
Abstract
lncRNA–protein interactions (LPIs) prediction can deepen the understanding of many important biological processes. Artificial intelligence methods have reported many possible LPIs. However, most computational techniques were evaluated mainly on one dataset, which may produce prediction bias. More importantly, they were validated only under cross validation on lncRNA–protein pairs, and did not consider the performance under cross validations on lncRNAs and proteins, thus fail to search related proteins/lncRNAs for a new lncRNA/protein. Under an ensemble learning framework (EnANNDeep) composed of adaptive k-nearest neighbor classifier and Deep models, this study focuses on systematically finding underlying linkages between lncRNAs and proteins. First, five LPI-related datasets are arranged. Second, multiple source features are integrated to depict an lncRNA–protein pair. Third, adaptive k-nearest neighbor classifier, deep neural network, and deep forest are designed to score unknown lncRNA–protein pairs, respectively. Finally, interaction probabilities from the three predictors are integrated based on a soft voting technique. In comparing to five classical LPI identification models (SFPEL, PMDKN, CatBoost, PLIPCOM, and LPI-SKF) under fivefold cross validations on lncRNAs, proteins, and LPIs, EnANNDeep computes the best average AUCs of 0.8660, 0.8775, and 0.9166, respectively, and the best average AUPRs of 0.8545, 0.8595, and 0.9054, respectively, indicating its superior LPI prediction ability. Case study analyses indicate that SNHG10 may have dense linkage with Q15717. In the ensemble framework, adaptive k-nearest neighbor classifier can separately pick the most appropriate k for each query lncRNA–protein pair. More importantly, deep models including deep neural network and deep forest can effectively learn the representative features of lncRNAs and proteins.
Graphic abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
1.1 Motivation
Long noncoding RNAs (lncRNAs) are a class of long endogenous noncoding RNAs with poor sequence conservation [1,2,3]. lncRNAs have close association with multiple key biological processes [4]. More importantly, increasing works imply that lncRNAs also densely linking with many complex diseases [5, 6], for example, brachydactyly syndrome and HELLP syndrome [7], facioscapulohumeral muscular dystrophy [8], fat [9], and cancers. For example, lncRNAs HOXA-AS2 and SNHG12 are identified as possible therapeutic targets and biomarkers in human cancers [10, 11], DLEU1 densely links with colorectal cancer progression through the activation of KPNA3 [12], HOTAIR’s expression is elevated in lung cancer [13], ZFAS1 has close relationship with cervical cancer cell chemosensitivity [14]. In summary, lncRNAs have been increasingly confirmed to be tumor-related biological molecules. However, to date, relationships between lncRNA and known tumor-suppressive entities remain largely elusive. Evidence indicates that lncRNAs exert their biological functions based on the linkages with RNA-binding proteins. Therefore, the identification of potential lncRNA–protein interactions (LPIs) contributes to understand many important biological processes and progression and metastasis of various complex diseases.
1.2 Related Work
Wet-lab experiments for LPI identification are time-consuming and waste of sources. Computational methods have been gradually explored for potential LPI discovery. Existing computation-based LPI prediction methods can be roughly categorized into network-based techniques and machine learning-based techniques. Network-based methods generally construct a few lncRNA/protein-related networks and then design a network algorithm to compute the probabilities of interactions between lncRNAs and proteins. Zhao et al. [15] and Ge et al. [16] designed two bipartite network-based recommended algorithm to score each lncRNA–protein pair. Zhou et al. [17] proposed a similarity kernel fusion method for LPI prediction (LPI-SKF). Zheng et al. [18] fused multiple protein similarity networks to uncover potential associations between lncRNAs and proteins.
Machine learning-based methods select features for lncRNAs and proteins to describe an lncRNA–protein pair, and use the extracted features as input to train a supervised learning model for possible LPI identification. The type of methods contain matrix factorization-based models, ensemble learning-based models, and deep learning-based models. To discover new LPIs, Liu et al. [19], Zhang et al. [20], and Ma et al. [21] explored neighborhood regularized logistic matrix factorization method, graph regularized nonnegative matrix factorization model, and projection-based neighborhood nonnegative matrix decomposition method (PMKDN), respectively.
