct target of miR-19a-3p. Overexpression of profilin in the hepatic cancer cell lines HCLM3 and HepG2 cell lines inhibited proliferation, migration and the development of invasive characteristics, and also prevents the cells from undergoing epithelial-mesenchymal transition (Wang et al. 2019). This is in contrast to a study in the murine cell line 4 T1, used as a model for triple negative breast cancers where the overexpression of miR-19a-3p, or its intra-tumour injection in explants were tumour-suppressive (Yang et al. 2014). In that experiment, low miR-19a-3p expression correlated with high VEGF/STAT3 activity. There is no published information on profilin expression levels in the 4 T1 cell line, but one experiment showed that profilin expression decreases when these cells were treated with a plant extract that disrupts glycolytic pathways and underwent apoptosis independently of mitochondrial pathways (Hernandez et al. 2014). Targeting miR-19-3p to regulate profilin expression in cancer to promote tumour regression may still be of therapeutic interest, but the type of cancer would need to be carefully evaluated in order to determine if profilin should be up- or downregulated.
The precise mechanisms that regulate profilin expression are complex, and there are cell line differences that are important in determining if changes to profilin expression drive a cell towards either survival or apoptosis. One possibility is that the status of other regulatory proteins in the cell such as the transcription factor p53 may determine the fate of the cells.
Profilin, apoptosis and p53 in cancer cell lines
Profilin is known to regulate the expression of p53 (Yao et al. 2013; Zaidi et al. 2016) and approximately half of human tumours contain mutations in the tumour suppressor p53 resulting in downregulation of the intrinsic apoptotic pathway (Yao et al. 2013). One of the differences between the hepatic cancer cell lines HCLM3 and HepG2, the breast cancer MDA-MB-231 cell line and 4T1, is the presence or mutation of the transcription factor p53. The 4T1 cell line lacks p53 expression (Yerlikaya et al. 2012) while HCLM3 and HepG2 cell lines express wild-type p53 (Leroy et al. 2014) and MDA-MB-231 cells contain a missense mutation of p53 that promote cell survival under conditions of serum starvation (Hui et al. 2006).
In the breast cancer cell line MDA-MB-468 and the pancreatic cancer line PANC-1 containing the same point mutation p53R27H, the overexpression of profilin by the drug staurosporine (STS) led to a restoration of apoptotic function to this mutant (Yao et al. 2013). The mechanism these authors propose is that STS facilitates the translocation of profilin to the nucleus. The interaction with profilin increases p53 mRNA expression, and stabilises p53, which then moves to the cytoplasm. Downstream of STS stimulation, profilin promotes the phosphorylation of p53 in the cytoplasm. Phosphorylated p53 locates to mitochondrial membrane, altering the membrane potential that initiates the caspase-9 apoptotic cascade and cell death. Profilin has been shown to act synergistically with chemotherapy drugs vinblastine, doxorubicin and benzofuran that upregulate p53 expression to increase cell death by apoptosis (Zaidi et al. 2016). As a regulator of p53 expression and function, profilin has a profound effect on major apoptotic pathways. Chemicals such as the small molecule triphostin A9 in MDA-MB-231 cells (Joy et al. 2014), all trans retinoic acid (Wu et al. 2006) and guttiferone K (Shen et al. 2016) in hepatocarcinoma upregulate profilin expression and inhibit cell proliferation and migration.
The study by Yang et al. (2014) did not examine the level of profilin expression in 4T1 cells, but under conditions of low miR-19a-3p expression, it would be expected to be high (Wang et al. 2019). Upregulation of p53 by profilin inhibits cell cycle progression in MDA-MB-231 cells by suppressing NF-κΒ as growth factors such as IL-8 and VEGF which are dependent on the activation of NF-κΒ (Zaidi et al. 2016). This suggested that profilin may act as a pro-apoptotic molecule in the presence of functional p53. However, in renal cell carcinomas, where high profilin expression is linked to tumour progression (Minamida et al. 2011), this is not the case as the renal cell carcinoma cell lines 769P, ACHN and Caki1 show wild-type p53 (Leroy et al. 2014). The elusive mechanisms that allow high levels of profilin to function to promote either apoptosis or cell cycle progression in different cell types still remains to be determined.
The upregulation of p53 and cell fate due to changes in cell culture conditions can be modelled in MDA-MB-231 cells. In MDA-MB-231 cells, suppression of profilin expression promotes a hypermotile phenotype that allows cells to migrate across the endothelial barriers but this same phenotype is detrimental to the establishment of metastatic tumours (Ding et al. 2014). Serum starvation provides a stress that mimics the nutrient poor niche of tumours that can promote the malignant potential of cancer cells (Tavaluc et al. 2007). One of the hallmarks of cancer cells is the ability to sustain proliferative signalling independently of extrinsic growth factors (Hanahan and Weinberg 2011). Serum starvation of MDA-MB-231 has been shown to result in the upregulation of EGFR and activate a switch in metabolism whereby phospholipase D2 (PLD2) is activated by Janus Kinase-3 (JAK3) (Ye et al. 2013). The activation of PLD2 by EGFR and JAK3 is a survival mechanism that corresponds to the increased migration and invasive behaviour that allows these cells to move from a solid tumour. The elevated activity of PLD2 suppresses apoptosis in serum starved MDA-MB-231 cells by stabilising the p53 mutant that is present in this cell line (Hui et al. 2006). Mutations in the tumour suppressor p53 result in downregulation of the intrinsic apoptotic pathway (Yao et al. 2013). The association of profilin and PTEN in the membrane has been shown to downregulate the PI3K/Akt signalling and cell survival pathways (Zaidi and Manna 2016). In MDA-MB-231 cells, profilin overexpression increases the sensitivity of the cells to apoptosis while a reduction of profilin expression allows cell cycle progression (Zaidi and Manna 2016).
Profilin expression levels in breast cancer
There are conflicting reports on the effects of profilin expression levels both clinically and in cell lines. The initial finding that aggressive and metastatic breast cancer cells under-expressed profilin was based on immunochemical staining of 40 clinical samples and low profilin levels in tumourogenic cell lines (Janke et al. 2000). Increased profilin expression in the invasive breast carcinoma cell line BT474 resulted in a reduction of the migration of these cells (Roy and Jacobson 2004) with similar results in MDA-MB-231 cells (Bae et al. 2010). This contrasts to a study by Rizwani et al. (2014) who showed that in the population studied, primary breast cancer tumours had high profilin expression. That study showed that in the cell line MCF7 stably transfected with profilin, motility increased with profilin expression. This demonstrates the importance of understanding the molecular context of profilin (Ding et al. 2012) as there are differences in how these cells are regulated (Noh et al. 2011). BT474 and MCF7 cell lines are from basal tumours and while both are oestrogen and progesterone receptor positive (ER+ PR+), BT474 has a high level of the human epidermal growth factor receptor 2 (HER2+) but MCF7 is HER2-. MDA-MB-231 is a triple negative (ER-, PR-, HER2-) breast cancer of luminal origin (Neve et al. 2006). In one study comparing MCF7 and MDA-MB-231 cell lines, both synthesized similar amounts of mRNA PTEN, but the ERα+ MCF7 cell line had an accelerated degradation and reduced phosphorylation of PTEN corresponding to an increase in Akt activation. In the MDA-MB-231 cells, the PTEN levels were higher than in the MCF7 cells and the PI3K/Akt pathways downregulated (Noh et al. 2011). The difference of stability of PTEN between the two cell lines may be due to a difference in pathways initiated downstream of ERα in MCF7 cells, as the downregulation by profilin of nuclear factor κΒ (NF-κB) in MDA-MB-231 cells was mediated by the stabilisation of PTEN by profilin (Zaidi and Manna 2016). Overexpression of profilin in MDA-MB-231 cells increased the membrane association of both proteins in the plasma membrane and sensitised these cells to chemotherapy drugs which raises the possibility of therapeutically modifying profilin expression in conjunction with breast cancer treatment (Zaidi and Manna 2016; Zaidi et al. 2016).
It is interesting to note that in breast cancer the Profilin 2 isoform is expressed in breast epithelium and is differentially expressed in human breast tumours (Mouneimne et al. 2012). These authors demonstrated that knocking down Profilin 2 in the SUM159 breast cancer cell line has an invasive but not metastatic phenotype promoting faster migration and an increase in the protrusive activities. The changes in the behaviour of these cells were linked to the Ena/VASP protein EVL that has a higher affinity for Profilin 2 than for Profilin 1 and tends to favour F-acting bundling. Clinically, low expression of EVL corresponded to poor prognosis, but both high and low Profilin 2 expressions were associated with poor outcomes. This suggests that Profilin 2 can promote an invasive phenotype in different contexts. The study by Mouneimne et al. (2012) is an important reminder that even though Profilin 2 is not present in high concentrations in most cells (Funk et al. 2019), it may contribute to pathologies such as breast cancer progression.
