
Abstract
Phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] is a minor lipid of the inner leaflet of the plasma membrane that controls the activity of numerous proteins and serves as a source of second messengers. This multifunctionality of PI(4,5)P2 relies on mechanisms ensuring transient appearance of PI(4,5)P2 clusters in the plasma membrane. One such mechanism involves phosphorylation of PI(4)P to PI(4,5)P2 by the type I phosphatidylinositol-4-phosphate 5-kinases (PIP5KI) at discrete membrane locations coupled with PI(4)P delivery/synthesis at the plasma membrane. Simultaneously, both PI(4)P and PI(4,5)P2 participate in anchoring PIP5KI at the plasma membrane via electrostatic bonds. PIP5KI isoforms are also selectively recruited and activated at the plasma membrane by Rac1, talin, or AP-2 to generate PI(4,5)P2 in ruffles and lamellipodia, focal contacts, and clathrin-coated pits. In addition, PI(4,5)P2 can accumulate at sphingolipid/cholesterol-based rafts following activation of distinct membrane receptors or be sequestered in a reversible manner due to electrostatic constrains posed by proteins like MARCKS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the five decades that have passed since the discovery of phosphoinositide turnover in the cell [1], it has been established that phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] is located mainly in the plasma membrane and serves as a source of second messengers: inositol trisphosphate (IP3), diacylglycerol (DAG), and phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3]. IP3 and DAG are generated by phospholipase C (PLC)-catalyzed hydrolysis of PI(4,5)P2, while PI(3,4,5)P3 arises by phosphorylation of PI(4,5)P2 by the class I phosphatidylinositol 3-kinases (PI3KI). Aside from the fundamental signaling role of its derivatives, PI(4,5)P2 itself controls the activity of several integral membrane proteins, like ion channels and transporters, and affects a myriad of proteins associating with the membrane due to PI(4,5)P2 binding. The latter group includes proteins of the actin cytoskeleton that recognize PI(4,5)P2 using either specific domains, like the pleckstrin homology (PH) domain or the band 4.1-ezrin-radixin-moesin homology (FERM) domain, or unfolded stretches of basic amino acids. Despite PI(4,5)P2 comprising only 1% of all plasma membrane phospholipids, its extraordinary versatility puts PI(4,5)P2 in the center of plasma membrane dynamics governing motility, cell adhesion, endo- and exocytosis. Some of these subjects have been discussed extensively in recent excellent reviews [2, 3].
The diversity of PI(4,5)P2 functions poses the question of the nature of the mechanisms governing the local and discrete character of PI(4,5)P2-dependent processes. This review covers work dealing with this puzzle and pointing to the existence of various pools of PI(4,5)P2 in the plasma membrane. The ideas proposed as a solution to the above problem include local control of PI(4,5)P2 synthesis and confinement of PI(4,5)P2 in the plasma membrane due to an involvement of membrane rafts and positively charged membrane proteins.
PI(4,5)P2 synthesis in the plasma membrane
Major role of type I phosphatidylinositol-4-phosphate 5-kinases
Phosphatidylinositol 4-monophosphate [PI(4)P] and PI(4,5)P2 are two major derivatives of phosphatidylinositol (PI), their amounts reaching about 5% each of the total cellular PI [4]. Due to the relative abundance of PI(4)P, its phosphorylation at the D-5 position of the inositol ring is the prevailing route of PI(4,5)P2 synthesis in the cell (Fig. 1). This reaction is catalyzed by kinases named the type I phosphatidylinositol-4-phosphate 5-kinases (PIP5KI) belonging to the family of phosphatidylinositol phosphate kinases (PIPK). Alternatively, minor amounts of PI(4,5)P2 arise from phosphorylation of phosphatidylinositol 5-monophosphate [PI(5)P] due to action of the type II phosphatidylinositol-5-phosphate 4-kinases (PIP4KII), a second PIPK subfamily (Fig. 1). Finally, PI(4,5)P2 can also be generated by PTEN- (phosphatase and tensin homologue on chromosome 10)-driven dephosphorylation of PI(3,4,5)P3 [5]. Although crucial for termination of PI(3,4,5)P3 signaling, the importance of this process for PI(4,5)P2 generation in the plasma membrane remains questionable. Of note, PI(4,5)P2 can also undergo dephosphorylation catalyzed either by inositol polyphosphate 5-phosphatases (IPP 5-Ptases) such as oculocerebrorenal syndrome of Lowe 1 phosphatase (OCRL1) and synaptojanins [6–8] or by PI(4,5)P2 4-phosphatases type I and II [9]. These enzymes dephosphorylate the D-5 and D-4 phosphoester linkage of PI(4,5)P2 yielding PI(4)P and PI(5)P, respectively. Activity of IPP 5-Ptases is important for controlling the PI(4,5)P2/PI(4)P balance at defined cellular locations and its disturbances lead to homeostatic PI(4,5)P2 defects bearing serious consequences to health. These are exemplified by the devastating human developmental disorder named Lowe syndrome caused by loss-of function for the OCRL1 [10, 11], mortality of synaptojanin 1-deficient mice [12] and possibly, Down’s syndrome connected with trisomy of the synaptojanin 1 gene [13]. On the other hand, PI(4,5)P2 4-phosphatases seem to play an essential role in the synthesis of PI(5)P since PI(4,5)P2 4-phosphatase I was shown to control nuclear levels of PI(5)P and thereby p53-dependent apoptosis [14].

PI(4,5)P2 metabolism and activity. PI(4,5)2 is generated mainly by phosphorylation of PI(4)P by PIP5KI (the pathway indicated by thicker arrows) and, to lower extend, by PIP4KII-catalyzed phosphorylation of PI(5)P. PI(4,5)2 directly affects activity of numerous proteins, serves as a source of second messengers: IP3, DAG, and PI(3,4,5)P3. The lipid can be also dephosphorylated at the D-4 and D-5 positions of the inositol ring
The third member of the PIPK family, the type III PIPK named PIKfyve in mammals, is a phosphatidylinositol-3-phosphate 5-kinase that phosphorylates phosphatidylinositol 3-monophosphate [PI(3)P] to PI(3,5)P2, a lipid that controls membrane trafficking in the endosomal pathway (see [15] for review). Of note, PIKfyve is also implicated in synthesis of PI(5)P either by direct phosphorylation of PI (see Fig. 1) or by production of PI(3,5)P2 which can be then converted into PI(5)P by PI(3)P phosphatases of the myotubularin family [16–18]. Both PIP5KI and PIP4PII also utilize PI(3)P as a substrate in vitro and are indicated to produce PI(3,4)P2, PI(3,5)P2, and PI(3,4,5)P3 in the cell [19, 20]. However, the physiological consequences of this substrate promiscuity of PIPKs are beyond the subject of this review.
The type I PIP5Ks, central to PI(4,5)P2 synthesis in the plasma membrane, are of three isoforms, α, β, and γ, encoded by distinct genes. PIP5KIα and Iβ of human and mouse origin were cloned simultaneously by two groups and given reciprocal names [21–23]. In this work, names adopted for the human enzymes by Anderson’s group are used. PIP5KIα1 consists of 549 amino acids while PIP5KIβ1 of 540 amino acids, and they represent one of three (PIP5KIα) and two (PIP5KIβ) splicing variants, all of molecular weight of about 62 kDa [22]. PIP5KIγ has three well-characterized splicing variants: PIP5KIγa of 668 amino acids (661 amino acids in murine PIP5KIγa), PIP5KIγb of 635 and PIP5KIγc of 688 amino acids (present in rodents), also called PIP5KIγ90, Iγ87 and Iγ93 based on their migration in gel electrophoresis [23, 24]. Although all these PIP5KI isoforms coexist in most tissues, PIP5KIα is highly expressed in skeletal muscles and PIP5KIβ in the heart, whereas PIP5KIγ is abundant in the brain, which suggests specific functions for these isoenzymes in distinct cells. The physiological importance of PIP5KIγ is indicated by early mortality of PIP5KIγ knockout mice [25]. The three PIP5KI isoforms display about 80% identity in the amino acid sequence of their catalytic domain consisting of 330–380 amino acids and located in the center of the molecule [22, 23]. Another unique feature of the PIP5KIs common to all the isoforms is the up-regulation (up to tenfold) of their enzymatic activity by phosphatidic acid (PA) [21, 23, 26]. Consequently, phospholipase D (PLD) and diacylglycerol kinase (DAGK), enzymes producing PA, can regulate PIP5KI activity, as discussed below. All PIP5KI isoforms undergo autophosphorylation and phosphorylation catalyzed by protein kinase A within the catalytic domain, which strongly suppresses their lipid kinase activity [27, 28]. The variable amino- and carboxy-terminal tails of PIP5KIα, Iβ, and Iγ are involved in the regulation of their activity [23] and are likely to participate in the targeting to distinct cellular locations. For example, the C-terminal 28 amino acids (26 in murine kinase) were found to govern interaction of PIP5KIγa with talin at focal contacts organized during adhesion of cells to the matrix [29, 30]. The C-terminus (amino acids 440–562) of PIP5KIα2 targets the enzyme to the nucleus [31]. A search of the human and mouse genome sequence databases for an amino acid motif conserved in all PIPK has revealed a PIPK homolog composed of 395 amino acids (about 40 kDa). This protein is abundant in brain and testis, has no intrinsic kinase activity but homodimerizes with PIP5KIα and Iβ, thus affecting their activity [32].
Studies on the functions of individual isoenzymes have yet to produce a comprehensive picture. However, it is becoming increasingly clear that the different PIP5KI isoforms play specific roles in individual cell types and one kinase isoform can not compensate for the loss of another [33–37]. PIP5KIα is implicated in actin remodeling that governs ruffling, expansion of the leading edge of migrating cells, and phagosome formation [38–40]. Nevertheless, participation of PIP5KIβ (murine PIP5KIα) in the uncapping of actin filaments leading to actin polymerization in thrombin-activated platelets [33], actin polymerization accompanying cell volume changes [34], and actin reorganization during oxidative stress should also be noted [41]. A growing body of evidence suggests that PIP5KIα and Iβ can shuttle between the plasma membrane and cytoplasm being recruited to the membrane upon activation of cell motility [39, 40, 42, 43]. Overexpression of PIP5KIα or Iβ induces various changes of the actin cytoskeleton including the appearance of unusual structures like actin-rich needles and comets [23, 44, 45]. Therefore, both these kinase isoforms have been postulated to generate PI(4,5)P2 affecting the actin cytoskeleton organization in the cell. However, since the cellular localization of the overexpressed kinases is often different than that of the endogenous ones (see below), and the final outcome of a kinase’s activity can be different depending on the cellular context including the activity of the Rho family members [45, 46], it is hard to ascribe a particular aspect of actin remodeling to one of the kinase isoforms based solely on those data. Besides the role in actin cytoskeleton reorganization, PIP5KIα is also found in the nucleus at sites of pre-mRNA processing where it interacts with non-canonical poly(A) polymerase to control the formation of selected mRNAs [31, 47]. On the other hand, PIP5KIβ is engaged in formation of clathrin-coated pits during receptor endocytosis [48]. Participation of PIP5KIγa in the formation of clathrin-coated pits has also been documented in great detail [49–51]. Additionally, the activity of PIP5KIγa-c emerges as crucial for assembly of focal contacts and cell–cell contacts and for association of the actin cytoskeleton with the plasma membrane mediated by proteins of these structures [29, 30, 35, 52–54].
Mammalian PIP5KIα and Iβ, but not PIP4KIIβ, were able to restore PI(4,5)P2 synthesis and rescue lethality of yeast mutants lacking Mss4p, the only yeast PIP5KI [55, 56]. The cells do not have PIP4KII but express the type III PIPK called Fab1p, which does not participate in PI(4,5)P2 synthesis and, hence, is not able to substitute for Mss4p [55]. The Mss4p kinase is localized to the plasma membrane and is required for yeast viability, actin organization, and cell wall integrity [57]. These data suggest that an involvement of PIP5KI-derived PI(4,5)P2 in actin skeleton remodeling is evolutionarily ancient.
PIP4KII provides a clue on PIP5KI–membrane interaction: role of PI(4)P
The second pathway for the production of PI(4,5)P2 relies on phosphorylation of PI(5)P on the D-4 position of the inositol ring catalyzed by type II PIP4K (Fig. 1). Three isoforms of PIP4KII (α, β, γ) have been identified in mammalian cells, with PIP4KIIα being the first PIPK cloned [58–60]. The distinct substrate specificity of PIP5KI and PIP4KII was established by Rameh et al. [61], who also found evidence for the presence of PI(5)P, a PIP4KII substrate, in fibroblasts. Kinases of the PIP4KII subfamily share about 35% amino acid identity with the enzymes of the PIP5KI subfamily within the kinase catalytic domain. A structure-based alignment of the catalytic domain of human PIP4KIIα and IIβ, PIP5KIα and Iβ, Mss4p (yeast PIP5KI), and Fab1p (yeast type III PIPK) as well as protein kinase A allowed three residues conserved in all these enzymes to be identified: Lys150, Asp278, and Asp369 (numbering as in PIP4KIIβ) engaged in ATP and Mg2+ binding [62]. Accordingly, mutation of the corresponding Lys138 in PIP5KIβ abolished the kinase activity [23]. Outside the core kinase domain, as well as in the so-called insert of the domain, very little amino acid identity is found between type I and type II PIPKs [22].