Ensemble learning-based techniques have been widely available for LPI identification. Hu et al. [22] presented a unified framework combining support vector machines, random forests, and extreme gradient boosting. Zhang et al. [23] designed a feature projection ensemble learning-based framework (SFPEL). Deng et al. [24] picked lncRNA and protein information including HeteSim features and diffusion features and constructed a gradient tree boosting algorithm (PLIPCOM). Fan et al. [25] explored a broad learning system-based ensemble classification model. Wekesa et al. [26] exploited a categorical boosting approach (LPI-CatBoost). Yi et al. [27] proposed a stacking ensemble learning algorithm.
Deep learning architectures can better learn hidden information in raw data and characterize data in each layer based on nonlinear transformations [28]. Therefore, deep learning has been a research hotspot in the area of bioinformatics [6, 29,30,31]. In LPI prediction, deep learning demonstrates also broad application, such as the works provided by [32,33,34,35]. Deng et al. [32] proposed a deep neural network for predicting binding site of RNA-binding proteins. Wei et al. [35] fused biological feature blocks via Deep Neural Network (DNN). Zhang et al. [33] presented an ensemble deep learning model for identifying interaction biomolecule types for lncRNAs. Wekesa et al. [34] explored a graph attention-based deep learning model to predict plant LPIs. Zhao et al. [36] developed a graph convolutional network-based method to prioritize target protein-coding genes of lncRNAs. Shaw et al. [37] exploited a multimodal deep learning model to identify relationships between lncRNAs and protein isoforms.
Computational methods effectively discovered many potential relevances between lncRNAs and proteins. However, network-based techniques fail to find possible proteins/lncRNAs for an orphan lncRNA/protein. Machine learning-based LPI prediction approaches remain the following problems to solve. First, most methods are measured on one dataset, which may result in prediction bias. Second, the majority of methods are validated under Cross Validation (CV) on lncRNA–protein pairs, ignored the performance under the other CVs, for example, CVs on lncRNAs or proteins. Finally, features of lncRNAs and proteins are required to further integration. The details are summarized in Table 1.
1.3 Study Contributions
In this manuscript, an ensemble learning framework (EnANNDeep) is developed to quantify the interplays between lncRNAs and proteins. EnANNDeep integrates diverse biological information, Adaptive k-nearest neighbor (AkNN) classifier, deep neural network, Deep forest, and ensemble learning theory to a unified framework. The work mainly has the following three contributions:
-
1.
An ensemble learning framework, composed of AkNN algorithm, DNN, and deep forest, is exploited to greatly learn labels of unknown lncRNA–protein pairs.
-
2.
The proposed AkNN classification model separately selects the right k for each neighborhood and provides an upper bound for the failure probability.
-
3.
Deep models including DNN and deep forest better represent biological features for each lncRNA–protein pair.
2 Materials and Methods
2.1 Data Preparation
In this study, five different LPI-related datasets are arranged. Table 2 shows the details of the five datasets. Datasets 1, 2, and 3 contain human LPI data and datasets 4 and 5 contain plant LPI data. Dataset 1 was provided by Li et al. [38]. We obtain 3479 correlations from 935 lncRNAs and 59 proteins after removing lncRNAs and proteins whose sequence information is unknown in the NPInter [39], NONCODE [40], and UniProt [41].
Dataset 2 was built by Zheng et al. [18]. We screen 3265 relationships from 885 lncRNAs and 84 proteins after the preprocessing similar to dataset 1. Dataset 3 was constructed by Zhang et al. [42] and contains 4158 interplays from 990 lncRNAs and 27 proteins.
Datasets 4 and 5 were from Arabidopsis thaliana and Zea mays, respectively. The former contains 948 interactions from 109 lncRNAs and 35 proteins and the latter provides 22,133 associations from 1704 lncRNAs and 42 proteins. Sequence data are extracted from the PlncRNADB database [43] and interaction data are obtained at http://bis.zju.edu.cn/PlncRNADB/.
We represent LPI network as a matrix Y with the element:
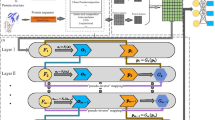
2.2 Overview of EnANNDeep
In this study, we develop an ensemble learning framework (EnANNDeep), composed of AkNN, DNN, and deep forest, to classify unknown lncRNA–protein pairs. Figure 1 describes the EnANNDeep framework.