Profilin expression and inflammation
Profilin has been linked to inflammatory disease processes (Romeo et al. 2013). One of the inflammatory pathways regulated by the levels of profilin expression is the activation of NF-κB by reactive oxygen species (ROS) (Fig. 4) (Li et al. 2013). In endothelial cells, advanced glycation end products (AGEs) produced in patients with long-term hyperglycaemia in diabetes, results in NF-κB activation and the upregulation of profilin (Li et al. 2013). The upregulation of profilin is initiated by stimulating the AGE receptor (RAGE) to produce excess reactive oxygen species (ROS), activating PKC and NF-κB pathways. Inhibiting either PKC or NF-kB reduced the protein and mRNA level of profilin expression. Silencing profilin in HUVECs did not affect PKC or NF-kB activity but it did protect the cells from abnormalities such as stress fibre formation, it reduced the production of intercellular adhesion molecule-1 and increased the synthesis of nitrous oxide (NO) (Li et al. 2013). Silencing profilin also protects HUVECs from the effect of AGEs and ROS by decreasing Rho/ROCK1 pathways (Li et al. 2018). In diabetic retinopathy, profilin is implicated in the development of microvascular endothelial dysfunction through RAGE signalling by multiple pathways downstream of RhoA, including NF-κB (Lu et al. 2015). In these examples of endothelial cell dysfunction, an increase in profilin expression was related to an increase in pathology. Profilin haploinsufficiency protects mice from the systemic effects of a high-fat diet including insulin resistance (Romeo et al. 2013). Nutrition is also linked to profilin expression and actin polymerisation though FOXO. In Drosophila, activation of insulin receptor initiates signalling down the insulin/insulin-like pathway. This relieves the repression of FOXO on the profilin homologue chickadee (chic), promotes the increase in chic/profilin expression and actin polymerisation (Ghiglione et al. 2018) showing the link between profilin, diet and inflammation. In the context of the inflammatory microenvironment of tumours, a basal high level of NF-κβ is maintained (Zaidi et al. 2016). The overexpression of profilin increases the interaction of profilin with PTEN in MDA-MB-231 cells preventing the phosphorylation cascade downstream from Akt (Das et al. 2009). This leads to the suppression of NF-κB and inflammatory pathways (Zaidi and Manna 2016) and demonstrates that profilin can have pro- or anti-inflammatory effects on cells depending on the context.

Advanced acetylation products (AGEs) activate the receptor for AGE (RAGE) and produce reactive oxygen species (ROS) that activate PKC/NF-κβ pathways that increase profilin expression. Angiotensin II (Ang II) activates the Ang II type 1 receptor (AT1) stimulating the Janus kinase/signal transducer and activator of transcription (JAK/STAT) and PI3K/Akt/ERK/eNOS pathways upregulating profilin expression. The increase in ROS and reactive nitrogen species (RNS) results in the nitration of profilin at T139 increasing actin polymerisation and contributing to vascular hypertrophy
Control of Profilin expression levels in other signalling pathways
Multiple feedback loops connect profilin expression levels to signalling pathways. This is seen in differentiating osteoblasts where the cytokine bone morphogenic protein (BMP) suppresses profilin mRNA while profilin is a negative regulator of BMP. Indeed, knockdown of profilin increased the activity of BMP (Lin et al. 2016). As BMP signalling is dependent on focal adhesion kinase (FAK) activation by the formation of focal adhesions (Tamura et al. 2001), the feedback loop between profilin and BMP gives profilin a role in connecting cell-matrix interactions to osteoblast differentiation (Lin et al. 2016). An actin/megakaryoblastic leukaemia (MKL)/serum response factor (SRF) transcription pathway is regulated by feedback loops involving profilin (Joy et al. 2017). Actin polymerisation triggers MKL release from G-actin that holds MKL in an inactive state. Once activated, MKL can shuffle between the nucleus and cytoplasm. In the nucleus, MKL activates SRF where the transcription targets include MKL, SRF and cytoskeletal structural and regulatory proteins including cofilin. Profilin protein concentration increases when MKL-STAT (signal transducer and activator of transcription) pathways are upregulated independently of SRF. The functional connections between profilin, MKL and SRF is seen as overexpression of profilin elevated MKL and SRF levels in MDA-MB-231 cells whereas profilin depletion led to their reduction (Fig. 3). Depletion of profilin by the knockdown of MKL led to a hypermotile phenotype in these cells that was rescued by the overexpression of profilin. As well as a role in modulating transcription pathways, profilin has a role in activating MKL by competing for G-actin and promoting F-actin polymerisation. In this way, profilin is both a positive regulator of MKL/SRF at the same time as being regulated by MKL itself (Joy et al. 2017).
Extracellular profilin may have autocrine/paracrine functions
MKL has a function in cellular retention of profilin as the MKL knockdown or its inhibition resulted in increased profilin released into extracellular space, decreasing profilin cellular protein concentration without altering profilin mRNA levels in MDA-MB-231 cells (Joy et al. 2017). Finding a specific pathway that increases extracellular profilin is significant as profilin has been found in the exosomes (Thery et al. 2001), the secretomes of pancreatic (Gronborg et al. 2006) and colon cancer (Ji et al. 2009), in serum (Caglayan et al. 2010) and urine (Zoidakis et al. 2012). While no specific profilin receptor has yet been identified (Caglayan et al. 2010; Pae and Romeo 2014; Tamura et al. 2000), extracellular profilin in glomerulonephritis triggers PKC activation inducing the transcription factor AP1 to increase DNA synthesis of genes involved in cell growth, differentiation and apoptosis (Tamura et al. 2000). In coronary artery disease, the level of serum profilin corresponded to the degree of aortic atherosclerosis and induced chemotaxis and proliferation through PI3K and ERK signalling cascades in cultured vascular smooth muscle cells (VSMCs) (Caglayan et al. 2010). This suggests that profilin released from apoptotic or intact endothelial cells acted as autocrine/paracrine factors to increase signalling cascades in VSMCs. In this experiment, a high-fat diet increased profilin expression in endothelial cells providing a link between profilin expression, nutrition and metabolism pathways to inflammatory responses (Caglayan et al. 2010). Intracellular profilin concentration in endothelial cells is regulated by MKL/SRF signalling as chemical inactivation of MKL by the small molecule CCG-1423 results in decreased profilin concentration and inhibited endothelial cell migration (Gau et al. 2017). As downregulation of MKL increases extracellular profilin in MDA-MB-231 cells (Joy et al. 2017), it seems possible that the profilin exported or secreted from endothelial cells to activate VSMCs (Caglayan et al. 2010) may also be regulated by MKL (Gau et al. 2017).
Extracellular profilin as an anti-inflammatory agent
In the context of atherosclerosis discussed above, extracellular profilin was reported to be pro-inflammatory but in the context of embryo implantation into the uterus profilin had the opposite effect (Menkhorst et al. 2017). Endometrial stromal cells undergo a process of differentiation called decidualisation that is hormonally regulated. If a blastocyst implants, the decidua further differentiates to become the maternal part of the placenta (Menkhorst et al. 2012). Factors released by endometrial stromal cells hormonally induced to differentiate into decidualised cells promoted the secretion of profilin by extravillous trophoblast (EVT) cells (Menkhorst et al. 2012). The secreted profilin has a role in facilitating further differentiation of endometrial stoma cells into the decidua in part by downregulating the pro-inflammatory cytokines downstream of arachidonate 5-lipoxygenase in both decidual cells and macrophages. This cross-talk between EVT, decidua and macrophages enables the blastocyst to implant into the uterus and the development of a healthy placenta. The extracellular profilin secreted by EVT is a key component of decidualisation (Menkhorst et al. 2012; Menkhorst et al. 2017). Defects in decidualisation reduce the amount of cross-talk between the decidual and EVT, reduce profilin secretion and change the cytokine profile to a pro-inflammatory environment associated with pre-eclampsia (Menkhorst et al. 2012; Menkhorst et al. 2017).
It is clear that extracellular profilin has physiological roles that go beyond its function in actin polymerisation and that specific factors control its release from the cells. In the breast cancer cell line MDA-MB-231 and in endothelial cells, this appears to be regulated by MKL/SRF pathways (Gau et al. 2017; Joy et al. 2017). Downregulation of profilin downstream of MKL knockdown in MDA-MB-231 cells led to a hypermotile phenotype (Joy et al. 2017) but in endothelial cells the decreased profilin levels led to inhibition of motility (Gau et al. 2017), emphasising the importance of cellular context as the downregulation of the same pathway has such opposite effects in different cell types. Extracellular profilin is associated with pro-inflammatory responses in vascular disease (Caglayan et al. 2010) but in the developing placenta, promotes an anti-inflammatory response that is essential for embryo implantation and maturation of the decidua (Menkhorst et al. 2017).
Profilin in oxidative stress pathways
Profilin contributes to the pathology of a number of conditions where cells are under oxidative stress (Fan et al. 2014; Jin et al. 2012; Li et al. 2013). One of the markers of oxidative damage in cells is the appearance of tyrosine nitrated proteins (Starr et al. 2011). Profilin and NO metabolism are linked as peroxynitrite, the product of NO and superoxide results in the nitration of the C-terminal T139 residue of profilin (Fig. 4) (Kasina et al. 2005). Overproduction of NO changes its roles from an essential signalling molecule with anti-inflammatory functions to a pro-inflammatory mediator (Sharma et al. 2007). The concentration of nitro-profilin increased in vitro with the level of peroxynitrite (Kasina et al. 2005) and in vivo with (iNOS) downstream from PI3K activation in platelets (Kasina et al. 2006). In vitro, nitration of profilin at T139 increased its affinity to PRD twenty-fold, decreased the critical concentration of actin from 250 nM to 150 nM but had no effect on PIP2 binding. Nitration will therefore profoundly alter profilin-PRD interactions and alter actin dynamics (Fig. 4) (Kasina et al. 2005). The result of this is seen in vivo in mice with systemic inflammation induced by lipopolysaccharide (LPS), superoxide released by neutrophils reacts with NO to produce peroxynitrite which breaks down to toxic ROS and reactive nitrogen species (Starr et al. 2011). Aged mice were more susceptible to oxidative/nitrosative damage than young ones and expressed less of the protective superoxide dismutase but more iNOS. Consequently, aged mice had higher levels of nitrated proteins including profilin in lung tissue. This suggested that loss of function to profilin and several other proteins involved in regulating the actin cytoskeleton due to nitration contributed to the pathology (Starr et al. 2011).