Type II PIP4Ks are found in the cytoplasm, nucleus, and endoplasmic reticulum and their cellular functions remain unclear (see [63] for review). In contrast to PIP5KI, both endogenous and overexpressed PIP4KII are only weakly associated with the plasma membrane and are insensitive to PA [56, 60, 64]. When first purified from erythrocytes, PIP4KIIα was found unable to phosphorylate the intrinsic PIP in isolated erythrocyte membrane [65]. This is in agreement with the finding that very small quantities of PI(5)P are present in the cell, reaching only about 2% of the PI(4)P level [61]. Accordingly, in cells subjected to radioactive labeling of phosphoinositides, phosphorylation at the D-5 position of the inositol ring was found to exceed that of the D-4 hydroxyl group two to threefold [66]. It seems, therefore, that PIP4KII activity does not contribute substantially to PI(4,5)P2 production in the plasma membrane.
Yet, it was the crystallization of PIP4KIIβ that shed light on the PIPK-membrane interaction [62]. The enzyme is a homo-dimer made up of subunits linked head-to-head at their amino-termini. In the dimer, the N-terminal fragments of the catalytic core of both subunits jointly form a highly basic flat surface proposed to provide a site for the kinase-membrane attachment. When modeled on a membrane bilayer, the dimer appeared as a peripheral protein not penetrating the membrane. Its membrane binding was thought to be directed by electrostatic interactions between the dimer’s flat positively charged surface and the anionic lipid head groups.
The importance of electrostatic interactions for plasma membrane binding was also shown for PIP5KIβ. The data presented by Anderson’s group indicated that amino acids involved in this linkage were located in the so-called activation loop of about 20 amino acids localized at the C-terminal part of the kinase catalytic core (Fig. 2a). After substitution of two adjacent lysine residues Lys359Lys360 of the activation loop with two asparagine residues, PIP5KIβ expressed in osteosarcoma cells no longer bound to the plasma membrane. On the other hand, substitution of the dilysine motif with positively charged arginine residues did not interfere with the membrane association of the protein [56]. The dilysine motif is conserved among all PIPK subfamilies [62], suggesting its key role in the membrane binding of the enzymes. However, the membrane association based solely on this dilysine motif cannot explain the targeting of PIP4KII and PIP5KI to diverse cellular membranes, with some PIP5KI isoforms recruited preferably to defined locations at the plasma membrane.

Organization of PIP5KI molecule. a Amino acid sequence of activation loop regions of human PIP5KIα, human Iβ, and murine Iγ (upper panel) and human PIP4KIIβ (lower panel). The first and the last amino acid of the regions as well as the total number of amino acids composing the proteins are indicated. Dark gray background marks amino acids conserved between PIP5KI and PIP4KII subfamilies; amino acids conserved only within the PIP5KI and PIP4KII subfamilies, but different between them are shown by the light gray background [22, 23, 59]. Bar below the alignment indicates regions exchanged between PIP5KIβ and PIP4KIIβ by Kunz et al. [56]. Asterisks indicate two lysine residues participating in membrane binding of PIPKs while glutamic acid residue and alanine residue determining substrate specificity of PIP5KI and PIP4KII, respectively, are marked by boxes. b A schematic representation of murine PIP5KIγa (661 amino acids) and PIP5KIγb (635 amino acids). PIP5KIγc of rodents is composed of 687 amino acids and contains a 26-amino-acid insert at Ph635 [23, 24]. AL, activation loop. In the lower panel, the amino acid composition of 26-amino-acid tail of murine PIP5KIγa is shown. The motifs engaged in the binding of talin, AP-2β and AP-2μ, are marked by bars. In human PIP5KIγa, two amino acids are inserted between Arg652 and Pro653 with the Arg652 exchanged for Glu652 [132]
A partial answer to this question lies in the amino acid sequence of the aforementioned activation loop. Aside from the amino acids common to all PIPKs, some amino acids are highly conserved only within the PIP5KI, PIP4KII, and type III PIPK subfamilies, but different between them. Further mutational studies confirmed the participation of the activation loop in the selective recognition of either PI(4)P or PI(5)P as well as proved its involvement in the association of PIP5KI with the plasma membrane. Swapping of the activation loop between PIP4KIIβ and PIP5KIβ resulted in a reversed substrate specificity of the obtained protein chimeras. Introduction of the PIP4KIIβ activation loop into PIP5KIβ converted the PIP5KIβ chimera into a kinase phosphorylating PI(5)P and simultaneously abolished its binding to the plasma membrane. Reciprocally, PIP4KIIβ with the activation loop of PIP5KIβ phosphorylated PI(4)P and gained the ability to localize to the plasma membrane [56]. A similar reversion of specificity was achieved after substitution of a single amino acid in the activation loop, A381E in PIP4KIIβ, and E362A in PIP5KIβ. Moreover, the E362A PIP5KIβ mutant, using PI(5)P as the preferred substrate, no longer associated with the plasma membrane [67]. Thus, the single amino acids determining the substrate specificity of PIP5KI and PIP4KII were identified (Fig. 2a). The data also suggested that the plasma membrane binding by PIP5KIβ is correlated with its ability to recognize and phosphorylate PI(4)P but not PI(5)P. However, a line of data argue against PI(4)P as the only factor recruiting PIP5KIβ to the plasma membrane. Firstly, analysis of the kinetic properties of the E362A PIP5KIβ mutant revealed that the protein still displayed a relatively high affinity towards PI(4)P. The significant reduction of the mutant kinase activity toward PI(4)P, below that toward PI(5)P, resulted from a low V max of PI(4)P phosphorylation by the mutated kinase. Secondly, other fragments of the PIP5KIβ activation loop were also shown to participate in an optimal plasma membrane binding by the kinase. Thirdly, the A381E PIP4KIIβ mutant, despite its efficient PI(4)P binding and phosphorylation, did not associate with the plasma membrane [67]. Therefore, the binding of PI(4)P is required but not sufficient to target PIP5KIβ, and presumably other PIP5KIs, to the plasma membrane.
PI(4,5)P2 also acts as an anchor for PIP5KI in the plasma membrane
Recent data obtained in several laboratories has underscored the idea about electrostatic interactions of PIP5KI with the plasma membrane pointing simultaneously to an involvement of PI(4,5)P2, the product of the kinases, in PIP5KI binding. Recombinant PIP5KIα fused with the GST tag interacted, apart from PI(4)P and PA, also with PI(4,5)P2, as shown by ELISA, protein-lipid overlay assay, and binding to PI(4,5)P2-containing liposomes [68, 69]. A fragment of PIP5KIα encompassing amino acids 374–440 and containing the activation loop of the kinase was found to interact preferably with PI(4,5)P2 over PI(4)P. Conversely, the affinity of the whole kinase for PI(4)P exceeded that for PI(4,5)P2 about 1.8-fold, indicating that multiple electrostatic interactions govern the membrane–kinase interaction [43, 69]. When expressed in cells, the aforementioned fragment of the kinase evoked a significant reduction of the PI(4,5)P2 level, affected the organization of actin filaments, and inhibited phagocytosis. Similar results were obtained after expression in cells of a recombinant protein containing the PH domain of PLCδ1, a probe known to bind PI(4,5)P2 with high affinity (K D ~ 1.2 μM, [70]). When looking for the mechanism of the probe action, it was found that both its expression and treatment of cells with the Ca2+ ionophore ionomycin, which activates PLC leading to PI(4,5)P2 hydrolysis, reduced the amount of endogenous PIP5KIα associated with the plasma membrane [43, 69]. Altogether, the data indicated that PI(4,5)P2 can serve as an anchor for PIP5KIα in the plasma membrane. Sequestration of PI(4,5)P2 by the lipid-binding probes or PI(4,5)P2 hydrolysis displaced the kinase from the plasma membrane, thus inhibiting PI(4,5)P2 synthesis and affecting the PI(4,5)P2-dependent actin reorganization. Taking into account the accumulation of PI(4,5)P2 in the plasma membrane [71], an involvement of the lipid in the binding of PIP5KI to the membrane can explain why various kinase isoforms, when overexpressed in the cell, all display submembraneous location [67, 69, 72]. The endogenous enzymes are located in different cellular compartments presumably because their distribution is under more subtle regulation by several other factors.
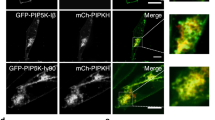
Results obtained for GFP-tagged PIP5KIα, Iβ and Iγa, Iγb expressed in RAW macrophages were in full agreement with the suggestion about the PI(4,5)P2 engagement in the binding of the kinases to the plasma membrane [72]. In addition to ionomycin treatment, also depletion of cellular ATP or termination of polyphosphoinositide synthesis released the PIP5KI isoforms from the plasma membrane. Partial dissociation of GFP-PIP5KIβ from the plasma membrane was also observed in cells co-expressing the kinase and synaptojanin 2, PI(4,5)P2-dephosphorylating enzyme [72]. A PIP5KIα mutant in which the positively charged arginine and leucine residues in the activation loop were substituted with the negatively charged aspartic acid residues failed to associate with the plasma membrane, while analogous mutations of amino acids located C-terminally from the activation loop were also effective but to a lower extent. Those data supported the suggestion that multiple electrostatic interactions of PIP5KI are crucial for its binding to anionic lipids of the plasma membrane. The authors took advantage of the crystal structure of PIP4IIβ described earlier [62] and proposed that PIP5KI isoforms form homo-dimers with a flat, positively charged surface of the enzyme oriented towards the plasma membrane. The amino acids contributing to the positive surface charge of PIP5KI are presumably present both outside and within the activation loop of the kinase and are crucial for the proper positioning of the loop for substrate recognition and of the catalytic center for substrate phosphorylation. In the plasma membrane, the negatively charged lipids including PI(4)P, PI(4,5)P2, PA, and phosphatidylserine can participate in PIP5KI binding. PA also strongly activates PIP5KI, possibly overcoming the substrate inhibition of PIP5KI [68] to create conditions for efficient local generation of PI(4,5)P2. Subsequent hydrolysis of the lipid by PLC can facilitate displacement of the kinase form the membrane, providing an “electrostatic switch”, which terminates local PI(4,5)P2 synthesis [72]. Such a local reduction of PI(4,5)P2 level, which follows its rapid generation, was observed during Fcγ receptor-mediated phagocytosis [73] and budding of clathrin-coated vesicles [12]. The existence of the “electrostatic switch” does not answer, however, the question about factors recruiting PIP5KI to distinct sites at the plasma membrane during cell activation. It is conceivable that an interaction of PIP5KIs with specific membrane proteins provides a key for the plasma membrane recruitment of the individual kinase isoforms.
Coupling between PI(4)P and PI(4,5)P2 synthesis at the plasma membrane
The considerations on PI(4)P and PI(4,5)P2 as plasma membrane anchors for PIP5KI need to be reconciled with the data on the subcellular location of these lipids. Thus, visualization of PI(4)P with specific antibodies indicated that about 47% of the lipid is accumulated in the Golgi apparatus. Very little PI(4)P was found to reside in the plasma membrane at steady state [74], as confirmed by studies with the PH domains of phosphatidylinositol-4-phosphate adaptor protein 1 (Fapp1) and oxysterol-binding protein (OSBP) as PI(4)P markers [75–77]. This PI(4)P distribution is opposite to the prevailing plasma membrane location of PI(4,5)P2 that is well documented by cell fractionation studies and image analysis [71, 78]. When combined with reports on PI(3)P enrichment in endosomes [79], these findings spark an idea that the organelle-specific composition of phosphoinositides can define the identity and functions of various cellular organelles by recruiting specific effector proteins and directing membrane traffic [3, 74]. In particular, PI(4)P at Golgi, besides serving as a source for a small local pool of PI(4,5)P2, controls the vesicular traffic towards distinct compartments like endo/lysosomes and plasma membrane by recruiting adaptor proteins including AP-1 complex or GGAs or FAPPs [74, 76, 80, 81], and participates in sphingolipid metabolism (see [82] for review). However, the analysis of PI(4)P distribution in the cell based solely on imaging of overexpressed PH domains of Fapp1 or OSBP can be biased to the Golgi at the expense of the plasma membrane staining since the both probes bind not only PI(4)P but also ARF1, a Golgi marker protein. Recently, a pool of PI(4)P has been revealed in the plasma membrane in experiments employing another probe, the PH domain of yeast oxysterol-binding protein homologue (OSH2), or an improved protocol of plasma membrane preservation for PI(4)P staining with antibodies [83, 84].
While the content of PI(4)P in the cellular membranes awaits direct estimation, it is well established that the activity of phosphatidylinositol 4-kinases (PI4K), which generate PI(4)P by PI phosphorylation, is present in the Golgi, the plasma membrane, and lysosomes [85]. Four PI4Ks have been identified in mammalian cells: PI4KIIα and IIβ, and PI4KIIIα and IIIβ, with these two classes being distinguished by the wortmannin-sensitivity of PI4KIIIs. PI4KIIIα is localized primarily to the endoplasmic reticulum. The Golgi apparatus contains PI4KIIIβ, PI4KIIα, and PI4KIIβ. Among the kinases, PI4KIIIβ contributes to production of PI(4)P in cis/medial Golgi, PI4KIIα in trans-Golgi network and PI4KIIβ displays more complex pattern of distribution as described below [86–90]. Silencing of PI4KIIα expression by RNA interference led to an about 50% reduction of PI(4)P synthesis and a 60% reduction of PI(4,5)P2 synthesis in HeLa cells, indicating that the activity of this Golgi-associated kinase significantly affects the overall level of PI(4,5)P2 [74]. Taken together, the data suggest that PI(4,5)P2 is generated by phosphorylation of PI(4)P “en route” from the Golgi to the plasma membrane. Alternatively, the Golgi-derived PI(4)P can be converted to PI(4,5)P2 immediately upon reaching the plasma membrane. In either case, the Golgi-plasma membrane trafficking emerges from these studies as an important factor controlling the plasma membrane level of PI(4,5)P2. This subject got a new input with the discovery of Balla’s group that substantial amounts of PI(4)P appear in the plasma membrane and in submembrane vesicles during the acute resynthesis of phosphoinositides following Ca2+-induced PLC activation and PI(4,5)P2 depletion [77]. Subsequent work of the group has indicated that also in the course of stimulation of cells with angiotensin II, an agonist of G protein-coupled receptor, PLC utilizes PI(4,5)P2 pool whose maintenance is limited by local PI(4)P synthesis [83]. Based on the wortmannin sensitivity and the results of siRNA application, the delivery of this plasma membrane pool of PI(4)P was attributed to PI4KIIIα, although the kinase itself was not detected at the plasma membrane [77, 83, 86]. Therefore, PI(4)P can reach the plasma membrane not only by exchange of vesicles between the plasma membrane and the Golgi but possibly also as a result of PI4KIIIα activity at sites of close apposition between the endoplasmic reticulum and the plasma membrane.