As shown in Fig. 1, EnANNDeep mainly contains three procedures after five different LPI datasets are arranged. (1) Feature selection—An ensemble method combining gapped k-mer [44], tri-nucleotide composition [45], reverse complement k-mer [46], and RNAfold [47] is available for lncRNA feature selection. SSpro [48] and binary profile are used to chose protein features. (2) Classification—AkNN, DNN, and deep forest are exploited to obtain labels of unknown lncRNA–protein pairs, respectively. (3) Ensemble—The results from the above three predictors are integrated based on a soft voting technique.

The flowchart of the LPI-DLDN framework: (1) Feature selection; (2) Classification; (3) Ensemble
2.3 Feature Selection
2.3.1 lncRNA Feature Selection
The integration of various lncRNA and protein features contributes to improve LPI prediction accuracy. In this work, an ensemble approach is explored to represent lncRNA features. For given an lncRNA sequence L with length a, where \({l_i} \in \{A, C, G, T\}\) and \(\{i = 1, 2, \ldots , a\}\). EnANNDeep utilizes gapped 3-mer [44], tri-nucleotide composition [45], reverse complement 2-mer [46], and RNAfold [47] to characterize an lncRNA.
The tri-nucleotide composition technique is used to obtain evolutionary features from L. The tri-nucleotide compositions are extracted by scanning the sequence using \(\{(1,2,3), (2,3,4), \ldots , (a-2, a-1, a)\}\), where \(\{1, 2, 3, \ldots , i, \ldots , a\}\) denotes the i-th nucleotide in L.
The gapped 3-mer method applies 3-mer with gap to obtain local and global information from L. Let b represent the number of non-gapped positions in L, and the number of gaps is \(g=3-b\). A feature vector of L can be denoted by Eq. (2):
where \(u_{\mathrm{i}}\) is the number of the i-th gapped 3-mer in L, M is the number of all gapped 3-mers and \(M = \left( {\begin{array}{*{20}{c}} 3\\ b \end{array}} \right) {4^3}\).
The reverse complement 2-mer method is used to extract regulatory features from L. First, 2-mer is generated. Second, reverse complement 2-length contiguous subsequences are eliminated. Finally, the computed occurrence frequencies of the remaining 2-length subsequences are calculated to build an lncRNA feature vector.
The RNA secondary structures have been validated to positively affect protein binding site selection. A dynamic programming technique, RNAfold, is used to infer RNA secondary structures according to its minimum free energy. Five features with high probability structures are extracted by counting occurrence frequency of each unique structure.
2.3.2 Protein Feature Selection
To depict a protein, first, its secondary structures are obtained based on \(\alpha\)-helix (H), \(\beta\)-sheet (E), and coil (C) conformation using SSpro [48]. Second, 20 amino acids are divided into three categories based on the computed secondary structures: \(\alpha\)-helix contains eight amino acids (E, A, L, M, Q, K, R, and H), \(\beta\)-sheet contains seven amino acids (V, I, Y, C, W, F, and T), and coil contains five amino acids (G, N, P, S, and D). Third, an amino acid can be replaced by its conformation and thus each protein sequence can be represented using H, E or C. 27 3-tuples are obtained from the permutation of the above three conformations. Fourth, 3-tuple is applied to the replaced sequences and the number of each 3-tuple is computed. Finally, the occurrence frequency of each 3-tuple can be calculated by Eq. (3):
where \({d_i}\) is the number of the i-th 3-tuples in L.
In addition, a binary profile describes composition and order of residues in a protein sequence. In this study, a binary profile with a \(20\times 16\) dimensions is produced based on a one-hot encoding of 20 amino acids. The details for lncRNA and protein feature extraction are described in Table 3. Thus an lncRNA–protein pair can be represented as a 554-dimensional vector \(\varvec{x}\) combining lncRNA and protein features.