Since the link between the overexpression of profilin, oxidative stress, increased vascular hypertrophy and hypertension was made (Moustafa-Bayoumi et al. 2007), work has been done in understanding the pathways by which profilin contributes to the pathology and contributes to endothelial nitrous oxide synthetase (eNOS) uncoupling. One pathway is mediated by activation of angiotensin (Ang) II type 1 receptor (AT1) by Ang II through Akt-ERK1/2 pathways. In rats, the activation of AT1 leads to increased profilin and eNOS expression, a decrease in angiotensin converting enzyme 2 (ACE2) activity, and an increase in of the activation of NADPH to produce superoxide that reacts with NO to produce peroxynitrite that contributes to vascular injury by oxidative and nitrosative reactions (Jin et al. 2012). The role of profilin in oxidative stress and eNOS uncoupling is further demonstrated in comparing wild type (WT) to ACE2 KO mice where the level of oxidative damage, profilin expression and downregulation of ACE2 were related to the dose of Ang II treatment (Jin et al. 2012). Treating the ACE2 knockout (KO) mice with Ang II further increased profilin expression, signalling through PI3K and Akt-ERK pathways to increase eNOS expression, NADPH oxidase activity to form superoxide and peroxynitrite, and increased inflammatory cytokines (Jin et al. 2012). In human umbilical artery smooth muscle cells, Ang II induced profilin expression was blocked by pharmacological treatment inhibiting either the ERK1/2 or the c-Jun N-terminal kinase (JNK) pathways abolishing profilin upregulation (Zhong et al. 2011). The Janus kinase 2 (JAK2)/STAT2 pathway has also been implicated in vascular hypertrophy (Cheng et al. 2011). In cultured rat aortic smooth muscle cells, treating cells with Ang II induced profilin expression in a dose-dependent manner, and silencing profilin reduced cell proliferation. Blockading JAK2/STAT3 pathway pharmacologically inhibited the profilin expression mediated by Ang II (Cheng et al. 2011).
Treating both epithelial and smooth muscle cells with Ang II changes PIP2 concentration and distribution in undifferentiated membrane and caveolae (Fujita et al. 2009) and this will change profilin localisation within the cell membrane (Senju et al. 2017). Caveolae are small invaginations of the plasma membrane composed of caveolins, cholesterol and sphingolipids that have a key role in signal transduction (Bastiani and Parton 2010). Caveolae are linked to the actin cytoskeleton (Head et al. 2014) and caveolins interact directly with eNOS (Sbaa et al. 2005) so profilin appears to indirectly modulate the eNOS/NO pathway by downregulating caveolin though disrupting the cytoskeleton (Xia et al. 2015). This relationship is not clear as vascular smooth muscle cells of spontaneously hypertensive rats (SHR) have increased profilin expression which accelerates actin turnover and promotes proliferation and hypertrophy (Cheng et al. 2011). In contrast, the cardiac tissues of SHR rats had increased profilin concentration but decreased F-actin content, increased collagen expression leading to fibrosis and had decreased levels of caveolae. Transiently overexpressing profilin in SHR reduced the number of caveolae which further disrupted the filament organisation (Zhao et al. 2013).
SHR are useful to study the relationship between profilin and cardiovascular disease as the SHR phenotype is associated with an overexpression of profilin, reduction of ACE2 and elevated ERK1/2 and JNK phosphorylation in vascular smooth muscle cells (Cheng et al. 2011). Further overexpression of profilin modulated by the activation of AT1 by Ang II in SHR and increased ERK1/2 and JNK signalling led to vascular hypertrophy in the aorta with an increase in systolic blood pressure (Zhong et al. 2011). Two studies used profilin silencing or overexpression in SHR rats via adenovirus constructs (Xia et al. 2015; Zhao et al. 2013) while three studies (Cheng et al. 2011; Jin et al. 2012; Zhong et al. 2011) used transient overexpression of profilin in SHR. The overexpression led to increased pathology whereas silencing profilin was seen as cardioprotective. In addition, overexpression of profilin in these experiments led to a decrease in eNOS activity, NO production and a downregulation of caveolin-3 in heart tissue (Xia et al. 2015; Zhao et al. 2013).
Function of profilin in the nucleus
Changing levels of profilin expression has a profound effect on the expression of a large number of genes (Coumans et al. 2014) and some of the mechanisms by which nuclear profilin impacts cell fate are beginning to be understood (Diamond et al. 2015; Kanaujiya et al. 2013). The interaction of profilin and multiple ligands at the plasma membrane are shown in Fig. 1. Extracellular profilin can act as an agonist of PI3K/Akt, Src and Ras-Raf-MEK-ERK pathways to initiate DNA synthesis, cell cycle progression and migration (Caglayan et al. 2010). In addition to this, profilin directly and indirectly regulates gene expression through its interaction with the transcription machinery of the cell (Soderberg et al. 2012).
Profilin is important in maintaining the transport of cargo across the nuclear membrane in transmitting the downstream effectors of signalling cascades to the nucleus by maintaining the GTP-binding nuclear protein Ran (Ran-GTP) gradients (Minakhina et al. 2005). While small molecules can efficiently diffuse in and out of the nucleus, most proteins and RNAs approaching the limit of 40 kDa cannot. At 15 kDa, profilin is small enough to diffuse into the nucleus (Birbach 2008). The translocation of large molecules across the nuclear pore is facilitated by the specialised transport receptors importins and exportins. The cargo proteins can be accumulated against steep gradients as there is an energy input into the system (Stuven et al. 2003). The Ran gradient is important in maintaining unidirectional transport of cargo, as importins bind their cargo in the cytoplasm and release it in the nucleus upon Ran-GTP binding. The importin-Ran-GTP complex returns to the cytoplasm where Ran-GTP is hydrolysed, and importin released. Exportins function in the opposite direction, forming ternary complexes of cargo, Ran-GTP and exportin at high Ran-GTP concentrations in the nucleus and dissociating in the cytoplasm upon hydrolysis of GTP. Nuclear transport factor 2 mediates the return of Ran-GDP to the nucleus (Minakhina et al. 2005; Stuven et al. 2003). The transport of actin from the nucleus by Exportin 6 and facilitated by profilin are only exported and this complex can include other actin binding proteins such as WASP, VASP, mDia and Mena (Stuven et al. 2003). Cargo of importins include phosphorylated ERK that is released by Ran-GTP in the nucleus to enable its interaction with transcription factors (Flores and Seger 2013). Therefore, the whole nuclear trafficking mechanisms that include the downstream effectors of cell signalling that depends on the Ran system are linked to profilin-exportin 6 regulating the Ran-GTP nuclear concentration. The in vivo effects of perturbing this system are seen in Drosophila where eye defects are the result of the downregulation of profilin and a disruption of the nuclear export system (Minakhina et al. 2005).
Another function of profilin and exportin 6 in the nucleus is the maintenance of the actin monomer concentration (Fig. 3) (Stuven et al. 2003). Actin in the nucleus is monomeric or forms dynamic short polymers and has many functions including chromatin remodelling and is a critical part of the gene transcription mechanism. Nuclear actin, and consequently gene transcription, is sensitive to changes in the ratio of G and F-actin in the cytoplasm to turn specific genes on or off (de Lanerolle 2012). Therefore, it is important that nuclear actin is tightly regulated. Knockdown of exportin-6 induces F-actin formation in the nucleus (Dopie et al. 2012) which disrupts nuclear organisation and is a hallmark of cellular stress in a number of disease states (Serebryannyy et al. 2016). The profilin-actin interaction is therefore essential in nuclear function.
Profilin has other specific functions in the nucleus including involvement in pre-mRNA splicing in nuclear gems (Giesemann et al. 1999). Profilin binds to a PRD on survival motor neurone protein, which is important for the biogenesis of small nuclear ribonucleoproteins (snRNPs) (Giesemann et al. 1999). Another ligand that binds profilin through a PRD is Myb-related transcription factor p42POP and profilin inhibits its activity as a repressor linking profilin directly to gene regulation (Lederer et al. 2005). Profilin is implicated in mRNA processing since being found in splicing speckles and Cajal bodies (Skare et al. 2003). In these bodies, profilin is associated with the heterogenous ribonucleoprotein (hnRNP) and is putatively involved in gene transcription. Profilin has also been found to associate with transcriptionally active genes using the polytene chromosomes of the insect Chironomus tentans (Soderberg et al. 2012). This group showed that profilin, snRNP and hnRNPs localise to transcriptionally active loci. HnRNP and snRNPs bind to nascent pre-mRNAs while still bound to the gene supporting the hypothesis that profilin is associated in transcription. Unlike actin, profilin is not dependent on RNA for its interaction with gene loci as profilin remains associated with the chromatin after RNase digestion and this suggests an actin-independent role for profilin (Soderberg et al. 2012).
A novel ligand for profilin in the nucleus
Intriguingly, a novel binding site has been identified on profilin as it acts as a corepressor of oestrogen receptor-α (ERα) (Kanaujiya et al. 2013). Once activated by oestrogen, ERs localise to the nucleus and bind to oestrogen response elements (EREs), specific DNA sequences upstream of oestrogen-responsive genes (Nicholson and Johnston 2005). ERα and ERβ interact with and modify the activity of one another; can modulate other transcription factors; bind endogenous ligands, oestrogen and anti-oestrogens with different specificities; take part in different signalling cascades; and also recruit different co-activators and corepressors to specific DNA sequences (Thomas and Gustafsson 2011). Profilin contains the amino acid sequence … IDNL…IRDSL.. which is a IXXL/H motif shared with other ERα corepressors. This forms a part of the extended [I/LXX(I/H/L] IXXX(I/L)] motif found in other corepressors of nuclear receptors (Kanaujiya et al. 2013). The arginine in the profilin sequence R74 is within the actin binding site (Lambrechts et al. 2002), so ERα is an addition to the canonical profilin ligands. In MCF7 breast cancer cells, tamoxifen, a selective ER modulator (SERM) used to treat ER+ breast cancer, has been found to upregulate the expression of profilin. Either treating the cells with tamoxifen or overexpressing profilin led to the downregulation of EREs reduced expression of oestrogen-induced genes including Cathepsin D, CyclinD1 and pS2 that have a role in proliferation. The inhibition of proliferation was followed by an increase in apoptosis (Fig. 3) (Kanaujiya et al. 2013). Gene expression is regulated by the recruitment of specific co-activators and corepressors (Kanaujiya et al. 2013). The novel binding site that allows profilin to function as a corepressor gives profilin another role to its repertoire as a gene regulator.