On the other hand, early biochemical studies detected significant activity of PI4Ks, particularly PI4KII, in the plasma membrane [85, 86]. This activity can likely be ascribed to PI4KIIβ, an enzyme cloned simultaneously by two groups [88, 89]. In a series of elegant studies combining immunofluorescence observations and biochemical fractionation it has been determined that PI4KIIβ is recruited to the plasma membrane from the cytosol. This redistribution is controlled by the monomeric small GTPase Rac1 in a GTP-dependent manner. Accordingly, the kinase was found in membrane ruffles evoked by stimulation of PDGF receptor which employs Rac1 as a downstream effector. It was established that the responsiveness of PI4KIIβ to Rac1 was mediated by the catalytic domain of the kinase. The recruitment of PI4KIIβ to the membrane stimulates its enzymatic activity 16-fold. The membrane-bound PI4KIIβ is palmitoylated and this acylation is a prerequisite for the high enzymatic activity of the kinase [88, 91]. Those data indicate that Rac1 controls the recruitment of PI4KIIβ to the plasma membrane and places it in a position allowing activation by other enzymes (palmitoyl acyltransferase?) or lipids present in the membrane.
Taken together, the studies on PI4KIIβ and PI4KIIIα support the suggestion that the synthesis of PI(4)P and PI(4,5)P2 in the plasma membrane can be coupled spatially and temporally providing a way for transient accumulation of PI(4,5)P2 at discrete plasma membrane locations. One can hypothesize that upon cell stimulation by agonists of G protein-coupled receptors PI(4)P is generated by PI4KIIIα residing in the endoplasmic reticulum adjacent to the plasma membrane. At the plasma membrane the lipid is rapidly converted into PI(4,5)P2 to be then hydrolyzed by PLC and trigger IP3/Ca+2 signaling. Thus far, PIP5KIα (murine Iβ) and PIP5KIγb have been shown to generate PI(4,5)P2 subsequently utilized for the IP3/Ca+2 signaling by G protein-coupled receptors [36, 92]. On the other hand, PI4KIIβ catalyzes the synthesis of PI(4)P converted into a poll of PI(4,5)P2 acting on cytoskeletal proteins. In the latter case, the coupling of PI(4)P and PI(4,5)P2 production can be achieved by Rac1, which recruits to the plasma membrane and thus facilitates activation of both PI4KIIβ [88] and PIP5KI, as discussed below.
Regulators of PIP5KI activity and PI(4,5)P2 synthesis at the plasma membrane
Rac1 recruits PIP5KI to the plasma membrane and facilitates its activation
Rac1 has been reported to associate with PIP5KIα and, to a lower extent, with PIP5KIβ and Iγ [33, 42, 93–95]. An association of RhoA, but not Cdc42, with all three isoforms of PIP5KI was also reported [94–96]. The association of Rac1 and RhoA with PIP5KI was independent of the GDP/GTP loading of the GTPases. In the cited reports, the binding of the two Rho family GTPases to PIP5KI was examined by the pull-down assays relying on incubation of immobilized Rho GTPases with homogenates prepared from rat brain or liver, or with cell lysates, and subsequent measurements of PIP5KI activity and/or kinase isoform identification. The data were supported by co-immunoprecipitation of Rac1 and RhoA with PIP5KI. Moreover, a direct binding of GDP/GTP-loaded Rac1 and RhoA to PIP5KIα and Iβ was also shown using recombinant proteins [33]. However, neither Rac1 nor RhoA activated PIP5KIβ in another in vitro assay [97], while activation of PIP5KIα, Iβ and Iγ by Rac1, RhoA, and Cdc42 was demonstrated in vivo. Only GTP-bound proteins activated PIP5KIs in cells since no increase in PIP5KI activity and no PI(4,5)P2 production was observed in cells expressing dominant negative mutants of Rac1, RhoA, and Cdc42 [46, 95, 98, 99]. Taken together, the data implicate that Rho family GTPases can interact with and control the activity of PIP5KI and also suggest that the activation can be indirect. Since the association of Rac1 and RhoA with PIP5KI does not depend on GDP/GTP binding to the GTPases, the complex of Rac1 or RhoA with PIP5KI can be maintained in both resting and activated cells. Therefore, the GTPases should be able to shift PIP5KI to the plasma membrane upon cell activation. The data on the participation of Rac1 and RhoA in PIP5KI regulation vary and point to either RhoA [46, 97, 100, 101] or Rac1 [33, 39, 42] or both [95, 99] as dominant factors affecting PIP5KI activity, probably depending on the cell type and stimulus used in those studies.
Among the GTPases of the Rho family, Rac1 is of special interest because of its ability to recruit to the plasma membrane and activate PI4KIIβ as well [88]. Hence, this GTPase could in principle coordinate the synthesis of PI(4)P and PI(4,5)P2 at distinct regions of the membrane. Rac1 affects actin organization and focal complex assembly contributing to ruffling and lamellipodia formation [102], which is in line with the crucial role of PI(4,5)P2 in those events. PIP5KI was shown to associate with the C-terminus of Rac1 in a nucleotide-independent fashion based on the ability of a recombinant peptide encompassing amino acids 166–188 of Rac1 to compete with full-length Rac1 for pull-down of PIP5KI activity from rat brain homogenate [42]. In further studies, the 185ArgLysArg187 sequence in the C-terminus of Rac1 was found crucial for the PIP5KI pull-down from fibroblast lysates [94]. The C-terminal fragment of Rac1 also associated with DAGK, catalyzing PA production. Similarly to PIP5KI, DAGK associated with both GDP- and GTP-bound Rac1. However, the association of the two lipid kinases with Rac1-GTP was enhanced in the presence of PA, phosphatidylserine, and PI(4)P. Combined with the detected association of PIP5KI and DAGK with RhoGDI the data suggested the existence of a multimolecular complex able to control PIP5KI recruitment to the plasma membrane [42]. In resting cells, RhoGDI is able to sequester the lipid moiety of Rac1 and prevent the association of the whole multimolecular complex containing Rac1, DAGK, and PIP5KI with the plasma membrane. Upon cell activation, RhoGDI releases Rac1, which can then anchor the protein complex to the plasma membrane leading to the synthesis of PA and, in turn, PI(4,5)P2 (Fig. 3a, d).

Rac1 forms multimolecular complex engaging PIP5KI. a In resting cells, PIP5KIα/β and DAGKζ are bound to the C-terminus of GDP-loaded Rac1. The interaction of Rac1 with the plasma membrane is blocked by RhoGDI, which masks the prenyl group attached to the C-terminal CAAX motif of Rac1. b Upon cell activation, PLC produces DAG. c DAG is subsequently phosphorylated by DAGKζ to PA. The PA activates PAK1, which phosphorylates RhoGDI at two sites. Phosphorylated RhoGDI dissociates from Rac1 exposing its prenyl group. d Rac1 binds to the membrane via the prenyl group and adjacent polybasic region and this facilitates replacement of GDP with GTP by membrane-bound exchange factors. PIP5KIα/β recruited to the plasma membrane together with Rac1 is activated by PA and phosphorylates PI(4)P to PI(4,5)P2
The above idea was developed further by Abramovici et al. [103], who identified ubiquitously expressed DAGKζ as a component of a multimolecular complex containing Rac1 and RhoGDI. Moreover, the activity DAGKζ was found to be a crucial factor controlling Rac1 activation: the PA produced by DAGKζ activates p21-activated kinase 1 (PAK1), which in turn phosphorylates RhoGDI leading to its dissociation from Rac1. In fibroblasts derived from DAGKζ−/− mice and stimulated with PDGF, a decreased Rac1 activation and inhibition of ruffling, spreading, and migration were found [103]. On the other hand, association of DAGKζ with PIP5KIα was also documented, and expression of DAGKζ significantly enhanced PIP5KIα activity in thrombin-stimulated cells [104]. Those data put DAGKζ and its product, PA, in the center of events controlling Rac1-mediated PIP5KI recruitment and activation at the plasma membrane. The PA generated by DAGKζ activates PAK1 enabling RhoGDI dissociation form Rac1, subsequent anchoring of Rac1 at the plasma membrane and activation of the Rac1-associated PIP5KI, leading eventually to local PI(4,5)P2 synthesis (Fig. 3c, d). The initial step of the cascade requires an appearance of DAG in the plasma membrane. The lipid arises most likely from hydrolysis of PI(4,5)P2 catalyzed by PLC at the onset of cell stimulation (Fig. 3b). Of note, Rac proteins were found to associate with and stimulate the activity of PLCβ2 and PLCγ2 isoforms [105–107].
The physiological importance of the Rac1-PIP5KI interaction for PI(4,5)P2 synthesis is well documented. Studies performed on permeabilized yet living platelets allowing introduction of recombinant proteins revealed that interaction of Rac1 with PIP5KIβ, but not PIP5KIα, determined the uncapping of actin filaments as a result of PI(4,5)P2 synthesis. A point mutation introduced in the C-terminus of Rac1, which abolished binding of PIP5KIβ, also inhibited actin assembly in thrombin-stimulated cells, while exogenous PIP5KIβ introduced to permeabilized platelets restored the process [33]. Those studies put the Iβ isoform of PIP5K downstream of Rac1 in the signaling pathway leading to PI(4,5)P2 synthesis and actin polymerization relying on the exposure of the barbed ends of microfilaments. Another line of data pointed to the Iα isoform of PIP5KI as the one acting downstream of Rac1 during stimulation of thrombin receptor [99]. In that case, Rac1-GTP was found to mediate recruitment of PIP5KIα to the plasma membrane, but this processes required Rho-GTP involvement as well. Both Rac-GTP and Rho-GTP increased the activity of PIP5KIα five to sevenfold when co-expressed with it [99]. Finally, PIP5KIα rather than PIP5KIβ was reported to act downstream of Rac1 during actin remodeling, which underlies ruffling in PDGF-stimulated MG-63 fibroblasts [39]. Taken together, the data indicate that local activation of Rac1 can result in the recruitment of PIP5KI and its activation at the plasma membrane, however, the kinase isoform involved and the mode of Rac1 action (direct or indirect) varies from cell to cell.
Another link between Rac1 and PIP5KIα seems to be provided by the LIM protein Ajuba. Ajuba on the one hand influences the activity of Rac1 at nascent adhesive sites in a migrating cell by promoting the assembly of the p130Cas-Crk complex that interacts with the DOCK180-ELMO complex, a guanine nucleotide exchange factor of Rac1 [108]. On the other hand, Ajuba was also found to associate with PIP4KIIβ, PIP5KIα, and PIP5KIβ, but not PIP5KIγ in vivo. The binding of PIP5KIα was enhanced 20-fold during cell migration. Further in vitro studies indicated that the interaction of PIP5KIα with Ajuba stimulated the activity of the lipid kinase sixfold [40]. Hence, the Ajuba-Rac1-PIP5KIα interactions can regulate PI(4,5)P2 synthesis in the course of assembly of adhesive complexes required for cell migration. Taking into account that PIP5KIγ, the kinase isoform neglected by Ajuba, is localized to focal contacts and activated at these sites by another protein—talin (see below), the assembly of focal contacts provides an example of complexity of mechanisms fine-tuning the functions of PIP5KI isoforms at the plasma membrane.
Besides the GTPases of the Rho family, an involvement of ARF6 from the ARF-family GTPases, often acting in cooperation with PLD, is also considered in activation of PIP5KI at the plasma membrane. Combined in vitro and in vivo studies have indicated that myristoylated membrane-bound ARF6 activates PIP5KIα, Iβ and Iγa, and PLD2 [97, 109–111]. Therefore, the ARF6-induced association of PIP5KI with the plasma membrane can increase its activity by bringing the kinase close to PI(4)P and PA and additionally facilitating the reciprocal stimulation of PIP5KI and PLD by the respective products of their activities, PI(4,5)P2 and PA [111–113]. The ternary ARF6-PLD-PIP5KI complex has been implicated in controlling Ca2+-regulated exocytosis, ruffling, and receptor endocytosis [97, 109, 114, 115]. In terms of actin cytoskeleton rearrangements, ARF6 cooperates with Rac1 and the cross-talk between the proteins can be coordinated at the level of guanine nucleotide exchange factors of the two GTPases [116, 117].