2.4 Problem Description
Given an LPI training set \(D=(X,Y)\) with labels \(\{+1,-1\}\), where a separable metric space (X, 554) denotes the sample space with 554 features and \(Y=\{+1,-1\}\) describes the label space. A training example \(\varvec{x}\) is a 554-dimensional feature vector applied to characterize an lncRNA–protein pair, \(\varvec{y}\in \{ +1, -1\}\) denotes its label. The label of \(\varvec{x}\) is 1 when there is an interaction between the lncRNA and the protein; the label is -1, otherwise. For any query lncRNA–protein pair \(\varvec{x}_i\), we aim to construct an ensemble model, EnANNDeep, to obtain its label.
2.5 Adaptive k-Nearest Neighbor
2.5.1 k-Nearest Neighbor
k-Nearest Neighbor (k-NN) classifier [49] is a simple but effective classification model. It is very appropriate to a classification task where there is lack of prior knowledge about data distribution. The classifier investigates label of a test sample based on the Euclidean distance between the test sample and all training samples.
Given n LPI samples, let \(\varvec{x}_i \,\,(i=1,2,\ldots ,n)\) denote the i-th sample with 554 features \(({x}_{i,1}, {x}_{i,2}, \ldots , {x}_{i,554})\). The Euclidean distance between two samples \(\varvec{x}_i\) and \(\varvec{x}_j\) is represented as:
Based on the theory provided by Voronoi [50], a Voronoi cell \(R_i\) for sample \(\varvec{x}_i\) encapsulates its all nearest neighbors and is defined by Eq. (5):
where \(\varvec{x}_a\) denotes all possible points (samples) within \(R_i\), that is, the nearest neighbors of the example \(\varvec{x}_i\).
For any LPI, k-NN classifier determines the nearest samples through the closest edges within the Voronoi cell \(R_i\). A test sample is assigned a label the same as the majority category label of its k nearest training samples based on k-NN classifier.
k-NN uses a fixed radius and can automatically adapt to the variation in marginal distribution. Therefore, it has been broadly applied to various areas. However, the choice of its nearest neighbor number k severely depends on features of each neighborhood and thus may greatly vary between different points. In the input space, for the regions where conditional expectation of \(\varvec{x}\) tends to 0, larger k is required for accurate prediction. For other regions where the conditional expectation is \(+1\) or \(-1\), smaller k can satisfy the requirement and larger k may result in incorrect classification due to the inconsistence of labels in the neighboring regions. Thus k-NN classifier has to select a single value for k to trade off the above two situations. To solve this problem, AkNN classifier is designed to separately select the right k for each neighborhood.
2.5.2 Adaptive k-Nearest Neighbor
Inspired by the AkNN algorithm proposed by Balsubramani et al. [51], we design an AkNN algorithm to compute interaction probability for each lncRNA–protein pairs. For a training set \({(\varvec{x}_1, \varvec{y}_1), (\varvec{x}_2, \varvec{y}_2)}\), \(\ldots\), \((\varvec{x}_n, \varvec{y}_n) \in {\mathcal {X}} \times {\mathcal {Y}}\), let all LPI data draw from an unobserved independent identically distribution P on \({\mathcal {X}} \times {\mathcal {Y}}\). Let \(\mu\) represent the marginal distribution on \({\mathcal {X}}\): if (X, Y) denotes a random draw from P, let \(\eta (\varvec{x}) = \mathrm{E}(Y|X = \varvec{x})\), then for any measurable set \(S \subseteq {\mathcal {X}}\):
For any given sample \(\varvec{x} \in {\mathcal {X}}\), conditional expectation of Y can be denoted by Eq. (7):
For any S where \(\mu (S)>0\) and given \(X \in S\), conditional expectation of Y can be described by Eq. (8):
Thus the error risk of k-NN classifier: \(g: {\mathcal {X}} \rightarrow \{-1, \, +1\}\) is the probability that it incorrectly classifies a query sample on the training set \((X,Y) \sim P\). The risk is denoted by Eq. (9):
For \(\varvec{x} \in {\mathcal {X}}\) and \(r>0\), let \(B(\varvec{x},r)\) represent the closed ball with radius r centered at \(\varvec{x}\):
For a query lncRNA–protein pair \(\varvec{x}\), AkNN classifier predicts its label based on the training lncRNA–protein pairs closest to \(\varvec{x}\). The empirical count is defined by Eq. (11):
The probability mass can be described by Eq. (12):
When the empirical count is non-zero, the empirical bias can be defined by Eq. (13).
where n indicates the number of all lncRNA–protein pairs. |Y| denotes the number of classes. In this manuscript, |Y| is 2.