Actin-regulator vs signalling protein
This review mainly focussed on the signalling role of profilin in the cell. However, a few studies have reported the total concentration of both actin and profilin in a range of cells (Funk et al. 2019; Kaiser et al. 1999; Kiuchi et al. 2011) and concluded that given the affinity of Profilin 1 for actin and the concentration of both proteins in the cell that most of the Profilin 1 is likely bound to actin. If most profilin is linked to actin and therefore involved in actin assembly regulation, how can it be involved in cell signalling? While a large proportion of profilin is likely to be associated to actin in the bulk of the cytoplasm, localised or compartmentalised conditions in the cell can modify the affinity of profilin for actin, allowing profilin to interact with other ligands and thus fulfil its role in cell signalling. Changes in pH near the plasma membrane have been shown to reduce the affinity of profilin for actin resulting in the dissociation of the proteins (McLachlan et al. 2007). Phosphorylation of profilin could readily change the affinity of the protein (Diamond et al. 2015; Gau et al. 2016; Lambrechts et al. 2002) which could then be involved in the various pathways described in this review. Local production of polyphosphoinositides creating areas of high local concentrations can also displace profilin from actin monomers (Janmey and Lindberg 2004; Moens and Bagatolli 2007). It is therefore possible that the role of profilin could shift from an actin regulator to a signalling molecule in function of the cell state and/or the local environment of the proteins.
Conclusion
This review has highlighted the interactions of profilin many different contexts. Through its active involvement in actin polymerisation, multiple cell signalling pathways, and gene transcription, profilin has a profound influence cell motility, differentiation, aspects of metabolism, and the differential expression of a host of other proteins. The interactions listed in this review are not exhaustive and will grow as more links are found, and information comes to hand of how this small protein is able to play a major part in modulating the life of a cell
References
Ali M, Heyob K, Jacob NK, Rogers LK (2016) Alterative expression and localization of Profilin 1/VASPpS157 and cofilin 1/VASPpS239 regulates metastatic growth and is modified by DHA supplementation. Mol Cancer Ther 15:2220–2231. https://doi.org/10.1158/1535-7163.MCT-16-0092
Babich M, Foti LR, Sykaluk LL, Clark CR (1996) Profilin forms tetramers that bind to G-actin. Biochem Biophys Res Commun 218:125–131. https://doi.org/10.1006/bbrc.1996.0022
Bader AG, Kang S, Zhao L, Vogt PK (2005) Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 5:921–929. https://doi.org/10.1038/nrc1753
Bae YH, Ding ZJ, Zou L, Wells A, Gertler F, Roy P (2009) Loss of profilin-1 expression enhances breast cancer cell motility by Ena/VASP proteins. J Cell Physiol 219:354–364. https://doi.org/10.1002/Jcp.21677
Bae YH, Ding Z, Das T, Wells A, Gertler F, Roy P (2010) Profilin1 regulates PI(3,4)P2 and lamellipodin accumulation at the leading edge thus influencing motility of MDA-MB-231 cells. Proc Natl Acad Sci U S A 107:21547–21552. https://doi.org/10.1073/pnas.1002309107
Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93:1019–1137. https://doi.org/10.1152/physrev.00028.2012
Balla T, Szentpetery Z, Kim YJ (2009) Phosphoinositide signaling: new tools and insights. Physiology (Bethesda, Md) 24:231–244. https://doi.org/10.1152/physiol.00014.2009
Bartolini F, Ramalingam N, Gundersen GG (2012) Actin-capping protein promotes microtubule stability by antagonizing the actin activity of mDia1. Mol Biol Cell 23:4032–4040. https://doi.org/10.1091/mbc.E12-05-0338
Bastiani M, Parton RG (2010) Caveolae at a glance. J Cell Sci 123:3831–3836. https://doi.org/10.1242/jcs.070102
Bezanilla M, Gladfelter AS, Kovar DR, Lee WL (2015) Cytoskeletal dynamics: a view from the membrane. J Cell Biol 209:329–337. https://doi.org/10.1083/jcb.201502062
Bhargavi V, Chari VB, Singh SS (1998) Phosphatidylinositol 3-kinase binds to profilin through the p85 alpha subunit and regulates cytoskeletal assembly. Biochem Mol Biol Int 46:241–248
Birbach A (2008) Profilin, a multi-modal regulator of neuronal plasticity. BioEssays : News Rev Molec Cell Dev Biol 30:994–1002. https://doi.org/10.1002/bies.20822
Bjorkegren C, Rozycki M, Schutt CE, Lindberg U, Karlsson R (1993) Mutagenesis of human profilin locates its poly(L-proline)-binding site to a hydrophobic patch of aromatic amino acids. FEBS Lett 333:123–126
Bjorkegren-Sjogren C, Korenbaum E, Nordberg P, Lindberg U, Karlsson R (1997) Isolation and characterization of two mutants of human profilin I that do not bind poly(L-proline). FEBS Lett 418:258–264
Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J (2014) Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94:235–263. https://doi.org/10.1152/physrev.00018.2013
Boczkowska M, Rebowski G, Kast DJ, Dominguez R (2014) Structural analysis of the transitional state of Arp2/3 complex activation by two actin-bound WCAs. Nat Commun 5:3308. https://doi.org/10.1038/ncomms4308
Boettner B, Govek EE, Cross J, Van Aelst L (2000) The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc Natl Acad Sci U S A 97:9064–9069
Caglayan E et al (2010) Profilin-1 is expressed in human atherosclerotic plaques and induces atherogenic effects on vascular smooth muscle cells. PLoS One 5:e13608. https://doi.org/10.1371/journal.pone.0013608
Campellone KG, Welch MD (2010) A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 11:237–251. https://doi.org/10.1038/nrm2867
Carlsson L, Nystrom LE, Sundkvist I, Markey F, Lindberg U (1977) Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J Mol Biol 115:465–483
Chakraborty J et al (2014) Profilin-2 increased expression and its altered interaction with beta-actin in the striatum of 3-nitropropionic acid-induced Huntington’s disease in rats. Neuroscience 281:216–228. https://doi.org/10.1016/j.neuroscience.2014.09.035
Chen Y, Wang X (2020) miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 48:D127–D131. https://doi.org/10.1093/nar/gkz757
Cheng JF et al (2011) Involvement of profilin-1 in angiotensin II-induced vascular smooth muscle cell proliferation. Vasc Pharmacol 55:34–41. https://doi.org/10.1016/j.vph.2011.04.003
Cheng YJ, Zhu ZX, Zhou JS, Hu ZQ, Zhang JP, Cai QP, Wang LH (2015) Silencing profilin-1 inhibits gastric cancer progression via integrin beta1/focal adhesion kinase pathway modulation. World J Gastroenterol 21:2323–2335. https://doi.org/10.3748/wjg.v21.i8.2323
Chou CH et al (2018) miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 46:D296–D302. https://doi.org/10.1093/nar/gkx1067
Coumans JV, Gau D, Poljak A, Wasinger V, Roy P, Moens PD (2014) Profilin-1 overexpression in MDA-MB-231 breast cancer cells is associated with alterations in proteomics biomarkers of cell proliferation, survival, and motility as revealed by global proteomics analyses. OMICS 18:778–791. https://doi.org/10.1089/omi.2014.0075
Coumans JVF, Davey RJ, Moens PDJ (2018) Cofilin and profilin: partners in cancer aggressiveness. Biophys Rev. https://doi.org/10.1007/s12551-018-0445-0
Das T, Bae YH, Wells A, Roy P (2009) Profilin-1 overexpression upregulates PTEN and suppresses AKT activation in breast cancer cells. J Cell Physiol 218:436–443. https://doi.org/10.1002/jcp.21618
Davidson AJ, Wood W (2016) Unravelling the actin cytoskeleton: a new competitive eedge? Trends Cell Biol 26:569–576. https://doi.org/10.1016/j.tcb.2016.04.001
de Lanerolle P (2012) Nuclear actin and myosins at a glance. J Cell Sci 125:4945–4949. https://doi.org/10.1242/jcs.099754
Delage E, Puyaubert J, Zachowski A, Ruelland E (2013) Signal transduction pathways involving phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate: convergences and divergences among eukaryotic kingdoms. Prog Lipid Res 52:1–14. https://doi.org/10.1016/j.plipres.2012.08.003
Di Paolo G, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. https://doi.org/10.1038/nature05185
Diamond MI et al (2015) Subcellular localization and Ser-137 phosphorylation regulate tumor-suppressive activity of profilin-1. J Biol Chem 290:9075–9086. https://doi.org/10.1074/jbc.M114.619874
Ding Z, Lambrechts A, Parepally M, Roy P (2006) Silencing profilin-1 inhibits endothelial cell proliferation, migration and cord morphogenesis. J Cell Sci 119:4127–4137. https://doi.org/10.1242/jcs.03178
Ding Z, Gau D, Deasy B, Wells A, Roy P (2009) Both actin and polyproline interactions of profilin-1 are required for migration, invasion and capillary morphogenesis of vascular endothelial cells. Exp Cell Res 315:2963–2973. https://doi.org/10.1016/j.yexcr.2009.07.004