PIP5KIγa and Iγc bind talin at focal contacts
Talin is a large, rod-shaped 270-kDa protein containing a globular head with the FERM domain, which binds PI(4,5)P2, PIP5KIγa/c, F-actin, focal adhesion kinase (FAK), β1- and β3-integrin. The flexible rod fragment of talin enables its dimerization and contains additional actin- and β-integrin-binding sites. The assembly of dimers engages the C-terminal domain of talin, which suggests that full-length talin can adopt variable conformations ranging from a parallel dimer to an extended one [118, 119]. The PIP5KIγa-binding site in talin was mapped to so-called subdomain F3 of the FERM domain, which structurally resembles the phosphotyrosine-binding (PTB) domain [29, 30]. The F3 subdomain of talin is also a site of β-integrin association, however, despite such overlapping, distinct amino acid motifs mediate the interactions of PIP5KIγa and β1-integrin with talin [29, 52, 120, 121]. In the kinase, the 642WVYSPLH648 motif located within the last 26 amino acids of the C-terminus (28 amino acids of human PIP5KIγa) was found to participate in talin binding [29, 52, 121] as shown in Fig. 2b. This amino acid sequence is also present in the rodent PIP5KIγc allowing its binding to talin as well, while PIP5KIγb, devoid of the C-terminal 26/28-amino acid extension does not interact with talin.
Phosphorylation of PIP5KIγa on Tyr644 within the 642WVYSPLH648 motif strengthens the interaction of the kinase with talin 15-fold and activates the kinase as well [29, 52]. Accordingly, the NMR structure of talin PTB in complex with the 641SWVpYSPLH648 peptide revealed an unusual electrostatic cluster involving the phospho-Tyr644 and basic amino acids of the PTB [122]. As shown in Fig. 4, the phosphorylation of PIP5KIγa is catalyzed by Src kinase, which associates with PIP5KIγa, and both these events are additionally upregulated by FAK kinase [30, 52]. It is assumed that the tyrosine phosphorylation releases autoinhibitory restraints within the PIP5KIγa molecule [121]. Of note, Src phosphorylates β-integrin as well, however, in contrast to phosphorylated PIP5KIγa, affinity of phosphorylated β1-integrin for talin is diminished. Eventually, this Src kinase activity weakens the interaction of β-integrin with talin [52]. The reciprocal effect exerted by tyrosine phosphorylation of PIP5KIγ and β1-integrin on their interactions with talin ensue from different amino acid composition of motifs involved in the binding [121].

Talin and PIP5KIγa/c interact during focal contact assembly. Upon cell activation, PIP5KIγa/c undergoes phosphorylation on Tyr644 by Src kinase, which also associates with it. Both events are positively regulated by FAK kinase. When phosphorylated, PIP5KIγa/c binds to the FERM domain located in the globular N-terminal part of talin. The kinase produces PI(4,5)P2, which binds to the FERM domain of talin and relieves autoinhibitory interaction of the talin head with rod. At these conditions, talin is able to bind β-integrins, inducing additionally their aggregation and activation. Intracellularly, talin binds actin filaments, which enables the formation of stress fibers. The Tyr644 can be dephosphorylated by Shp-1, which associates with PIP5KIγ. The activity of Shp-1 is inhibited by PI(4,5)P2 favoring the lipid accumulation. On the other hand, the binding of β-integrin to talin can be inhibited by phosphorylation of β-integrin by Src kinase
The enzymatic activity of PIP5KIγa is a prerequisite for the formation of talin-containing focal adhesions [30]. This is because PI(4,5)P2 produced locally by the kinase can bind to the FERM domain and relieve the autoinhibitory head–tail association within the talin molecule, thus exposing the integrin-binding site [123, 124]. As a result, PI(4,5)P2 provides a positive signal for the interaction of talin with β1- and β3-integrin, also promoting integrin clustering [125] and enabling their interaction with the extracellular matrix (Fig. 4). Simultaneously, talin interacts with actin filaments, thereby initiating the assembly of focal complexes. In addition to talin, PI(4,5)P2 also binds to other proteins of focal complexes, like vinculin, α-actinin, and syndecan-4, promoting their association with the complexes. PI(4,5)P2 can also support its own generation in a positive-feedback manner (as shown in Fig. 4) by inhibition of Shp-1 tyrosine phosphatase, which associates with and dephosphorylates PIP5KIγa [126]. Besides the phosphorylation-based activation of PIP5KIγa/c, stimulation of PIP5KIγb by PLD2 also adds to the synthesis of PI(4,5)P2, which facilitates cell adhesion [53]. All of these mechanisms enhance the activity of PIP5KIγ and production of PI(4,5)P2 during formation and maturation of focal contacts. Recently, PIP5KIγa was found to associate with PLCγ1 and the association was inhibited by phosphorylation of Tyr634 in PIP5KIγa. This phosphorylation was catalyzed by the receptor for epidermal growth factor (EGF), which in this way can diminish PI(4,5)P2 hydrolysis and favor PI(4,5)P2 accumulation at the focal contacts required for cell migration [127].
On the other hand, a line of data indicates that PIP5KIγa and β-integrin compete for binding to talin [52, 120], which suggests that PI(4,5)P2, generated by tyrosine-phosphorylated talin-bound PIP5KIγa can limit its own production. The lipid promotes the interaction of talin with β1-integrin, therefore facilitating the displacement of PIP5KIγa from talin. This fine-tuned interaction of talin with β1-integrin, strengthened by PI(4,5)P2 and weakened by the competitive binding of phosphorylated PIP5KIγa and by β-integrin phosphorylation, has been proposed to confer a dynamic character on focal adhesions, enabling their assembly and disassembly [52]. Taking into account that talin forms dimers and that recently a second β-integrin-binding site in the talin rod has been characterized [117, 128, 129], possible combinations of talin interactions during focal contact assembly/disassembly are complex.
Interaction of PIP5KIγa with AP-2 adaptor at clathrin-coated pits
PIP5KIγ is the major PIP5KI producing PI(4,5)P2 in synapses where the lipid controls both exocytosis of synaptical vesicles and subsequent clathrin-mediated enodocytosis of membranes [23, 25, 130]. A combination of co-immunoprecipitation, pull-out and yeast two-hybrid studies has indicated that PIP5KIγa binds to the AP-2 adaptor of the clathrin-based coat [49–51]. The AP-2 adaptor is a heterotetramer composed of large α and β2 subunits, a medium-sized μ2 subunit recognizing cargo proteins, and a small σ2 subunit. The subunits form a core with two appendage domains of α and β2 subunits linked to them via flexible linkers (Fig. 5). Besides the cargo recognition, the complex acts a scaffold binding an array of clathrin-accompanying proteins.

PIP5KIγa interacts with AP-2 at the onset of receptor endocytosis. a, b Initially, GTP-loaded ARF6 activates PIP5KIγa leading to synthesis of PI(4,5)P2. The lipid provides an anchor for the α subunit of the AP-2 complex. Concomitant dephosphorylation of Ser645 of PIPKIγa by calcineurin (CalN) is required for binding of the kinase C-terminus to the β2 appendage of the AP-2 complex. c Conformational changes of the μ subunit of the AP-2 complex allow recognition of a cargo receptor. Besides the binding of the cargo, μ2 also interacts with PIPKIγa catalytic core, activating the kinase and leading to PI(4,5)P2 production. The lipid serves as an anchor for the μ2 chain of the AP-2 complex. d The C-terminus of the kinase can also shift from β2 to the μ2 and binds to the μ2 chain via the 644YSPL647 motif. This interaction promotes the recognition of cargo receptors devoid of the canonical YXXØ motif; it also activates PIP5KIγa facilitating local PI(4,5)P2 accumulation
The interaction between PIP5KIγa and AP-2 involves multiple sites (see Fig. 5): the μ2 subunit binds to the catalytic core of the kinase [50] and to the 644YSPL647 motif of the 26-amino-acid C-terminal extension of the kinase [49, 131]. In addition, the C-terminus of PIP5KIγa interacts with the β2 chain of AP-2 [51]. PIP5KIγa is the only PIP5KIγ isoform interacting with AP-2 [132]. Further site-directed mutagenesis studies and structural analysis indicated that the amino acid motifs of the kinase C-terminus involved in the binding of the μ2 and β2 chains partially overlap (Fig. 2b). Therefore, the two interactions of the kinase C-terminus are mutually exclusive [131, 132]. Important contacts formed between the PIP5KIγa C-terminal tail and the β2 chain involve aromatic residues (Tyr635, Phe636, Trp642, and Tyr644) and Ser645 of the kinase [51, 131, 132]. Phosphorylation of Ser645 by cdk5 kinase inhibits the interaction between PIP5KIγa and AP-2 β, while dephosphorylation of Ser645, catalyzed presumably by calcineurin, allows the binding. It is striking that the amino acid sequences involved in AP-2 β and talin binding overlap (Fig. 2b). Notably, while Ser645 phosphorylation inhibits the association of PIP5KIγa with both talin and AP-2, phosphorylation of Tyr644 facilitates association of PIP5KIγa with talin but inhibits the association of the kinase with the AP-2 complex. These data suggest that depending on the phosphorylation state of Tyr644, PIP5KIγa can shuttle between sites of cell adhesion and endocytosis [131].
The complex nature of the interactions between PIP5KIγa and the AP-2 adaptor creates an elaborate molecular mechanism controlling rapid PI(4,5)P2 synthesis during assembly of clathrin-based coat. In resting neurons, PIP5KIγa is located on the presynaptic membrane but, being phosphorylated on Ser645, it does not interact with AP-2. Excitation of cells and depolarization of the presynaptic membrane leads to dephosphorylation of the kinase allowing AP-2 binding, as shown in hippocampal neurons [51]. There are data suggesting that initially PIP5KIγa is activated by ARF6 [109, 133], producing a pool of PI(4,5)P2 that anchors AP-2 at the plasma membrane by binding to the α subunit of the complex (Fig. 5a, b). This pool of AP-2 can, in turn, interact with dephosphorylated PIP5KIγa, most likely by engaging the β2 appendage and the kinase C-terminus via relatively weak interactions (Fig. 5b). Subsequent conformational changes of the μ2 chain induced by its phosphorylation allow binding of cargo proteins containing the conventional YXXØ sorting motif. They can also facilitate the interaction of the μ2 chain with the PIP5KIγa catalytic core leading to activation of the kinase and thus to local production and accumulation of PI(4,5)P2 (Fig. 5c). A shift of the C-terminus of the kinase from the β2 to the μ2 chain (Fig. 5d) also stimulates the PIP5KIγa activity [130]. This displacement of the kinase tail from the β2 appendage can be facilitated by clathrin in the course of coat assembly [132]. Eventually, local production of PI(4,5)P2 enables recruitment of the components of the clathrin coat, including AP-2 itself, epsin, AP180, Dab2 and dynamin 2. As the 644YSPL647 sequence of the PIP5KIγa C-terminus corresponds to the conventional YXXØ sorting motif of cargo receptors, the occupation of μ2 by the kinase tail may also favor sorting of cargo lacking the YXXØ motif. This mechanism can be characteristic to internalization of proteins of presynaptic vesicles helping to sort them away from constitutively internalized cargo receptors [134].
It is noteworthy that PIP5KIβ rather than PIP5KIγ has been implicated in the production of PI(4,5)P2 required for AP-2 recruitment and transferrin receptor uptake in HeLa and CV-1 cells [48], while PIP5KIα (murine PIP5KIβ) was indicated to participate in endocytosis of EGF receptor in NR6 fibroblasts [135]. These data led to a suggestion that clathrin-mediated endocytosis in nonneuronal cells, especially endocytosis of nutrient receptors like transferrin receptor, can be regulated differently from endocytosis taking place in synapses [48]. However, thus far no specific structural basis for the binding of PIP5KIα and Iβ isoforms to AP-2 or other components of the clathrin-containing coat has been proposed. In addition, the above-mentioned studies were based on overexpression of the kinases or their knockdown by RNA interference and the results discussed above may reflect the effects of perturbations of cellular PI(4,5)P2 level on clathrin coat assembly. The PIP5KIγa participation in AP-2 recruitment at synapses is well documented and this may be the major function of this kinase isoform in the brain, judging from the high expression of PIP5KIγa there. However, an engagement of PIP5KIγa in receptor endocytosis in other cell types is conceivable [49, 50].
The termination of PI(4,5)P2 signals at clathrin-coated vesicles in synapses comes from cessation of its synthesis due to phosphorylation of Ser645 of PIP5KIγa [51]. Equally important is the dephosphorylation of PI(4,5)P2 by synaptojanin 1. The reduction of PI(4,5)P2 level shortly after scission of clathrin-coated vesicles is required for the coat turnover, judging from an excessive coat association in neurons in synaptojanin 1-null mice [12]. Such rapid and transient production of PI(4,5)P2 accompanies phagocytosis as well. Similarly to the clathrin-based coat assembly, production of PI(4,5)P2 by PIP5KIα is required during phagocytosis for actin polymerization and particle internalization. The subsequent actin depolymerization and phagosome maturation requires, in turn, reduction of the PI(4,5)P2 level in the phagosome membrane by PI(4,5)P2 hydrolysis and phosphorylation to PI(3,4,5)P3 [73, 136].
Are PI(4,5)P2 clusters formed in the plasma membrane?