AkNN classification model is described in Algorithm 1. The label of a query lncRNA–protein pair \(\varvec{x}\) can be predicted through expanding a ball around \(\varvec{x}\) until it produces a significant bias based on Algorithm 1.

In Algorithm 1, \(\Delta (n,k,\delta )\) denotes a confidence interval of average labels in the region closest to the query sample \(\varvec{x}\). \(c_1\) represents a constant.
Algorithm 1 infinitely makes many parameter selection. It picks k for each query point and asks for a single failure probability to measure how to assign its confidence intervals. In comparing to standard k-NN classifier, the AkNN classification algorithm seems to merely replace the parameter k with another parameter \(\delta\). However, it is not accurate. \(\delta\), a customary confident level parameter, provides an upper bound upon the failure probability for Algorithm 1.
To simplify the parameters, we replace \(\Delta\) with \(\Delta =\frac{A}{\sqrt{k}}\) in \(\Delta (n,k,\delta ) = {c_1}\sqrt{\frac{{\log n + \log (1/\delta )}}{k}}\). The parameter A is used to control conservations in Algorithm 1 and A → 0 denotes the most aggressive setting where Algorithm 1 never abstains. The detailed discussion is provided by Balsubramani et al. [51].
2.5.3 Deep Neural Network
The rapid development of machine learning models and computer hardware promotes the birth of DNNs. DNN is a feed-forward artificial neural networks. A DNN consists of one input layer, multiple hidden layers composed of nonlinear hidden units, and one larger output layer. The input layer achieves the original data. Each hidden unit j in a hidden layer uses an activation function to map the input \(x_j\) from the input layer to a scalar state. The output layer accommodates multiple hidden Markov model states.
DNNs have been already broadly applied to various association prediction [28]. For example, Zhao et al. [52] identified drug–target interactions combining graph convolutional network and DNN. Chu et al. [29] developed an optimized DNN to screen epidermal growth factor receptor inhibitors. Wang et al. [53] exploited a deep convolutional neural network-based drug–target interactions algorithm. Wei et al. [35] designed a DNN-based lncRNA-disease association prediction approach.
In this study, we utilize DNN to reveal possible LPIs. The DNN-based LPI prediction framework is shown in Fig. 2.

The flowchart of DNN-based LPI prediction algorithm
In the DNN model, the input layer has 554 neurons and achieves the input LPI samples with 554-dimensional features. The following two layers are hidden layers. The two layers are full connection layers containing 128 and 64 neurons, respectively. And each hidden layer follows by a dropout layer with the rate of 0.5 to avoid over-fitting by setting the output of 50% units to 0. Exponential Linear Unit (ELU) is considered as an activation function in the hidden layers. ELU can alleviate gradient vanishing, make the average output of an activation unit closer to 0 to achieve the effect of batch normalization and reduce the computation time. In addition, ELU is only qualitative but not quantitative for the input characteristics because it is an exponential function when it is negative. More importantly, ELU contributes to faster learning and better generalization ability on DNNs. It is denoted by Eq. (15):
Our objective is to quantify how many the predicted labels differ from the real ones by minimizing the binary cross-entropy in the process of training by Eq. (16):
where \({\varvec{y}_i}\) is the true label and \({\widehat{\varvec{y}}_i}\) denotes the probability that the i-th sample is predicted to be positive LPI. The training is implemented with 100 epochs and each epoch has a mini-batch with the size of 128 to update its weights. We use the Adam algorithm [54] as the optimization technique to train DNN.
The final output layer contains a single neuron to output an interaction probability for each query lncRNA–protein pair based on a sigmoid function defined by Eq. (17):
The sigmoid function can map a real number to the interval of (0,1). It is smooth and easy to derivation and is thus used as an activation function in the output layer of DNN to compute interaction score for each lncRNA–protein pair.