Ding Z, Bae YH, Roy P (2012) Molecular insights on context-specific role of profilin-1 in cell migration. Cell Adhes Migr 6:442–449. https://doi.org/10.4161/cam.21832
Ding Z et al (2014) Profilin-1 downregulation has contrasting effects on early vs late steps of breast cancer metastasis. Oncogene 33:2065–2074. https://doi.org/10.1038/onc.2013.166
Dominguez R (2010) Structural insights into de novo actin polymerization. Curr Opin Struct Biol 20:217–225. https://doi.org/10.1016/j.sbi.2009.12.012
Dominguez R (2016) The WH2 domain and actin nucleation: necessary but insufficient. Trends Biochem Sci 41:478–490. https://doi.org/10.1016/j.tibs.2016.03.004
Dopie J, Skarp KP, Rajakyla EK, Tanhuanpaa K, Vartiainen MK (2012) Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci U S A 109:E544–E552. https://doi.org/10.1073/pnas.1118880109
Fan Y et al (2012) Stimulus-dependent phosphorylation of profilin-1 in angiogenesis. Nat Cell Biol 14:1046–1056. https://doi.org/10.1038/ncb2580
Fan Y et al (2014) Profilin-1 phosphorylation directs angiocrine expression and glioblastoma progression through HIF-1alpha accumulation. Nat Cell Biol 16:445–456. https://doi.org/10.1038/ncb2954
Flores K, Seger R (2013) Stimulated nuclear import by ?-like importins. In: F1000Prime Rep, vol 5. doi:https://doi.org/10.12703/p5-41
Frantzi M et al (2016) Silencing of Profilin-1 suppresses cell adhesion and tumor growth via predicted alterations in integrin and Ca2+ signaling in T24M-based bladder cancer models. Oncotarget 7:70750–70768. https://doi.org/10.18632/oncotarget.12218
Fujita A, Cheng J, Tauchi-Sato K, Takenawa T, Fujimoto T (2009) A distinct pool of phosphatidylinositol 4,5-bisphosphate in caveolae revealed by a nanoscale labeling technique. Proc Natl Acad Sci U S A 106:9256–9261. https://doi.org/10.1073/pnas.0900216106
Fukami K, Inanobe S, Kanemaru K, Nakamura Y (2010) Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog Lipid Res 49:429–437. https://doi.org/10.1016/j.plipres.2010.06.001
Funk J et al (2019) Profilin and formin constitute a pacemaker system for robust actin filament growth. Elife 8. https://doi.org/10.7554/eLife.50963
Gao X, Lowry PR, Zhou X, Depry C, Wei Z, Wong GW, Zhang J (2011) PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc Natl Acad Sci U S A 108:14509–14514. https://doi.org/10.1073/pnas.1019386108
Gau D, Veon W, Zeng X, Yates N, Shroff SG, Koes DR, Roy P (2016) Threonine 89 is an important residue of profilin-1 that is phosphorylatable by protein kinase A. PLoS One 11:e0156313. https://doi.org/10.1371/journal.pone.0156313
Gau D, Veon W, Capasso TL, Bottcher R, Shroff S, Roman BL, Roy P (2017) Pharmacological intervention of MKL/SRF signaling by CCG-1423 impedes endothelial cell migration and angiogenesis. Angiogenesis. https://doi.org/10.1007/s10456-017-9560-y
Gericke A, Leslie NR, Losche M, Ross AH (2013) PtdIns(4,5)P2-mediated cell signaling: emerging principles and PTEN as a paradigm for regulatory mechanism. Adv Exp Med Biol 991:85–104. https://doi.org/10.1007/978-94-007-6331-9_6
Ghiglione C, Jouandin P, Cerezo D, Noselli S (2018) The Drosophila insulin pathway controls Profilin expression and dynamic actin-rich protrusions during collective cell migration. Development 145. https://doi.org/10.1242/dev.161117
Giesemann T, Rathke-Hartlieb S, Rothkegel M, Bartsch JW, Buchmeier S, Jockusch BM, Jockusch H (1999) A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J Biol Chem 274:37908–37914
Goldschmidt-Clermont PJ, Kim JW, Machesky LM, Rhee SG, Pollard TD (1991) Regulation of phospholipase C-gamma 1 by profilin and tyrosine phosphorylation. Science 251:1231–1233
Grecco HE, Schmick M, Bastiaens PI (2011) Signaling from the living plasma membrane. Cell 144:897–909. https://doi.org/10.1016/j.cell.2011.01.029
Grenklo S, Johansson T, Bertilson L, Karlsson R (2004) Anti-actin antibodies generated against profilin:actin distinguish between non-filamentous and filamentous actin, and label cultured cells in a dotted pattern. Eur J Cell Biol 83:413–423. https://doi.org/10.1078/0171-9335-00400
Gronborg M et al (2006) Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol Cell Proteomics 5:157–171. https://doi.org/10.1074/mcp.M500178-MCP200
Guttilla IK, White BA (2009) Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem 284:23204–23216. https://doi.org/10.1074/jbc.M109.031427
Haarer BK, Petzold AS, Brown SS (1993) Mutational analysis of yeast profilin. Mol Cell Biol 13:7864–7873
Hammond GRV, Burke JE (2020) Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr Opin Cell Biol 63:57–67. https://doi.org/10.1016/j.ceb.2019.12.007
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hansen SD, Mullins RD (2010) VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol 191:571–584. https://doi.org/10.1083/jcb.201003014
Hansson A, Skoglund G, Lassing I, Lindberg U, Ingelman-Sundberg M (1988) Protein kinase C-dependent phosphorylation of profilin is specifically stimulated by phosphatidylinositol bisphosphate (PIP2). Biochem Biophys Res Commun 150:526–531
Hartwig JH, Chambers KA, Hopcia KL, Kwiatkowski DJ (1989) Association of profilin with filament-free regions of human leukocyte and platelet membranes and reversible membrane binding during platelet activation. J Cell Biol 109:1571–1579
Head BP, Patel HH, Insel PA (2014) Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta Biomembr 1838:532–545. https://doi.org/10.1016/j.bbamem.2013.07.018
Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701. https://doi.org/10.1038/nrm2476
Henty-Ridilla JL, Juanes MA, Goode BL (2017) Profilin directly promotes microtubule growth through residues mutated in amyotrophic lateral sclerosis. Curr Biol 27(3535-3543):e3534. https://doi.org/10.1016/j.cub.2017.10.002
Hernandez JF et al (2014) A Petiveria alliacea standardized fraction induces breast adenocarcinoma cell death by modulating glycolytic metabolism. J Ethnopharmacol 153:641–649. https://doi.org/10.1016/j.jep.2014.03.013
Hornbeck PV et al (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40:D261–D270. https://doi.org/10.1093/nar/gkr1122
Hu E, Chen Z, Fredrickson T, Zhu Y (2001) Molecular cloning and characterization of profilin-3: a novel cytoskeleton-associated gene expressed in rat kidney and testes. Exp Nephrol 9:265–274
Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA (2006) Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase. D Oncog 25:7305–7310. https://doi.org/10.1038/sj.onc.1209735
Janke J et al (2000) Suppression of tumorigenicity in breast cancer cells by the microfilament protein profilin 1. J Exp Med 191:1675–1686
Janmey PA, Lindberg U (2004) Cytoskeletal regulation: rich in lipids. Nat Rev Mol Cell Biol 5:658–666. https://doi.org/10.1038/nrm1434
Ji H, Greening DW, Kapp EA, Moritz RL, Simpson RJ (2009) Secretome-based proteomics reveals sulindac-modulated proteins released from colon cancer cells. Proteomics Clin Appl 3:433–451. https://doi.org/10.1002/prca.200800077
Jiang C, Veon W, Li H, Hallows KR, Roy P (2015) Epithelial morphological reversion drives Profilin-1-induced elevation of p27(kip1) in mesenchymal triple-negative human breast cancer cells through AMP-activated protein kinase activation. Cell Cycle 14:2914–2923. https://doi.org/10.1080/15384101.2015.1069929
Jin HY et al (2012) ACE2 deficiency enhances angiotensin II-mediated aortic profilin-1 expression, inflammation and peroxynitrite production. PLoS One 7:e38502. https://doi.org/10.1371/journal.pone.0038502
Jockusch BM, Murk K, Rothkegel M (2007) The profile of profilins. Rev Physiol Biochem Pharmacol 159:131–149. https://doi.org/10.1007/112_2007_704
Joy ME et al (2014) A high-content, multiplexed screen in human breast cancer cells identifies profilin-1 inducers with anti-migratory activities. PLoS One 9:e88350. https://doi.org/10.1371/journal.pone.0088350
Joy M, Gau D, Castellucci N, Prywes R, Roy P (2017) The myocardin-related transcription factor MKL co-regulates the cellular levels of two profilin isoforms. J Biol Chem 292:11777–11791. https://doi.org/10.1074/jbc.M117.781104
Kaiser DA, Vinson VK, Murphy DB, Pollard TD (1999) Profilin is predominantly associated with monomeric actin in Acanthamoeba. J Cell Sci 112(Pt 21):3779–3790
Kanaujiya JK et al (2013) Proteomic identification of Profilin1 as a corepressor of estrogen receptor alpha in MCF7 breast cancer cells. Proteomics 13:2100–2112. https://doi.org/10.1002/pmic.201200534
Kasina S, Rizwani W, Radhika KV, Singh SS (2005) Nitration of profilin effects its interaction with poly (L-proline) and actin. J Biochem 138:687–695. https://doi.org/10.1093/jb/mvi163