Type I PIP5Ks can efficiently increase the local concentration of PI(4,5)P2 when the lateral mobility of the lipid is restricted. If the diffusion coefficient (D) of PI(4,5)P2 was comparable to that of freely diffusible membrane lipids, PI(4,5)P2 would rapidly move away from the sites of its synthesis. This subject was thoroughly studied by McLaughlin and colleges [137] who microinjected micelles of long-chain BODIPY-labeled PI(4,5)P2 into fibroblasts and epithelial cells and measured the diffusion coefficient of the lipid in the inner leaflet of the plasma membrane using confocal imaging and fluorescence correlation spectroscopy. The averaged D value of PI(4,5)P2 in the examined cells was 0.8 ± 0.2 μm2/s in comparison to 1.5 ± 0.9 μm2/s found for rhodamine-phosphatidylethanolamine (25°C). The diffusion coefficient of the fluorescent PI(4,5)P2 was significantly higher when the lipid was incorporated into the outer leaflet of the plasma membrane of various cells or into giant unilamellar vesicles. The D values of PI(4,5)P2 in those membranes reached 2 ± 1.3 μm2/s and 3.7 ± 0.8 μm2/s, respectively, with the latter value characteristic also of other lipids in model membranes [137–139]. The estimated values of the diffusion coefficient of PI(4,5)P2 in the outer leaflet was in agreement with earlier FRAP (fluorescence recovery after photobleaching) measurements for BODIPY-labeled PI(4,5)P2 incorporated into the outer leaflet of the plasma membrane of N1E-155 neuroblastoma cells [140]. The over twofold lower D value of PI(4,5)P2 in the cytoplasmic than in the extracellular leaflet of the plasma membrane suggests that the lateral diffusion of the lipid in the inner leaflet is indeed restricted.
Notably, the diffusion coefficient of PI(3,4,5)P3 estimated at 0.5 μm2/s by analysis of translocations of the PH domain of Akt kinase, a molecular sensor of PI(3,4,5)P3 expressed in NIH 3T3 fibroblasts [141], is in approximate agreement with the D of PI(4,5)P2 found by McLaughlin’s group [137]. When combined with the short lifetime of PI(3,4,5)P3 (<1 min) this rate of PI(3,4,5)P3 mobility allows a polar gradient of the lipid to be formed in moving cells. The gradient is generated by localized synthesis of PI(3,4,5)P3 at the cell front coupled with its degradation toward the rear and enables efficient cell motility. Therefore, accumulation of PI(4,5)P2 at particular plasma membrane locations, like forming phagosomes and ruffles (1–10 μm in length) is also conceivable [137, 142], although contradictory opinions should be noted [140].
Some studies indicate even lower lateral mobility of PI(4,5)P2 [143, 144]. It was shown that long-chain NBD-labeled PI(4,5)P2 mixed with a cationic shuttle and delivered to the plasma membrane of mouse atrial myocytes through a micropipette attached to the cells failed to leave the patch of the membrane surrounded by the pipette walls. Accompanying FRAP measurements confirmed that PI(4,5)P2 moved extremely slowly in the plane of the myocyte plasma membrane with the D calculated at 0.00039 μm2/s [143]. This is 104 time less than the D values found in the other studies discussed above. A similar severe restriction of lateral mobility of NBD-labeled PI(4,5)P2 was found in the plasma membrane of HEK293 cells [144]. The confinement of PI(4,5)P2 was abrogated by depolymerization of actin pointing to the cytoskeleton as a factor responsible for the low mobility of the lipid. On the other hand, NBD-PI and NBD-PI(4)P rapidly crossed the boundaries of micropipettes attached to the cell surface [143, 144]. Notably, the same approach has revealed that in CHO cells PI(4,5)P2 is as mobile as PI(4)P [144] leaving open the question as to how common is the extremely slow diffusion of PI(4,5)P2 in cells.
Several mechanisms can limit the lateral mobility of PI(4,5)P2 in the plasma membrane [145]. One can envisage the existence of boundaries or “fences” imposed by the submembrane cytoskeleton and strong bending of the plasma membrane, which could hinder PI(4,5)P2 diffusion, for example, at the neck of the phagosome. Recently, confinement of PI(4,5)P2 within so-called lipid rafts or its sequestration by proteins due to electrostatic interactions are being considered, as discussed below.
Lipid rafts and raft-related caveolae are plasma membrane microdomains enriched in sphingolipids and cholesterol revealed in the 1990s [146–150]. Since this discovery, several attempts have been undertaken to examine whether PI(4,5)P2 can be accumulated within these membrane domains. Rafts/caveolae-derived membranes are resistant to detergent solubilization, and during ultracentrifugation of detergent cell lysates over density gradients they float to fractions of low density. After fractionation of lysates derived from cells depleted of cholesterol, rafts/caveolae-residing lipids and proteins shift to medium- and high-density fractions reflecting disintegration of the microdomains [151–154]. Based on such biochemical analysis, accumulation of PI(4,5)P2 has been postulated in rafts isolated from epithelial Madin-Darby kidney cells (MDCK), epidermal A431 cells, and neuroblastoma cells [155–159]. Those early biochemical data indicated also that rafts harbored enzymes governing PI(4)P and PI(4,5)P2 synthesis, hydrolysis, and dephosphorylation [155, 158, 160]. Accumulation of PI(4,5)P2 and production of PI(3,4,5)P3 was also implicated to take place in platelet rafts [161]. However, fractionation of detergent cell lysates may lead to an enrichment of bona-fide raft-originating membranes in non-raft proteins and lipids [162–164]. Moreover, the dependence of the PI(4,5)P2 distribution and activity on cholesterol level in the plasma membrane does not necessarily prove an association of PI(4,5)P2 with rafts [165, 166]. Due to these limitations, in another approach the PH domain of PLCδ1 was used as a probe to localize PI(4,5)P2 in frozen ultrasections of fibroblasts and HEK293 cells by immunoelectron microscopy and for in vivo analysis of PI(4,5)P2 clustering by the FRET technique [71, 166]. No indications of PI(4,5)P2 clusters have been found in those studies. This is in agreement with calculations based on the diffusion coefficient value of PI(4,5)P2, suggesting that PI(4,5)P2 patches of nanometer scale should rapidly dissipate (within milliseconds according to [142]; see also [140, 167]). One should bear in mind, however, that the PI(4,5)P2-binding probes can have a limited access to PI(4,5)P2 already pre-occupied by other proteins at steady state, leaving the problem of PI(4,5)P2 distribution in the plasma membrane unsolved.
Despite the technical difficulties of the raft studies, a line of data indicates that during activation of a distinct class of plasma membrane receptors named immunoreceptors rafts can contribute to PI(4,5)P2 synthesis and clustering. All these receptors have a tyrosine-based activation motif whose phosphorylation is catalyzed by kinases of the Src family known to reside in rafts. Upon activation, the immunoreceptors merge with the rafts, which enables their phosphorylation and triggers signaling cascades. Concomitantly, the rafts merge into spatially organized signaling platforms accommodating proteins and lipids of the cascades [168–171]. Upon activation of B cell antigen receptor, PIP5KI has been found to become recruited to plasma membrane rafts by Bruton’s tyrosine kinase (BTK) catalyzing local synthesis of PI(4,5)P2, which could then be used by PI3K and PLCγ to produce PI(3,4,5)P3, DAG, and IP3 [172]. During T cell antigen receptor stimulation, rafts assemble into a structure known as the immunological synapse at the T cell/antigen-presenting cell contact site. The integrity of the immunological synapse is maintained through anchoring to the underlying actin cytoskeleton, possibly via PI(4,5)P2-binding proteins. Among these, vinculin and talin were identified by mass spectroscopic analysis in raft fractions isolated from T cells [173]. Accordingly, clusters of PI(4,5)P2 were visualized in vivo in T cells transfected with the PH domain of PLCδ1, following induction of raft coalescence by cross-linking of GM1 ganglioside. PI(3,4,5)P3 and DAG were also found in the GM1-enriched domains, indicating that rafts can serve as sites of PI(4,5)P2 phosphorylation and hydrolysis [174]. Activation of another immunoreceptor, Fcγ receptor II involved in phagocytosis, induces an enrichment of raft fractions in PIP5KIα and PI(4,5)P2. Further ultrastructural studies performed on sheets of plasma membrane obtained by mechanical cleavage of cells revealed that PI(4,5)P2 accumulated at the margins of raft conglomerates [43]. Such structures form during Fcγ receptor II activation and accommodate the receptor and proteins of its signaling cascade [175, 176]. At these particular locations, PI(4,5)P2 may function in tethering the actin cytoskeleton to the plasma membrane (as proposed for T cell receptor) and govern internalization of particles. Taken together, the data suggest that PI(4,5)P2 can accumulate at margins of raft conglomerates whose formation accompanies activation of immunoreceptors.
It has also been hypothesized that the spatial confinement of PI(4,5)P2 in the plasma membrane can result from the lateral sequestration of the lipid by proteins. Indeed, a significant fraction of the lipid is bound to proteins since PI(4,5)P2 controls the adhesion between the plasma membrane and the submembrane cytoskeleton [177]. This finding is in agreement with the results of studies on lipid composition of microvesicles released from human erythrocytes [178]. The microvesicles were depleted by half in PI(4,5)P2 and PI(4)P despite having a composition of the major phospholipids very similar to that of native erythrocyte membrane. Based on those data it was proposed that about 50% of PI(4,5)P2 in the erythrocyte membrane was bound to the integral membrane proteins glycophorin and proteins of the spectrin-based membrane skeleton [178]. Such interactions could slow the diffusion of PI(4,5)P2 into budding microvesicles. Subsequent measurements of the diffusion of PI(4,5)P2 in the plasma membrane of Rat1 cells indicated that the erythrocyte results were representative of other cells as well [137]. The diffusion coefficient of long-chain BODIPY-labeled PI(4,5)P2 in the inner leaflet of the plasma membrane (0.9 ± 0.2 μm2/s) was 2.8-fold lower that the D value of PI(4,5)P2 in blebs formed by the cells (2.5 ± 0.8 μm2/s) and presumably lacking most of the cytoskeleton and integral membrane proteins interacting with the cytoskeleton. The authors interpreted those results to mean that two-thirds of PI(4,5)P2 in Rat1 plasma membrane were bound reversibly to the cytoskeleton or other proteins which, in turn, hindered the lateral diffusion of the lipid [137]. Further theoretical calculations support this hypothesis. A hypothetical spherical cell 10 μm in diameter contains about 30,000 PI(4,5)P2/μm2 of the plasma membrane, accounting for 30 μM PI(4,5)P2 concentration (assuming PI(4,5)P2 is dispersed uniformly in the interior of the cells) [137, 142]. Out of this, 20 μM PI(4,5)P2 is sequestered while 10 μM diffuses freely, as indicated by the diffusion measurements [137]. Accordingly, a predominant fraction of the PH domain of PLCδ1 expressed in cells is bound to the plasma membrane (K D ~ 1.2 μM) while the PH domain of pleckstrin (K D ~ 30 μM) remains in the cytoplasm, suggesting that the concentration of free PI(4,5)P2 is in the range between 2 and 30 μM [137, 142].
Ample data indicate that PI(4,5)P2 can be sequestered by a group of integral and peripheral membrane proteins that contain stretches of positively charged amino acids. Thus far, the ability to sequester and cluster PI(4,5)P2 has been ascribed to a ubiquitously expressed protein named myristoylated alanine-rich C kinase substrate (MARCKS). The highly conserved effector domain of MARCKS contains three serine residues whose phosphorylation is catalyzed by protein kinase C; the domain also contains sites of calmodulin and actin binding. MARCKS binds to actin filaments and cross-links them, and this cross-linking activity is disrupted by both phosphorylation of MARCKS and by Ca2+-loaded calmodulin. Most importantly, the effector domain of MARCKS also contains 15 basic amino acid residues that can bind three PI(4,5)P2 molecules inducing the lipid clustering in the plane of the membrane, as indicated by biochemical and biophysical studies. Physiological levels of MARCKS (10 μM in neuronal tissue) correspond to the estimated cellular PI(4,5)P2 concentration and, thus, MARCKS can sequester a significant pool of the lipid [142]. The sequestration of PI(4,5)P2 by MARCKS can be reversed by interaction of the effector domain with Ca2+-loaded calmodulin or by phosphorylation of the serine residues of the domain by protein kinase C. Under these conditions, MARCKS detaches from the plasma membrane despite myristoylation of its N-terminus. MARCKS-bound PI(4,5)P2 is less susceptible to hydrolysis by PLCδ1, however, dissociation of the protein from the plasma membrane exposes PI(4,5)P2 for hydrolysis and, possibly, for interactions with other effector proteins [179, 180]. Some transmembrane receptors, like EGF receptor and membrane-binding proteins structurally resembling MARCKS, have also been proposed to reversibly sequester PI(4,5)P2 in the plasma membrane in a calcium-dependent manner [180]. Among the latter, neuronal growth-associated protein 43 (GAP43) is of special interest. Unlike MARCKS, GAP43 undergoes dual palmitoylation at the N-terminus, which predisposes the protein for anchoring in the inner leaflet of rafts. Studies performed on large unilamellar vesicles composed of lipids mimicking the chemical make-up of rafts indicate that palmitoylated GAP43 can facilitate association of PI(4,5)P2 with rafts. Considering that PI(4,5)P2 usually has an unsaturated fatty acyl residue at the n-2 position of the glycerol backbone, its partition to lipid rafts is unlikely. However, after binding to palmitoylated GAP43, the free energy of transfer of PI(4,5)P2 from non-raft to raft environment is lowered [181]. GAP43 can act as a reversible PI(4,5)P2 sink in growth cones on neurons responding to changes in the level of Ca2+-loaded calmodulin. In accordance with the in vitro studies, an involvement of MARCKS and GAP43 in the formation of PI(4,5)P2 clusters engaged in cytoskeleton rearrangement at the leading edge of motile cells was indicated by Caroni’s group [182, 183]. Hence, distinct proteins can attract and concentrate PI(4,5)P2 in clusters in the plane of the plasma membrane to release the lipid reservoir in response to the rise of free Ca2+ concentration.
Conclusions
Stimulation of cells with a variety of agents triggers signaling cascades that involve PI(4,5)P2. PI(4,5)P2 contributes to these processes by being converted into second messengers or by controlling the activity of PI(4,5)P2-binding proteins. The accumulation of PI(4,5)P2 at sites of cell stimulation can ensue from two general mechanisms: induction of local PI(4,5)P2 synthesis by PIP5KI and revealing of PI(4,5)P2 clusters pre-existing in the plasma membrane. PIP5KI can be anchored at the plasma membrane via electrostatic interactions with PI(4)P and PI(4,5)P2 being subject to substrate and product inhibition until the appearance of activating factors like PA. PIP5KI can be also recruited and activated at the plasma membrane by proteins including Rac1, talin, the AP-2 complex, and BTK. These interactions allow selective engagement of PIP5KI isoforms. Thus far, the molecular basis underlying such selectivity is only known for the interaction of PIP5KIγa with talin and the AP-2 complex. In the case of activation of receptors that utilize assemblies of rafts as “signaling platforms”, the synthesis of PI(4,5)P2 can be confined to these membrane structures. Finally, proteins like MARCKS can sequester PI(4,5)P2 into clusters in the plane of the plasma membrane of resting cells and release the lipid allowing its interaction with effector proteins in response to changes of free Ca2+ level accompanying cell activation.
Abbreviations
- FERM:
-
Band 4.1-ezrin-radixin-moesin homology
- BTK:
-
Bruton’s tyrosine kinase
- DAG:
-
Diacylglycerol
- DAGK:
-
Diacylglycerol kinase
- EGF:
-
Epidermal growth factor
- IP3 :
-
Inositol trisphosphate
- MDCK:
-
Madin-Darby kidney cells
- MARCKS:
-
Myristoylated alanine-rich C kinase substrate
- GAP43:
-
Growth-associated protein 43
- OCRL1:
-
Oculocerebrorenal syndrome of Lowe 1 phosphatase
- OSBP:
-
Oxysterol-binding protein
- PTEN:
-
Phosphatase and tensin homologue on chromosome 10
- PA:
-
Phosphatidic acid
- PI:
-
Phosphatidylinositol
- PIPK:
-
Phosphatidylinositol phosphate kinase
- PI3K:
-
Phosphatidylinositol 3-kinase
- PI4K:
-
Phosphatidylinositol 4-kinase
- PI(3)P:
-
Phosphatidylinositol 3-monophosphate
- PI(4)P:
-
Phosphatidylinositol 4-monophosphate
- PI(5)P:
-
Phosphatidylinositol 5-monophosphate
- PI(4,5)P2 :
-
Phosphatidylinositol 4,5-bishosphate
- PI(3,4,5)P3 :
-
Phosphatidylinositol 3,4,5-trisphosphate
- Fapp:
-
Phosphatidylinositol-4-phosphate adaptor protein
- IPP 5-Ptase:
-
Inositol polyphosphate 5-phosphatase
- PLC:
-
Phospholipase C
- PLD:
-
Phospholipase D
- PTB:
-
Phosphotyrosine-binding
- PH:
-
Pleckstrin homology
- PIP5KI:
-
Type I phosphatidylinositol-4-phosphate 5-kinase
- PIP4KII:
-
Type II phosphatidylinositol-5-phosphate 4-kinase
- OSH2:
-
Yeast oxysterol-binding protein homologue
References
Hokin MR, Hokin LE (1953) Enzyme secretion and the incorporation of P32 into phospholipides of pancreas slices. J Biol Chem 203:967–977
Yin HL, Janmey PA (2003) Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol 65:761–789
Di Paolo G, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657
Rameh LE, Cantley LC (1999) The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem 274:8347–8350
Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273:13375–13378
McPherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, Sossin WS, Bauerfeind R, Nemoto Y, De Camilli P (1996) A presynaptic inositol-5-phosphatase. Nature 379:353–357
Nemoto Y, Arribas M, Haffner C, DeCamilli P (1997) Synaptojanin 2, a novel synaptojanin isoform with a distinct targeting domain and expression pattern. J Biol Chem 272:30817–30821
Lowe M (2005) Structure and function of the Lowe syndrome protein OCRL. Traffic 6:711–719
Ungewickell A, Hugge C, Kisseleva M, Chang SC, Zou J, Feng Y, Galyov EE, Wilson M, Majerus PW (2005) The identification and characterization of two phosphatidylinositol-4,5-bisphosphate 4-phosphatases. Proc Natl Acad Sci USA 102:18854–18859
Lowe CU, Terrey M, MacLachlan EA (1952) Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child 83:164–184
Leahey AM, Charnas LR, Nussbaum RL (1993) Nonsense mutations in the OCRL-1 gene in patients with the oculocerebrorenal syndrome of Lowe. Hum Mol Genet 2:461–463
Cremona O, Di Paolo G, Wenk MR, Lüthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99:179–188
Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, Arancio O, Davisson MT, Antonarakis SE, Gardiner K, De Camilli P, Di Paolo G (2008) Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc Natl Acad Sci USA 105:9415–9420
Zou J, Marjanovic J, Kisseleva MV, Wilson M, Majerus PW (2007) Type I phosphatidylinositol-4,5-bisphosphate 4-phosphatase regulates stress-induced apoptosis. Proc Natl Acad Sci USA 104:16834–16839
Dove SK, Dong K, Kobayashi T, Williams FK, Michell RH (2009) Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. Biochem J 419:1–13
Sbrissa D, Ikonomov OC, Deeb R, Shisheva A (2002) Phosphatidylinositol 5-phosphate biosynthesis is linked to PIKfyve and is involved in osmotic response pathway in mammalian cells. J Biol Chem 277:47276–47284
Coronas S, Lagarrigue F, Ramel D, Chicanne G, Delsol G, Payrastre B, Tronchere H (2008) Elevated levels of PtdIns5P in NPM-ALK transformed cells: implication of PIKfyve. Biochem Biophys Res Commun 372:351–355
Shisheva A (2008) PIKfyve: partners, significance, debates, paradoxes. Cell Biol Int 32:591–604
Tolias KF, Rameh LE, Ishihara H, Shibasaki Y, Chen J, Prestwich GD, Cantley LC, Carpenter C (1998) Type I phosphatidylinositol-4-phosphate 5-kinases synthesize the novel lipids phosphatidylinositol 3,5-bisphosphate and phosphatidylinositol 5-phosphate. J Biol Chem 273:18040–18046
Ikonomov OC, Sbrissa D, Mlak K, Kanzaki M, Pessin J, Shisheva A (2002) Functional dissection of lipid and protein kinase signals of PIKfyve reveals the role of PtdIns 3,5-P2 production for endomembrane integrity. J Biol Chem 277:9206–9211
Ishihara H, Shibasaki Y, Kizuki N, Katagiri H, Yazaki Y, Asano T, Oka Y (1996) Cloning of cDNAs encoding two isoforms of 68-kDa type I phosphatidylinositol-4-phosphate 5-kinase. J Biol Chem 271:23611–23614
Loijens JC, Anderson RA (1996) Type I phosphatidylinositol-4-phosphate 5-kinases are distinct members of this novel lipid kinase family. J Biol Chem 271:32937–32943
Ishihara H, Shibasaki Y, Kizuki N, Wada T, Yazaki Y, Asano T, Oka Y (1998) Type I phosphatidylinositol-4-phosphate 5-kinases. Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J Biol Chem 273:8741–8748
Giudici ML, Emson PC, Irvine RF (2004) A novel neuronal-specific splice variant of type I phosphatidylinositol 4-phosphate 5-kinase isoform γ. Biochem J 379:489–496
Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431:415–422
Jenkins GH, Fisette PL, Anderson RA (1994) Type I phosphatidylinositol 4-phosphate 5-kinase isoforms are specifically stimulated by phosphatidic acid. J Biol Chem 269:11547–11554
Itoh T, Ishihara H, Shibasaki Y, Oka Y, Takenawa T (2000) Autophosphorylation of type I phosphatidylinositol phosphate kinase regulates its lipid kinase activity. J Biol Chem 275:19389–19394
Park SJ, Itoh T, Takenawa T (2001) Phosphatidylinositol 4-phosphate 5-kinase type I is regulated through phosphorylation response by extracellular stimuli. J Biol Chem 276:4781–4787
Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P (2002) Recruitment and regulation of phosphatidylinositol phosphate kinase type 1γ by the FERM domain of talin. Nature 420:85–89
Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA (2002) Type Iγ phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 420:89–93
Mellman DL, Gonzales ML, Song C, Barlow CA, Wang P, Kendziorski C, Anderson RA (2008) A PtdIns(4,5)P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 451:1013–1017
Chang JD, Field SJ, Rameh LE, Carpenter CL, Cantley LC (2004) Identification and characterization of a phosphoinositide phosphate kinase homolog. J Biol Chem 279:11672–11679
Tolias KF, Hartwig JH, Ishihara H, Shibasaki Y, Cantley LC, Carpenter CL (2000) Type Iα phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr Biol 10:153–156
Yamamoto M, Chen MZ, Wang YJ, Sun HQ, Wei Y, Martinez M, Yin HL (2006) Hypertonic stress increases phosphatidylinositol 4,5-bisphosphate levels by activating PIP5KIβ. J Biol Chem 281:32630–32638
Wang Y, Litvinov RI, Chen X, Bach TL, Lian L, Petrich BG, Monkley SJ, Kanaho Y, Critchley DR, Sasaki T, Birnbaum MJ, Weisel JW, Hartwig J, Abrams CS (2008) Loss of PIP5KIγ, unlike other PIP5KI isoforms, impairs the integrity of the membrane cytoskeleton in murinsmegakaryocytes. J Clin Invest 118:812–881
Wang Y, Chen X, Lian L, Tang T, Stalker TJ, Sasaki T, Kanaho Y, Brass LF, Choi JK, Hartwig JH, Abrams CS (2008) Loss of PIP5KIβ demonstrates that PIP5KI isoform-specific PIP2 synthesis is required for IP3 formation. Proc Natl Acad Sci USA 105:14064–14069
Mao YS, Yamaga M, Zhu X, Wei Y, Sun H, Wang J, Yun M, Wang Y, Di Paolo G, Bennett M, Mellman I, Abrams CS, De Camilli P, Lu CY, Yin HL (2009) Essential and unique roles of PIP5K-γ and -α in Fcγ receptor-mediated phagocytosis. J Cell Biol 184:281–296
Coppolino MG, Dierckman R, Loijens J, Collins RF, Pouladi M, Jongstra-Bilen J, Schreiber AD, Trimble WS, Anderson R, Grinstein S (2002) Inhibition of phosphatidylinositol-4-phosphate 5-kinase Iα impairs localized actin remodeling and suppresses phagocytosis. J Biol Chem 277:43849–43857
Doughman RL, Firestone AJ, Wojtasiak ML, Bunce MW, Anderson RA (2003) Membrane ruffling requires coordination between type Iα phosphatidylinositol phosphate kinase and Rac signaling. J Biol Chem 278:23036–23045
Kisseleva M, Feng Y, Ward M, Song C, Anderson RA, Longmore GD (2005) The LIM protein Ajuba regulates phosphatidylinositol 4,5-bisphosphate levels in migrating cells through an interaction with and activation of PIPKIα. Mol Cell Biol 25:3956–3966
Chen MZ, Zhu X, Sun HQ, Mao YS, Wei Y, Yamamoto M, Yin HL (2009) Oxidative stress decreases phosphatidylinositol 4,5-bisphosphate levels by deactivating phosphatidylinositol-4-phosphate 5-kinase β in a Syk-dependent manner. J Biol Chem 284:23743–23753
Tolias KF, Couvillon AD, Cantley LC, Carpenter CL (1998) Characterization of a Rac1- and RhoGDI-associated lipid kinase signaling complex. Mol Cell Biol 18:762–770
Szymanska E, Korzeniowski M, Raynal P, Sobota A, Kwiatkowska K (2009) Contribution of PIP-5 kinase Iα to raft-based FcγRIIA signaling. Exp Cell Res 315:981–995
Shibasaki Y, Ishihara H, Kizuki N, Asano T, Oka Y, Yazaki Y (1997) Massive actin polymerization induced by phosphatidylinositol-4-phosphate 5-kinase in vivo. J Biol Chem 272:7578–7581
Rozelle AL, Machesky LM, Yamamoto M, Driessens MH, Insall RH, Roth MG, Luby-Phelps K, Marriott G, Hall A, Yin HL (2000) Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr Biol 10:311–320
Yamamoto M, Hilgemann DH, Feng S, Bito H, Ishihara H, Shibasaki Y, Yin HL (2001) Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J Cell Biol 152:867–876
Boronenkov IV, Loijens JC, Umeda M, Anderson RA (1998) Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol Biol Cell 9:3547–3560
Padron D, Wang YJ, Yamamoto M, Yin H, Roth MG (2003) Phosphatidylinositol phosphate 5-kinase Iβ recruits AP-2 to the plasma membrane and regulates rates of constitutive endocytosis. J Cell Biol 162:693–701
Bairstow SF, Ling K, Su X, Firestone AJ, Carbonara C, Anderson RA (2006) Type Iγ661 phosphatidylinositol phosphate kinase directly interacts with AP2 and regulates endocytosis. J Biol Chem 281:20632–20642
Krauss M, Kukhtina V, Pechstein A, Haucke V (2006) Stimulation of phosphatidylinositol kinase type I-mediated phosphatidylinositol (4,5)-bisphosphate synthesis by AP-2 μ-cargo complexes. Proc Natl Acad Sci USA 103:11934–11939
Nakano-Kobayashi A, Yamazaki M, Unoki T, Hongu T, Murata C, Taguchi R, Katada T, Frohman MA, Yokozeki T, Kanaho Y (2007) Role of activation of PIP5Kγ661 by AP-2 complex in synaptic vesicle endocytosis. EMBO J 26:1105–1116