Kasina S, Wasia R, Fasim A, Radhika KV, Singh SS (2006) Phorbol ester mediated activation of inducible nitric oxide synthase results in platelet profilin nitration. Nitric Oxide Biol Chem 14:65–71. https://doi.org/10.1016/j.niox.2005.09.008
Khadka DK, Liu W, Habas R (2009) Non-redundant roles for Profilin2 and Profilin1 during vertebrate gastrulation. Dev Biol 332:396–406. https://doi.org/10.1016/j.ydbio.2009.06.008
Kim JG et al (2012) Ras-related GTPases Rap1 and RhoA collectively induce the phagocytosis of serum-opsonized zymosan particles in macrophages. J Biol Chem 287:5145–5155. https://doi.org/10.1074/jbc.M111.257634
Kiuchi T, Nagai T, Ohashi K, Mizuno K (2011) Measurements of spatiotemporal changes in G-actin concentration reveal its effect on stimulus-induced actin assembly and lamellipodium extension. J Cell Biol 193:365–380. https://doi.org/10.1083/jcb.201101035
Komiya Y, Habas R (2008) Wnt signal transduction pathways. Organogenesis 4:68–75. https://doi.org/10.4161/org.4.2.5851
Korupolu RV et al (2009) Profilin oligomerization and its effect on poly (L-proline) binding and phosphorylation. Int J Biol Macromol 45:265–273. https://doi.org/10.1016/j.ijbiomac.2009.06.001
Krauss M, Haucke V (2007) Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep 8:241–246. https://doi.org/10.1038/sj.embor.7400919
Krishnan K, Moens PDJ (2009) Structure and functions of profilins. Biophys Rev 1:71–81. https://doi.org/10.1007/s12551-009-0010-y
Kwiatkowska K (2010) One lipid, multiple functions: how various pools of PI(4,5)P(2) are created in the plasma membrane. Cell Mol Life Sci 67:3927–3946. https://doi.org/10.1007/s00018-010-0432-5
Lafuente EM et al (2004) RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev Cell 7:585–595. https://doi.org/10.1016/j.devcel.2004.07.021
Lambrechts A, Jonckheere V, Dewitte D, Vandekerckhove J, Ampe C (2002) Mutational analysis of human profilin I reveals a second PI(4,5)-P2 binding site neighbouring the poly(L-proline) binding site. BMC Biochem 3:12
Lassing I, Lindberg U (1985) Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature 314:472–474
Lassing I, Lindberg U (1988) Specificity of the interaction between phosphatidylinositol 4,5-bisphosphate and the profilin: actin complex. J Cell Biochem 37:255–267. https://doi.org/10.1002/jcb.240370302
Lederer M, Jockusch BM, Rothkegel M (2005) Profilin regulates the activity of p42POP, a novel Myb-related transcription factor. J Cell Sci 118:331–341. https://doi.org/10.1242/jcs.01618
Leroy B, Anderson M, Soussi T (2014) TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat 35:672–688. https://doi.org/10.1002/humu.22552
Levental I, Christian DA, Wang YH, Madara JJ, Discher DE, Janmey PA (2009) Calcium-dependent lateral organization in phosphatidylinositol 4,5-bisphosphate (PIP2)- and cholesterol-containing monolayers. Biochemistry 48:8241–8248. https://doi.org/10.1021/bi9007879
Li H, Marshall AJ (2015) Phosphatidylinositol (3,4) bisphosphate-specific phosphatases and effector proteins: A distinct branch of PI3K signaling. Cell Signal 27:1789–1798. https://doi.org/10.1016/j.cellsig.2015.05.013
Li ZQ, Yang T, Xie X, Chen M (2013) The role of profilin-1 in endothelial cell injury induced by advanced glycation end products (AGEs). Cardiovasc Diabetol 12:141. https://doi.org/10.1186/1475-2840-12-141
Li X, Liu J, Chen B, Fan L (2018) A positive feedback loop of profilin-1 and RhoA/ROCK1 promotes endothelial dysfunction and oxidative stress. Oxidative Med Cell Longev 2018:4169575. https://doi.org/10.1155/2018/4169575
Lin H et al (2010) Unregulated miR-96 induces cell proliferation in human breast cancer by downregulating transcriptional factor FOXO3a. PLoS One 5:e15797. https://doi.org/10.1371/journal.pone.0015797
Lin HY et al (2012) Identification of direct forkhead box O1 targets involved in palmitate-induced apoptosis in clonal insulin-secreting cells using chromatin immunoprecipitation coupled to DNA selection and ligation. Diabetologia 55:2703–2712. https://doi.org/10.1007/s00125-012-2643-9
Lin W et al (2016) Profilin expression is regulated by bone morphogenetic protein (BMP) in osteoblastic cells. J Cell Biochem 117:621–628. https://doi.org/10.1002/jcb.25310
Liu W, Wang X (2019) Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol 20:18. https://doi.org/10.1186/s13059-019-1629-z
Liu H et al (2013) Expression and regulatory function of miRNA-182 in triple-negative breast cancer cells through its targeting of profilin 1. Tumour Biol 34:1713–1722. https://doi.org/10.1007/s13277-013-0708-0
Lorente G, Syriani E, Morales M (2014) Actin filaments at the leading edge of cancer cells are characterized by a high mobile fraction and turnover regulation by profilin I. PLoS One 9:e85817. https://doi.org/10.1371/journal.pone.0085817
Lu PJ, Shieh WR, Rhee SG, Yin HL, Chen CS (1996) Lipid products of phosphoinositide 3-kinase bind human profilin with high affinity. Biochemistry 35:14027–14034. https://doi.org/10.1021/bi961878z
Lu Q, Lu L, Chen W, Chen H, Xu X, Zheng Z (2015) RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy Graefes. Arch Clin Exp Ophthalmol 253:669–680. https://doi.org/10.1007/s00417-015-2985-3
Ma CY et al (2011) Decreased expression of profilin 2 in oral squamous cell carcinoma and its clinicopathological implications. Oncol Rep 26:813–823. https://doi.org/10.3892/or.2011.1365
Machesky LM, Goldschmidt-Clermont PJ, Pollard TD (1990) The affinities of human platelet and Acanthamoeba profilin isoforms for polyphosphoinositides account for their relative abilities to inhibit phospholipase C. Cell Regul 1:937–950
Mahoney NM, Rozwarski DA, Fedorov E, Fedorov AA, Almo SC (1999) Profilin binds proline-rich ligands in two distinct amide backbone orientations. Nat Struct Biol 6:666–671. https://doi.org/10.1038/10722
McLachlan GD, Cahill SM, Girvin ME, Almo SC (2007) Acid-induced equilibrium folding intermediate of human platelet profilin. Biochemistry 46:6931–6943. https://doi.org/10.1021/bi0602359
McLaughlin S, Murray D (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438:605–611. https://doi.org/10.1038/nature04398
McLaughlin S, Wang J, Gambhir A, Murray D (2002) PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct 31:151–175. https://doi.org/10.1146/annurev.biophys.31.082901.134259
Menkhorst EM et al (2012) Decidual-secreted factors alter invasive trophoblast membrane and secreted proteins implying a role for decidual cell regulation of placentation. PLoS One 7:e31418. https://doi.org/10.1371/journal.pone.0031418
Menkhorst EM, Van Sinderen ML, Rainczuk K, Cuman C, Winship A, Dimitriadis E (2017) Invasive trophoblast promote stromal fibroblast decidualization via Profilin 1 and ALOX5. Sci Rep 7:8690. https://doi.org/10.1038/s41598-017-05947-0
Metzler WJ, Bell AJ, Ernst E, Lavoie TB, Mueller L (1994) Identification of the poly-L-proline-binding site on human profilin. J Biol Chem 269:4620–4625
Michaelsen K, Murk K, Zagrebelsky M, Dreznjak A, Jockusch BM, Rothkegel M, Korte M (2010) Fine-tuning of neuronal architecture requires two profilin isoforms. Proc Natl Acad Sci U S A 107:15780–15785. https://doi.org/10.1073/pnas.1004406107
Minakhina S, Myers R, Druzhinina M, Steward R (2005) Crosstalk between the actin cytoskeleton and Ran-mediated nuclear transport. BMC Cell Biol 6:32. https://doi.org/10.1186/1471-2121-6-32
Minamida S et al (2011) Profilin 1 overexpression in renal cell carcinoma. Int J Urol 18:63–71. https://doi.org/10.1111/j.1442-2042.2010.02670.x
Mittermann I, Fetrow JS, Schaak DL, Almo SC, Kraft D, Herberle-Bors E, Valenta R (1998) Oligomerization of profilins from birch, man and yeast. Profilin, a ligand for itself? Sex Plant Reprod 11:183–191
Moens PD, Bagatolli LA (2007) Profilin binding to sub-micellar concentrations of phosphatidylinositol (4,5) bisphosphate and phosphatidylinositol (3,4,5) trisphosphate. Biochim Biophys Acta 1768:439–449. https://doi.org/10.1016/j.bbamem.2006.12.012
Mouneimne G et al (2012) Differential remodeling of actin cytoskeleton architecture by profilin isoforms leads to distinct effects on cell migration and invasion. Cancer Cell 22:615–630. https://doi.org/10.1016/j.ccr.2012.09.027
Moustafa-Bayoumi M et al (2007) Vascular hypertrophy and hypertension caused by transgenic overexpression of profilin 1. J Biol Chem 282:37632–37639. https://doi.org/10.1074/jbc.M703227200
Navakauskiene R, Treigyte G, Gineitis A, Magnusson KE (2004) Identification of apoptotic tyrosine-phosphorylated proteins after etoposide or retinoic acid treatment. Proteomics 4:1029–1041. https://doi.org/10.1002/pmic.200300671