Ling K, Doughman RL, Iyer VV, Firestone AJ, Bairstow SF, Mosher DF, Schaller MD, Anderson RA (2003) Tyrosine phosphorylation of type Iγ phosphatidylinositol phosphate kinase by Src regulates an integrin-talin switch. J Cell Biol 163:1339–1349
Powner DJ, Payne RM, Pettitt TR, Giudici ML, Irvine RF, Wakelam MJ (2005) Phospholipase D2 stimulates integrin-mediated adhesion via phosphatidylinositol 4-phosphate 5-kinase Iγb. J Cell Sci 118:2975–2986
El Sayegh TY, Arora PD, Ling K, Laschinger C, Janmey PA, Anderson RA, McCulloch CA (2007) Phosphatidylinositol-4,5 bisphosphate produced by PIP5KIγ regulates gelsolin, actin assembly, and adhesion strength of N-cadherin junctions. Mol Biol Cell 18:3026–3038
McEwen RK, Dove SK, Cooke FT, Painter GF, Holmes AB, Shisheva A, Ohya Y, Parker PJ, Michell RH (1999) Complementation analysis in PtdInsP kinase-deficient yeast mutants demonstrates that Schizosaccharomyces pombe and murine Fab1p homologues are phosphatidylinositol 3-phosphate 5-kinases. J Biol Chem 274:33905–33912
Kunz J, Wilson MP, Kisseleva M, Hurley JH, Majerus PW, Anderson RA (2000) The activation loop of phosphatidylinositol phosphate kinases determines signaling specificity. Mol Cell 5:1–11
Homma K, Terui S, Minemura M, Qadota H, Anraku Y, Kanaho Y, Ohya Y (1998) Phosphatidylinositol-4-phosphate 5-kinase localized on the plasma membrane is essential for yeast cell morphogenesis. J Biol Chem 273:15779–15786
Boronenkov IV, Anderson RA (1995) The sequence of phosphatidylinositol-4-phosphate 5-kinase defines a novel family of lipid kinases. J Biol Chem 270:2881–2884
Castellino AM, Parker GJ, Boronenkov IV, Anderson RA, Chao MV (1997) A novel interaction between the juxtamembrane region of the p55 tumor necrosis factor receptor and phosphatidylinositol-4-phosphate 5-kinase. J Biol Chem 272:5861–5870
Itoh T, Ijuin T, Takenawa T (1998) A novel phosphatidylinositol-5-phosphate 4-kinase (phosphatidylinositol-phosphate kinase IIγ) is phosphorylated in the endoplasmic reticulum in response to mitogenic signals. J Biol Chem 273:20292–20299
Rameh LE, Tolias KF, Duckworth BC, Cantley LC (1997) A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature 390:192–196
Rao VD, Misra S, Boronenkov IV, Anderson RA, Hurley JH (1998) Structure of type IIβ phosphatidylinositol phosphate kinase: a protein kinase fold flattened for interfacial phosphorylation. Cell 94:829–839
Clarke JH, Richardson JP, Hinchliffe KA, Irvine RF (2007) Type II PtdInsP kinases: location, regulation and function. Biochem Soc Symp 74:149–159
Ciruela A, Hinchliffe KA, Divecha N, Irvine RF (2000) Nuclear targeting of the β isoform of type II phosphatidylinositol phosphate kinase (phosphatidylinositol 5-phosphate 4-kinase) by its α-helix 7. Biochem J 346:587–591
Bazenet CE, Ruano AR, Brockman JL, Anderson RA (1990) The human erythrocyte contains two forms of phosphatidylinositol-4-phosphate 5-kinase which are differentially active toward membranes. J Biol Chem 265:18012–18022
Whiteford CC, Brearley CA, Ulug ET (1997) Phosphatidylinositol 3,5-bisphosphate defines a novel PI 3-kinase pathway in resting mouse fibroblasts. Biochem J 323:597–601
Kunz J, Fuelling A, Kolbe L, Anderson RA (2002) Stereo-specific substrate recognition by phosphatidylinositol phosphate kinases is swapped by changing a single amino acid residue. J Biol Chem 277:5611–5619
Jarquin-Pardo M, Fitzpatrick A, Galiano FJ, First EA, Davis JN (2007) Phosphatidic acid regulates the affinity of the murine phosphatidylinositol 4-phosphate 5-kinase-Iβ for phosphatidylinositol-4-phosphate. J Cell Biochem 100:112–128
Szymanska E, Sobota A, Czurylo E, Kwiatkowska K (2008) Expression of PI(4,5)P2-binding proteins lowers the PI(4,5)P2 level and inhibits FcγRIIA-mediated cell spreading and phagocytosis. Eur J Immunol 38:260–272
Lemmon MA (2008) Membrane recognition by phospholipids-binding domains. Nature 9:99–111
Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM (2002) Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase Cδ1. Biochem J 363:657–666
Fairn GD, Ogata K, Botelho RJ, Stahl PD, Anderson RA, De Camilli P, Meyer T, Wodak S, Grinstein S (2009) An electrostatic switch displaces phosphatidylinositol phosphate kinases from the membrane during phagocytosis. J Cell Biol 187:701–714
Scott CC, Dobson W, Botelho RJ, Coady-Osberg N, Chavrier P, Knecht DA, Heath C, Stahl P, Grinstein S (2005) Phosphatidylinositol-4,5-bisphosphate hydrolysis directs actin remodeling during phagocytosis. J Cell Biol 169:139–149
Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin HL (2003) Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114:293–299
Levine TP, Munro S (2002) Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr Biol 12:695–704
Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA (2004) FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nature Cell Biol 6:393–404
Balla A, Tuymetova G, Tsiomenko A, Varnai P, Balla T (2005) A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-IIIα: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell 16:1282–1295
Tran D, Gascard P, Berthon B, Fukami K, Takenawa T, Giraud F, Claret M (1993) Cellular distribution of polyphosphoinositides in rat hepatocytes. Cell Signal 5:565–581
Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H (2000) Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J 19:4577–4588
Vieira OV, Verkade P, Manninen A, Simons K (2005) FAPP2 is involved in the transport of apical cargo in polarized MDCK cells. J Cell Biol 170:521–526
Wang J, Sun HQ, Macia E, Kirchhausen T, Watson H, Bonifacino JS, Yin HL (2007) PI4P promotes the recruitment of the GGA adaptor proteins to the trans-Golgi network and regulates their recognition of the ubiquitin sorting signal. Mol Biol Cell 18:2646–2655
D’Angelo G, Vicinanza M, Di Campli A, De Matteis MA (2008) The multiple roles of PtdIns(4)P—not just the precursor of PtdIns(4,5)P2. J Cell Sci 121:1955–1963
Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM, Balla T (2008) Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIα. Mol Biol Cell 19:711–721
Hammond GR, Schiavo G, Irvine RF (2009) Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P2. Biochem J 422:23–35
Cockcroft S, Taylor JA, Judah JD (1985) Subcellular localisation of inositol lipid kinases in rat liver. Biochim Biophys Acta 845:163–170
Wong K, Meyers R, Cantley LC (1997) Subcellular locations of phosphatidylinositol 4-kinase isoforms. J Biol Chem 272:13236–13241
Barylko B, Gerber SH, Binns D, Grichine N, Khvotchev M, Südhof TC, Albanesi JP (2001) A novel family of phosphatidylinositol 4-kinases conserved from yeast to humans. J Biol Chem 276:7705–7708
Wei YJ, Sun HQ, Yamamoto M, Wlodarski P, Kunii K, Martinez M, Barylko B, Albanesi JP, Yin HL (2002) Type II phosphatidylinositol 4-kinase β is a cytosolic and peripheral membrane protein that is recruited to the plasma membrane and activated by Rac-GTP. J Biol Chem 277:46586–46593
Balla A, Tuymetova G, Barshishat M, Geiszt M, Balla T (2002) Characterization of type II phosphatidylinositol 4-kinase isoforms reveals association of the enzymes with endosomal vesicular compartments. J Biol Chem 277:20041–20050
Weixel KM, Blumental-Perry A, Watkins SC, Aridor M, Weisz OA (2005) Distinct Golgi populations of phosphatidylinositol 4-phosphate regulated by phosphatidylinositol 4-kinases. J Biol Chem 280:10501–10508
Jung G, Wang J, Wlodarski P, Barylko B, Binns DD, Shu H, Yin HL, Albanesi JP (2008) Molecular determinants of activation and membrane targeting of phosphoinositol 4-kinase IIβ. Biochem J 409:501–509
Wang YJ, Li WH, Wang J, Xu K, Dong P, Luo X, Yin HL. Critical role of PIP5KIγ87 in InsP3-mediated Ca2+ signaling. J Cell Biol 167:1005–1010
Tolias KF, Cantley LC, Carpenter CL (1995) Rho family GTPases bind to phosphoinositide kinases. J Biol Chem 270:17656–17699
van Hennik PB, ten Klooster JP, Halstead JR, Voermans C, Anthony EC, Divecha N, Hordijk PL (2003) The C-terminal domain of Rac1 contains two motifs that control targeting and signaling specificity. J Biol Chem 278:39166–39175
Oude Weernink PA, Meletiadis K, Hommeltenberg S, Hinz M, Ishihara H, Schmidt M, Jakobs KH (2004) Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J Biol Chem 279:7840–7849
Ren XD, Bokoch GM, Traynor-Kaplan A, Jenkins GH, Anderson RA, Schwartz MA (1996) Physical association of the small GTPase Rho with a 68-kDa phosphatidylinositol 4-phosphate 5-kinase in Swiss 3T3 cells. Mol Biol Cell 7:435–442
Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, Kanaho Y (1999) Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99:521–532
Chong LD, Traynor-Kaplan A, Bokoch GM, Schwartz MA (1994) The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell 79:507–513
Chatah NE, Abrams CS (2001) G-protein-coupled receptor activation induces the membrane translocation and activation of phosphatidylinositol-4-phosphate 5-kinase Iα by a Rac- and Rho-dependent pathway. J Biol Chem 276:34059–34065
Yamazaki M, Miyazaki H, Watanabe H, Sasaki T, Maehama T, Frohman MA, Kanaho Y (2002) Phosphatidylinositol 4-phosphate 5-kinase is essential for ROCK-mediated neurite remodeling. J Biol Chem 277:17226–17230
Yang SA, Carpenter CL, Abrams CS (2004) Rho and Rho-kinase mediate thrombin-induced phosphatidylinositol 4-phosphate 5-kinase trafficking in platelets. J Biol Chem 279:42331–42336
Heasman SJ, Ridley AJ (2009) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701
Abramovici H, Mojtabaie P, Parks RJ, Zhong XP, Koretzky GA, Topham MK, Gee SH (2009) Diacylglycerol kinase ζ regulates actin cytoskeleton reorganization through dissociation of Rac1 from RhoGDI. Mol Biol Cell 20:2049–2059
Luo B, Prescott SM, Topham MK (2004) Diacylglycerol kinase ζ regulates phosphatidylinositol 4-phosphate 5-kinase Iα by a novel mechanism. Cell Signal 16:91–897
Illenberger D, Walliser C, Nurnberg B, Diaz Lorente M, Gierschik P (2003) Specificity and structural requirements of phospholipase C-β stimulation by Rho GTPases versus G protein βγ dimers. J Biol Chem 278:3006–3014
Piechulek T, Rehlen T, Walliser C, Vatter P, Moepps B, Gierschik P (2005) Isozyme-specific stimulation of phospholipase C-γ2 by Rac GTPases. J Biol Chem 280:38923–38931
Bunney TD, Opaleye O, Roe SM, Vatter P, Baxendale RW, Walliser C, Everett KL, Josephs MB, Christow C, Rodrigues-Lima F, Gierschik P, Pearl LH, Katan M (2009) Structural insights into formation of an active signaling complex between Rac and phospholipase Cγ2. Mol Cell 34:223–233
Pratt SJ, Epple H, Ward M, Feng Y, Braga VM, Longmore GD (2005) The LIM protein Ajuba influences p130Cas localization and Rac1 activity during cell migration. J Cell Biol 168:813–824
Krauss M, Kinuta M, Wenk MR, De Camilli P, Takei K, Haucke V (2003) ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Iγ. J Cell Biol 162:113–124
Hiroyama M, Exton JH (2005) Localization and regulation of phospholipase D2 by ARF6. J Cell Biochem 95:149–164
Perez-Mansilla B, Ha VL, Justin N, Wilkins AJ, Carpenter CL, Thomas GM (2006) The differential regulation of phosphatidylinositol 4-phosphate 5-kinases and phospholipase D1 by ADP-ribosylation factors 1 and 6. Biochim Biophys Acta 1761:1429–1442
Divecha N, Roefs M, Halstead JR, D’Andrea S, Fernandez-Borga M, Oomen L, Saqib KM, Wakelam MJ, D’Santos C (2000) Interaction of the type Iα PIPkinase with phospholipase D: a role for the local generation of phosphatidylinositol 4,5-bisphosphate in the regulation of PLD2 activity. EMBO J 19:5440–5449
Skippen A, Jones DH, Morgan CP, Li M, Cockcroft S (2002) Mechanism of ADP ribosylation factor-stimulated phosphatidylinositol 4,5-bisphosphate synthesis in HL60 cells. J Biol Chem 277:5823–5831
Aikawa Y, Martin TF (2003) ARF6 regulates a plasma membrane pool of phosphatidylinositol(4,5)bisphosphate required for regulated exocytosis. J Cell Biol 162:647–659
Begle A, Tryoen-Toth P, de Barry J, Bader MF, Vitale N (2009) ARF6 regulates the synthesis of fusogenic lipids for calcium-regulated exocytosis in neuroendocrine cells. J Biol Chem 284:4836–4845
Santy LC, Ravichandran KS, Casanova JE (2005) The DOCK180/Elmo complex couples ARNO-mediated Arf6 activation to the downstream activation of Rac1. Curr Biol 15:1749–1754
Myers KR, Casanova JE (2008) Regulation of actin cytoskeleton dynamics by Arf-family GTPases. Trends Cell Biol 18:184–192
Gingras AR, Bate N, Goult BT, Hazelwood L, Canestrelli I, Grossmann JG, Liu H, Putz NS, Roberts GC, Volkmann N, Hanein D, Barsukov IL, Critchley DR (2008) The structure of the C-terminal actin-binding domain of talin. EMBO J 27:458–469
Gingras AR, Ziegler WH, Bobkov AA, Joyce MG, Fasci D, Himmel M, Rothemund S, Ritter A, Grossmann JG, Patel B, Bate N, Goult BT, Emsley J, Barsukov IL, Roberts GC, Liddington RC, Ginsberg MH, Critchley DR (2009) Structural determinants of integrin binding to the talin rod. J Biol Chem 284:8866–8876
Barsukov IL, Prescot A, Bate N, Patel B, Floyd DN, Bhanji N, Bagshaw CR, Letinic K, Di Paolo G, De Camilli P, Roberts GC, Critchley DR (2003) Phosphatidylinositol phosphate kinase type 1γ and β1-integrin cytoplasmic domain bind to the same region in the talin FERM domain. J Biol Chem 278:31202–31209
de Pereda JM, Wegener KL, Santelli E, Bate N, Ginsberg MH, Critchley DR, Campbell ID, Liddington RC (2005) Structural basis for phosphatidylinositol phosphate kinase type Iγ binding to talin at focal adhesions. J Biol Chem 280:8381–8386
Kong X, Wang X, Misra S, Qin J (2006) Structural basis for the phosphorylation-regulated focal adhesion targeting of type Iγ phosphatidylinositol phosphate kinase (PIPKIγ) by talin. J Mol Biol 359:47–54
Hamada K, Shimizu T, Matsui T, Tsukita S, Hakoshima T (2000) Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J 19:4449–4462
Martel V, Racaud-Sultan C, Dupe S, Marie C, Paulhe F, Galmiche A, Block MR, Albiges-Rizo C (2001) Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem 276:21217–21227
Saltel F, Mortier E, Hytonen VP, Jacquier MC, Zimmermann P, Vogel V, Liu W, Wehrle-Haller B (2009) New PI(4,5)P2- and membrane proximal integrin-binding motifs in the talin head control β3-integrin clustering. J Cell Biol 187:715–731
Bairstow SF, Ling K, Anderson RA (2002) Phosphatidylinositol phosphate kinase type Iγdirectly associates with and regulates Shp-1 tyrosine phosphatase. J Biol Chem 280:23884–23891
Sun Y, Ling K, Wagoner MP, Anderson RA (2007) Type Iγ phosphatidylinositol phosphate kinase is required for EGF-stimulated directional cell migration. J Cell Biol 178:297–308
Tanentzapf G, Brown NH (2006) An interaction between integrin and the talin FERM domain mediates integrin activation but not linkage to the cytoskeleton. Nat Cell Biol 8:601–606
Cheung TY, Fairchild MJ, Zarivach R, Tanentzapf G, Van Petegem F (2009) Crystal structure of the talin integrin binding domain 2. J Mol Biol 387:787–793
Wenk MR, Pellegrini L, Klenchin VA, Di Paolo G, Chang S, Daniell L, Arioka M, Martin TF, De Camilli P (2001) PIP kinase Iγ is the major PI(4,5)P2 synthesizing enzyme at the synapse. Neuron 32:79–88
Kahlfeldt N, Vahedi-Faridi A, Koo SJ, Schäfer JG, Krainer G, Keller S, Saenger W, Krauss M, Haucke V (2010) Molecular basis for association of PIPKIγ-p90 with clathrin adaptor AP-2. J Biol Chem 285:2734–2749
Thieman JR, Mishra SK, Ling K, Doray B, Anderson RA, Traub LM (2009) Clathrin regulates the association of PIPKIγ661 with the AP-2 adaptor β2 appendage. J Biol Chem 284:13924–13939
Paleotti O, Macia E, Luton F, Klein S, Partisani M, Chardin P, Kirchhausen T, Franco M (2005) The small G-protein ARF6GTP recruits the AP-2 adaptor complex to membranes. J Biol Chem 280:21661–21666
Jung N, Haucke V (2007) Clathrin-mediated endocytosis at synapses. Traffic 81129-11136
Barbieri MA, Heath CM, Peters EM, Wells A, Davis JN, Stahl PD (2001) Phosphatidylinositol-4-phosphate 5-kinase-1β is essential for epidermal growth factor receptor-mediated endocytosis. J Biol Chem 276:47212–47216
Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, Grinstein S (2000) Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol 151:1353–1368
Golebiewska U, Nyako M, Woturski W, Zaitseva I, McLaughlin S (2008) Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol Biol Cell 19:1663–1669
Wagner ML, Tamm LK (2001) Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J 81:266–275
Golebiewska U, Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Radler J, McLaughlin S (2006) Membrane-bound basic peptides sequester multivalent (PIP2), but not monovalent (PS), acidic lipids. Biophys J 91:588–599
van Rheenen J, Jalink K (2002) Agonist-induced PIP2 hydrolysis inhibits cortical actin dynamics: regulation at a global but not at a micrometer scale. Mol Biol Cell 13:3257–3267
Haugh JM, Codazzi F, Teruel M, Meyer T (2000) Spatial sensing in fibroblasts mediated by 3’ phosphoinositides. J Cell Biol 151:1269–1280
McLaughlin S, Wang J, Gambhir A, Murray D (2002) PIP2 and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct 31:151–175
Cho H, Kim YA, Yoon JY, Lee D, Kim JH, Lee SH, Ho WK (2005) Low mobility of phosphatidylinositol 4,5-bisphosphate underlies receptor specificity of Gq-mediated ion channel regulation in atrial myocytes. Proc Natl Acad Sci USA 102:15241–15246
Cho H, Kim YA, Ho WK (2006) Phosphate number and acyl chain length determine the subcellular location and lateral mobility of phosphoinositides. Mol Cells 22:97–103
Hilgemann DW (2007) Local PIP2 signals: when, where, and how? Pflugers Arch 455:55–67
Stefanova I, Horejsi V, Ansotegui IJ, Knapp W, Stockinger H (1991) GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science 254:1016–1019
Fra AM, Williamson E, Simons K, Parton RG (1994) Detergent-insoluble glycolipid microdomains in lymphocytes in the absence of caveolae. J Biol Chem 269:30745–30748
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572
Brown DA, London E (1997) Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? Biochem Biophys Res Commun 240:1–7
Fridriksson EK, Shipkova PA, Sheets ED, Holowka D, Baird B, McLafferty FW (1999) Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry 38:8056–8063
Brown DA, Rose JK (1992) Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68:344–533
Ahmed SN, Brown DA, London E (1997) On the origin of sphingolipid/cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of a detergent-insoluble, liquid-ordered lipid phase in model membranes. Biochemistry 36:10944–10953
Montixi C, Langlet C, Bernard AM, Thimonier J, Dubois C, Wurbel MA, Chauvin JP, Pierres M, He HT (1998) Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J 17:5334–5348
Sheets ED, Holowka D, Baird B (1999) Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcεRI and their association with detergent-resistant membranes. J Cell Biol 145:877–887
Hope HR, Pike LJ (1996) Phosphoinositides and phosphoinositide-utilizing enzymes in detergent-insoluble lipid domains. Mol Biol Cell 7:843–851
Pike LJ, Casey L (1996) Localization and turnover of phosphatidylinositol 4,5-bisphosphate in caveolin-enriched membrane domains. J Biol Chem 271:6453–26456
Pike LJ, Miller JM (1998) Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J Biol Chem 273:22298–22304
Waugh MG, Lawson D, Tan SK, Hsuan JJ (1998) Phosphatidylinositol 4-phosphate synthesis in immunoisolated caveolae-like vesicles and low buoyant density non-caveolar membranes. J Biol Chem 273:17115–17121
Liu Y, Casey L, Pike LJ (1998) Compartmentalization of phosphatidylinositol 4,5-bisphosphate in low-density membrane domains in the absence of caveolin. Biochem Biophys Res Commun 245:684–690
Hur EM, Park YS, Lee BD, Jang IH, Kim HS, Kim TD, Suh PG, Ryu SH, Kim KT (2004) Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. Implications for a role of lipid microdomains. J Biol Chem 279:5852–5860
Bodin S, Giuriato S, Ragab J, Humbel BM, Viala C, Vieu C, Chap H, Payrastre B (2001) Production of phosphatidylinositol 3,4,5-trisphosphate and phosphatidic acid in platelet rafts: evidence for a critical role of cholesterol-enriched domains in human platelet activation. Biochemistry 40:15209–15290
Heerklotz H (2002) Triton promotes domain formation in lipid raft mixtures. Biophys J 83:2693–2701
Korzeniowski M, Kwiatkowska K, Sobota A (2003) Insights into the association of FcγRII and TCR with detergent-resistant membrane domains: isolation of the domains in detergent-free density gradients facilitates membrane fragment reconstitution. Biochemistry 42:5358–5367
Shogomori H, Brown DA (2003) Use of detergents to study membrane rafts: the good, the bad, and the ugly. J Biol Chem 384:1259–1263
Kwik J, Boyle S, Fooksman D, Margolis L, Sheetz MP, Edidin M (2003) Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci USA 100:13964–13969
Van Rheenen J, Achame EM, Janssen H, Calafat J, Jalink K (2005) PIP2 signaling in lipid domains: a critical re-evaluation. EMBO J 24:1664–1673
Yaradanakul A, Hilgemann DW (2007) Unrestricted diffusion of exogenous and endogenous PIP2 in baby hamster kidney and Chinese hamster ovary cell plasmalemma. J Membr Biol 220:53–67
Janes PW, Ley SC, Magee AI (1999) Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J Cell Biol 147:447–461
Holowka D, Sheets ED, Baird B (2000) Interactions between FcεRI and lipid raft components are regulated by the actin cytoskeleton. J Cell Sci 113:1009–1019
Wilson BS, Pfeiffer JR, Surviladze Z, Gaudet EA, Oliver JM (2001) High-resolution mapping of mast cell membranes reveals primary and secondary domains of FcεRI and LAT. J Cell Biol 154:645–658
Kwiatkowska K, Frey J, Sobota A (2003) Phosphorylation of FcγRIIA is required for the receptor-induced actin rearrangement and capping: the role of membrane rafts. J Cell Sci 116:537–550
Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, Scharenberg A, Rawlings DJ, Kinet JP, Carpenter CL (2003) BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19:669–678
Foster LJ, De Hoog CL, Mann M (2003) Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc Natl Acad Sci USA 100:5813–5818
Parmryd I, Adler J, Patel R, Magee AI (2003) Imaging metabolism of phosphatidylinositol 4,5-bisphosphate in T-cell GM1-enriched domains containing Ras proteins. Exp Cell Res 285:27–38
Strzelecka-Kiliszek A, Korzeniowski M, Kwiatkowska K, Mrozinska K, Sobota A (2004) Activated FcγRII and signalling molecules revealed in rafts by ultra-structural observations of plasma-membrane sheets. Mol Membr Biol 21:101–108
Korzeniowski M, Shakor AB, Makowska A, Drzewiecka A, Bielawska A, Kwiatkowska K, Sobota A (2007) FcγRII activation induces cell surface ceramide production which participates in the assembly of the receptor signaling complex. Cell Physiol Biochem 20:347–356
Raucher D, Stauffer T, Chen W, Shen K, Guo S, York JD, Sheetz MP, Meyer T (2000) Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell 100:221–228
Hagelberg C, Allan D (1990) Restricted diffusion of integral membrane proteins and polyphosphoinositides leads to their depletion in microvesicles released from human erythrocytes. Biochem J 271:831–834
Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S (2004) Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J 86:2188–2207
McLaughlin S, Murray D (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 43:605–611
Tong J, Nguyen L, Vidal A, Simon SA, Skene JH, McIntosh TJ (2008) Role of GAP-43 in sequestering phosphatidylinositol 4,5-bisphosphate to raft bilayers. Biophys J 94:125–133
Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P (2000) GAP43, MARCKS, and CAP23 modulate PI(4,5)P2 at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol 149:455–1471
Golub T, Caroni P (2005) PI(4,5)P2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. J Cell Biol 169:151–165
Acknowledgments
I gratefully thank Prof. Andrzej Sobota, Nencki Institute of Experimental Biology, Warsaw, for support and excellent discussion. I also thank Prof. Jan Fronk, Warsaw University, for valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kwiatkowska, K. One lipid, multiple functions: how various pools of PI(4,5)P2 are created in the plasma membrane. Cell. Mol. Life Sci. 67, 3927–3946 (2010). https://doi.org/10.1007/s00018-010-0432-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0432-5