Nejedla M et al (2016) Profilin connects actin assembly with microtubule dynamics. Mol Biol Cell 27:2381–2393. https://doi.org/10.1091/mbc.E15-11-0799
Neve RM et al (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10:515–527. https://doi.org/10.1016/j.ccr.2006.10.008
Nicholson RI, Johnston SR (2005) Endocrine therapy--current benefits and limitations. Breast Cancer Res Treat 93(Suppl 1):S3–S10. https://doi.org/10.1007/s10549-005-9036-4
Noh EM, Lee YR, Chay KO, Chung EY, Jung SH, Kim JS, Youn HJ (2011) Estrogen receptor alpha induces down-regulation of PTEN through PI3-kinase activation in breast cancer cells. Mol Med Rep 4:215–219. https://doi.org/10.3892/mmr.2011.412
Nolle A et al (2011) The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum Mol Genet 20:4865–4878. https://doi.org/10.1093/hmg/ddr425
O'Bryan S, Dong S, Mathis JM, Alahari SK (2017) The roles of oncogenic miRNAs and their therapeutic importance in breast cancer. Eur J Cancer 72:1–11. https://doi.org/10.1016/j.ejca.2016.11.004
Olson EN, Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11:353–365. https://doi.org/10.1038/nrm2890
Osaki M, Oshimura M, Ito H (2004) PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 9:667–676. https://doi.org/10.1023/B:APPT.0000045801.15585.dd
Ostrander DB, Gorman JA, Carman GM (1995) Regulation of profilin localization in Saccharomyces cerevisiae by phosphoinositide metabolism. J Biol Chem 270:27045–27050
Pae M, Romeo GR (2014) The multifaceted role of profilin-1 in adipose tissue inflammation and glucose homeostasis. Adipocyte 3:69–74. https://doi.org/10.4161/adip.26965
Pandey DK, Chaudhary B (2017) Evolutionary expansion and structural functionalism of the ancient family of profilin proteins. Gene 626:70–86. https://doi.org/10.1016/j.gene.2017.05.024
Parri M, Chiarugi P (2010) Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal 8:23. https://doi.org/10.1186/1478-811X-8-23
Pecar Fonovic U, Kos J (2015) Cathepsin X Cleaves Profilin 1 C-Terminal Tyr139 and Influences Clathrin-Mediated Endocytosis. PLoS One 10:e0137217. https://doi.org/10.1371/journal.pone.0137217
Pecar Fonovic U, Jevnikar Z, Rojnik M, Doljak B, Fonovic M, Jamnik P, Kos J (2013) Profilin 1 as a target for cathepsin X activity in tumor cells. PLoS One 8:e53918. https://doi.org/10.1371/journal.pone.0053918
Pernier J, Shekhar S, Jegou A, Guichard B, Carlier MF (2016) Profilin interaction with actin filament barbed end controls dynamic instability, capping, branching, and motility. Dev Cell 36:201–214. https://doi.org/10.1016/j.devcel.2015.12.024
Pinto-Costa R, Sousa MM (2020) Profilin as a dual regulator of actin and microtubule dynamics. Cytoskeleton 77:76–83. https://doi.org/10.1002/cm.21586
Popova JS, Greene AK, Wang J, Rasenick MM (2002) Phosphatidylinositol 4,5-bisphosphate modifies tubulin participation in phospholipase Cbeta1 signaling. J Neurosci 22:1668–1678
Quinlan MP (2004) Vinculin, VASP, and profilin are coordinately regulated during actin remodeling in epithelial cells, which requires de novo protein synthesis and protein kinase signal transduction pathways. J Cell Physiol 200:277–290. https://doi.org/10.1002/jcp.20009
Rebowski G, Boczkowska M, Drazic A, Ree R, Goris M, Arnesen T, Dominguez R (2020) Mechanism of actin N-terminal acetylation. Sci Adv 6:eaay8793. https://doi.org/10.1126/sciadv.aay8793
Rennella E, Sekhar A, Kay LE (2017) Self-assembly of human profilin-1 detected by Carr-Purcell-Meiboom-Gill nuclear magnetic resonance (CPMG NMR) spectroscopy. Biochemistry. https://doi.org/10.1021/acs.biochem.6b01263
Richer SM, Stewart NK, Tomaszewski JW, Stone MJ, Oakley MG (2008) NMR investigation of the binding between human profilin I and inositol 1,4,5-triphosphate, the soluble headgroup of phosphatidylinositol 4,5-bisphosphate. Biochemistry 47:13455–13462. https://doi.org/10.1021/bi801535f
Rizwani W, Fasim A, Sharma D, Reddy DJ, Bin Omar NA, Singh SS (2014) S137 phosphorylation of profilin 1 is an important signaling event in breast cancer progression. PLoS One 9:e103868. https://doi.org/10.1371/journal.pone.0103868
Romeo GR, Kazlauskas A (2008) Oxysterol and diabetes activate STAT3 and control endothelial expression of profilin-1 via OSBP1. J Biol Chem 283:9595–9605. https://doi.org/10.1074/jbc.M710092200
Romeo GR, Pae M, Eberle D, Lee J, Shoelson SE (2013) Profilin-1 haploinsufficiency protects against obesity-associated glucose intolerance and preserves adipose tissue immune homeostasis. Diabetes 62:3718–3726. https://doi.org/10.2337/db13-0050
Romero S, Le Clainche C, Didry D, Egile C, Pantaloni D, Carlier MF (2004) Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell 119:419–429. https://doi.org/10.1016/j.cell.2004.09.039
Roy P, Jacobson K (2004) Overexpression of profilin reduces the migration of invasive breast cancer cells. Cell Motil Cytoskeleton 57:84–95. https://doi.org/10.1002/cm.10160
Saarikangas J, Zhao H, Lappalainen P (2010) Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol Rev 90:259–289. https://doi.org/10.1152/physrev.00036.2009
Sathish K et al (2004) Phosphorylation of profilin regulates its interaction with actin and poly (L-proline). Cell Signal 16:589–596
Sbaa E, Frerart F, Feron O (2005) The double regulation of endothelial nitric oxide synthase by caveolae and caveolin: a paradox solved through the study of angiogenesis. Trends Cardiovasc Med 15:157–162. https://doi.org/10.1016/j.tcm.2005.05.006
Schlett K (2017) More than a mere supply of monomers: G-Actin pools regulate actin dynamics in dendritic spines. J Cell Biol 216:2255–2257. https://doi.org/10.1083/jcb.201705216
Schluter K, Jockusch BM, Rothkegel M (1997) Profilins as regulators of actin dynamics. Biochim Biophys Acta 1359:97–109
Schoppmeyer R et al (2017) Human profilin 1 is a negative regulator of CTL mediated cell-killing and migration. Eur J Immunol 47:1562–1572. https://doi.org/10.1002/eji.201747124
Senju Y, Kalimeri M, Koskela EV, Somerharju P, Zhao H, Vattulainen I, Lappalainen P (2017) Mechanistic principles underlying regulation of the actin cytoskeleton by phosphoinositides. Proc Natl Acad Sci U S A 114:E8977–E8986. https://doi.org/10.1073/pnas.1705032114
Serebryannyy LA, Yuen M, Parilla M, Cooper ST, de Lanerolle P (2016) The effects of disease models of nuclear actin polymerization on the nucleus. Front Physiol 7:454. https://doi.org/10.3389/fphys.2016.00454
Shao J, Diamond MI (2012) Protein phosphatase 1 dephosphorylates profilin-1 at Ser-137. PLoS One 7:e32802. https://doi.org/10.1371/journal.pone.0032802
Shao J, Welch WJ, Diprospero NA, Diamond MI (2008) phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol Cell Biol 28:5196–5208. https://doi.org/10.1128/MCB.00079-08
Sharma JN, Al-Omran A, Parvathy SS (2007) Role of nitric oxide in inflammatory diseases. Inflammopharmacology 15:252–259. https://doi.org/10.1007/s10787-007-0013-x
Shen K et al (2016) Guttiferone K suppresses cell motility and metastasis of hepatocellular carcinoma by restoring aberrantly reduced profilin 1. Oncotarget 7:56650–56663. https://doi.org/10.18632/oncotarget.10992
Simons M, Schwartz MA (2012) Profilin phosphorylation as a VEGFR effector in angiogenesis. Nat Cell Biol 14:985–987. https://doi.org/10.1038/ncb2596
Singh SS, Chauhan A, Murakami N, Chauhan VP (1996a) Profilin and gelsolin stimulate phosphatidylinositol 3-kinase activity. Biochemistry 35:16544–16549. https://doi.org/10.1021/bi9609634
Singh SS, Chauhan A, Murakami N, Styles J, Elzinga M, Chauhan VP (1996b) Phosphoinositide-dependent in vitro phosphorylation of profilin by protein kinase C. Phospholipid specificity and localization of the phosphorylation site. Recept Signal Transduct 6:77–86
Sit ST, Manser E (2011) Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci 124:679–683. https://doi.org/10.1242/jcs.064964
Siton-Mendelson O, Bernheim-Groswasser A (2017) Functional actin networks under construction: the cooperative action of actin nucleation and elongation factors. Trends Biochem Sci 42:414–430. https://doi.org/10.1016/j.tibs.2017.03.002
Skare P, Kreivi JP, Bergstrom A, Karlsson R (2003) Profilin I colocalizes with speckles and Cajal bodies: a possible role in pre-mRNA splicing. Exp Cell Res 286:12–21
Skruber K, Read TA, Vitriol EA (2018) Reconsidering an active role for G-actin in cytoskeletal regulation. J Cell Sci 131. https://doi.org/10.1242/jcs.203760
Skruber K, Warp PV, Shklyarov R, Thomas JD, Swanson MS, Henty-Ridilla JLR, Read TA, Vitriol EA (2020) Arp2/3 and Mena/VASP require profilin 1 for actin network assembly at the leading edge. Curr Biol. https://doi.org/10.1016/j.cub.2020.04.085
Soderberg E, Hessle V, von Euler A, Visa N (2012) Profilin is associated with transcriptionally active genes. Nucleus 3:290–299. https://doi.org/10.4161/nucl.20327
Sohn RH, Goldschmidt-Clermont PJ (1994) Profilin: at the crossroads of signal transduction and the actin cytoskeleton. BioEssays : News Rev Molec Cell Dev Biol 16:465–472. https://doi.org/10.1002/bies.950160705
Sorci G, Riuzzi F, Giambanco I, Donato R (2013) RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta 1833:101–109. https://doi.org/10.1016/j.bbamcr.2012.10.021
Starr ME, Ueda J, Yamamoto S, Evers BM, Saito H (2011) The effects of aging on pulmonary oxidative damage, protein nitration, and extracellular superoxide dismutase down-regulation during systemic inflammation. Free Radic Biol Med 50:371–380. https://doi.org/10.1016/j.freeradbiomed.2010.11.013
Stuven T, Hartmann E, Gorlich D (2003) Exportin 6: a novel nuclear export receptor that is specific for profilin.actin complexes. EMBO J 22:5928–5940. https://doi.org/10.1093/emboj/cdg565
Sun Y, Thapa N, Hedman AC, Anderson RA (2013) Phosphatidylinositol 4,5-bisphosphate: targeted production and signaling. BioEssays : News Rev Molec Cell Dev Biol 35:513–522. https://doi.org/10.1002/bies.201200171
Takenawa T (2010) Phosphoinositide-binding interface proteins involved in shaping cell membranes. Proc Jpn Acad Ser B Phys Biol Sci 86:509–523
Tamura M et al (2000) Activation of DNA synthesis and AP-1 by profilin, an actin-binding protein, via binding to a cell surface receptor in cultured rat mesangial cells. J Am Soc Nephrol 11:1620–1630
Tamura Y, Takeuchi Y, Suzawa M, Fukumoto S, Kato M, Miyazono K, Fujita T (2001) Focal adhesion kinase activity is required for bone morphogenetic protein--Smad1 signaling and osteoblastic differentiation in murine MC3T3-E1 cells. J Bone Miner Res 16:1772–1779. https://doi.org/10.1359/jbmr.2001.16.10.1772
Tavaluc RT, Hart LS, Dicker DT, El-Deiry WS (2007) Effects of low confluency, serum starvation and hypoxia on the side population of cancer cell lines. Cell Cycle 6:2554–2562. https://doi.org/10.4161/cc.6.20.4911
Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J, Amigorena S (2001) Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol 166:7309–7318
Thomas C, Gustafsson JA (2011) The different roles of ER subtypes in cancer biology and therapy Nature reviews. Cancer 11:597–608. https://doi.org/10.1038/nrc3093
Valenzuela-Iglesias A et al (2015) Profilin1 regulates invadopodium maturation in human breast cancer cells. Eur J Cell Biol 94:78–89. https://doi.org/10.1016/j.ejcb.2014.12.002
Vemuri B, Singh SS (2001) Protein kinase C isozyme-specific phosphorylation of profilin. Cell Signal 13:433–439
Viaud J et al (2016) Phosphoinositides: Important lipids in the coordination of cell dynamics. Biochimie 125:250–258. https://doi.org/10.1016/j.biochi.2015.09.005
Wang J, Richards DA (2012) Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol Open 1:857–862. https://doi.org/10.1242/bio.20122071
Wang W et al (2004) Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res 64:8585–8594. https://doi.org/10.1158/0008-5472.CAN-04-1136
Wang Z, Shi Z, Zhang L, Zhang H, Zhang Y (2019) Profilin 1, negatively regulated by microRNA-19a-3p, serves as a tumor suppressor in human hepatocellular carcinoma. Pathol Res Pract 215:499–505. https://doi.org/10.1016/j.prp.2018.12.012
Watanabe N et al (1997) p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J 16:3044–3056. https://doi.org/10.1093/emboj/16.11.3044
Welf ES, Ahmed S, Johnson HE, Melvin AT, Haugh JM (2012) Migrating fibroblasts reorient directionality by a metastable, PI3K-dependent mechanism. J Cell Biol 197:105–114. https://doi.org/10.1083/jcb.201108152
Witke W (2004) The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol 14:461–469. https://doi.org/10.1016/j.tcb.2004.07.003
Witke W, Sutherland JD, Sharpe A, Arai M, Kwiatkowski DJ (2001) Profilin I is essential for cell survival and cell division in early mouse development. Proc Natl Acad Sci U S A 98:3832–3836. https://doi.org/10.1073/pnas.051515498
Wittenmayer N et al (2004) Tumor suppressor activity of profilin requires a functional actin binding site. Mol Biol Cell 15:1600–1608. https://doi.org/10.1091/mbc.E03-12-0873
Wu N et al (2006) Profilin 1 obtained by proteomic analysis in all-trans retinoic acid-treated hepatocarcinoma cell lines is involved in inhibition of cell proliferation and migration. Proteomics 6:6095–6106. https://doi.org/10.1002/pmic.200500321
Wu CY, Lin MW, Wu DC, Huang YB, Huang HT, Chen CL (2014) The role of phosphoinositide-regulated actin reorganization in chemotaxis and cell migration. Br J Pharmacol 171:5541–5554. https://doi.org/10.1111/bph.12777
Xia LH, Chen T, Zhang B, Chen M (2015) Mechanism of Profilin-1 in regulating eNOS/NO signaling pathway and its role in hypertensive myocardial hypertension. Asian Pac J Trop Med 8:399–404. https://doi.org/10.1016/S1995-7645(14)60351-5
Yang C, Czech L, Gerboth S, Kojima S, Scita G, Svitkina T (2007) Novel roles of formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol 5:e317. https://doi.org/10.1371/journal.pbio.0050317
Yang J et al (2014) MicroRNA-19a-3p inhibits breast cancer progression and metastasis by inducing macrophage polarization through downregulated expression of Fra-1 proto-oncogene. Oncogene 33:3014–3023. https://doi.org/10.1038/onc.2013.258
Yao W et al (2013) Profilin 1 potentiates apoptosis induced by staurosporine in cancer cells. Curr Mol Med 13:417–428
Yao W et al (2014) Profilin-1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3-HIF1alpha axis. Mol Cancer 13:187. https://doi.org/10.1186/1476-4598-13-187
Yap KL et al (2012) NAC1 is an actin-binding protein that is essential for effective cytokinesis in cancer cells. Cancer Res 72:4085–4096. https://doi.org/10.1158/0008-5472.CAN-12-0302
Ye Q, Kantonen S, Gomez-Cambronero J (2013) Serum deprivation confers the MDA-MB-231 breast cancer line with an EGFR/JAK3/PLD2 system that maximizes cancer cell invasion. J Mol Biol 425:755–766. https://doi.org/10.1016/j.jmb.2012.11.035
Yerlikaya A, Okur E, Ulukaya E (2012) The p53-independent induction of apoptosis in breast cancer cells in response to proteasome inhibitor bortezomib. Tumour Biol 33:1385–1392. https://doi.org/10.1007/s13277-012-0386-3
Yoshinaga S et al (2012) A phosphatidylinositol lipids system, lamellipodin, and Ena/VASP regulate dynamic morphology of multipolar migrating cells in the developing cerebral cortex. J Neurosci 32:11643–11656. https://doi.org/10.1523/JNEUROSCI.0738-12.2012
Yu J, Li A, Hong SM, Hruban RH, Goggins M (2012) MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res 18:981–992. https://doi.org/10.1158/1078-0432.CCR-11-2347
Zaidi AH, Manna SK (2016) Profilin-PTEN interaction suppresses NF-kappaB activation via inhibition of IKK phosphorylation. Biochem J 473:859–872. https://doi.org/10.1042/BJ20150624
Zaidi AH, Raviprakash N, Mokhamatam RB, Gupta P, Manna SK (2016) Profilin potentiates chemotherapeutic agents mediated cell death via suppression of NF-kappaB and upregulation of p53. Apoptosis 21:502–513. https://doi.org/10.1007/s10495-016-1222-9
Zhang H et al (2018) Loss of profilin 2 contributes to enhanced epithelial-mesenchymal transition and metastasis of colorectal cancer. Int J Oncol. https://doi.org/10.3892/ijo.2018.4475
Zhao SH et al (2013) Profilin-1 promotes the development of hypertension-induced cardiac hypertrophy. J Hypertens 31:576–586; discussion 586. https://doi.org/10.1097/HJH.0b013e32835d6a56
Zhong JC et al (2011) Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul Pept 166:90–97. https://doi.org/10.1016/j.regpep.2010.09.005
Zoidakis J et al (2012) Profilin 1 is a potential biomarker for bladder cancer aggressiveness. Mol Cell Proteomics 11(M111):009449. https://doi.org/10.1074/mcp.M111.009449
Zou L, Jaramillo M, Whaley D, Wells A, Panchapakesa V, Das T, Roy P (2007) Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br J Cancer 97:1361–1371. https://doi.org/10.1038/sj.bjc.6604038
Zou L, Ding Z, Roy P (2010) Profilin-1 overexpression inhibits proliferation of MDA-MB-231 breast cancer cells partly through p27kip1 upregulation. J Cell Physiol 223:623–629. https://doi.org/10.1002/jcp.22058
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Rhonda J Davey declares that she has no conflicts of interest. Pierre DJ Moens declares that he has no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Davey, R.J., Moens, P.D. Profilin: many facets of a small protein. Biophys Rev 12, 827–849 (2020). https://doi.org/10.1007/s12551-020-00723-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-020-00723-3