2.5.4 Deep Forest
To tackle complicated tasks, learning models gradually go deep [55]. However, traditional deep algorithms are always designed based on neural networks. Non-neural network style-based deep models will demonstrate great learning ability if they can go deep, especially when neural networks are multi-layered deep models with parameterized differentiable nonlinear modules. Considering this feature of neural networks, deep forest [56, 57], a non-neural network style deep model, is built upon multi-grained cascade framework.
Deep forest is a novel ensemble algorithm. Its feature learning capability is further boosted by multi-grained scanning the input data. Second, its complexity can be automatically set. Third, it performs better even on small-scale data. Finally, the training costs can be controlled based on available computational resources. Deep forest only needs to train much fewer hyper-parameters in comparing to other deep learning models. Therefore, deep forest obtains highly competitive classification ability while its training time drops sharply.
In this manuscript, deep forest with no more than 20 layers is utilized to classify unobserved lncRNA–protein pairs. Random forest [58, 59] and Extra trees [60] are chosen as basic classifiers. The random forest technique [58, 59] is a general-purpose, nonparametric, and interpretable classification model. It is an ensemble of a few randomized decision trees and can return measurements of variable importance. It has unique characteristics in dealing with complex data structures, small sample size, and high-dimensional feature space. In particular, it demonstrates excellent performance when the number of variables is far more than the number of samples.
The Extra tree model [60] is an ensemble of unpruned decision trees based on the classical top–down procedure. Extra tree has three advantages: First, it splits nodes by fully randomly selecting cut-points and contributes to more strongly reduce variance than the weaker randomization algorithms. Second, it utilizes the whole learning samples rather than a bootstrap replicas to minimize classification bias. Finally, it contains a node splitting scheme to obtain much smaller constant factor during cut-point optimization.
In the proposed deep forest model, each cascade layer consists of two random forests and two Extra trees. Each estimator consists of 100 decision trees. In each layer, for a given LPI feature, each classifier calculates the ratio of the feature belonging to positive class or negative class. The predicted class probability from all classifiers forms a class vector. The vector is concatenated with the raw LPI feature vector as input in the next level.
As illustrated in Fig. 3, a 554-dimensional vector is taken as input of deep forest. After training four basic classifiers, an 8-dimensional class vector is produced and concatenated with the 554-dimensional vector to generate a 562-dimensional feature vector. The produced vector is considered as input in the second layer. Similar to the first layer, the second layer of deep forest also generates another 562-dimensional vector applied to the third layer. If the estimated performance outperforms all previously-constructed layers, deep forest continues to increase a new layer. The model will terminate training when its performance fails to improve in the successive two layers. Finally, in the output layer, for each lncRNA–protein pair, its predicted interaction probability belonging to positive class and negative class is averaged, respectively. The class that the lncRNA–protein pair has higher average interaction probability is chosen as the final class.

The flowchart of deep forest
In particular, similar to DNN, deep forest utilizes a cascade structure. In the structure, each level receives features from its preceding level, and outputs the results to next level. Therefore, although the proportion of an 8-dimensional class vector in the input layer may be relatively smaller, its proportion in an LPI feature vector will continuing increase with the deepening of the number of layers. Therefore, in our model, the 8-dimensional class vector cannot be drown out.
2.5.5 Ensemble Learning
Ensemble learning demonstrates better prediction accuracy of a single model through training multiple classifiers and integrating their predictions [27, 61, 62]. Chen et al. [63] exploited a decision tree ensemble algorithm to uncover possible miRNA-disease associations. Zhang et al. [23] designed a sequence feature projection-based ensemble learning model to identify LPI candidates. Yi et al. [27] exploited a stacking ensemble learning algorithm to discover ncRNA–protein interactions.
Although AkNN, DNN, and deep forest can effectively predict LPIs, their predictive performance remains improvement. In this study, we present a soft voting-based ensemble learning framework, composed of AkNN, DNN, and deep forest, to enhance the classification ability of existing single model. Let \(S_{AkNN}\), \(S_{DNN}\), and \(S_{DF}\) denote association probability of an lncRNA–protein pair obtained by AkNN, DNN, and deep forest, respectively, its final relevance score is defined by Eq. (18) based on a soft voting technique:
An lncRNA–protein pair is labeled as positive class if its score is larger than 0.5 based on Eq. (18); otherwise, the lncRNA–protein pair is classified to negative.
3 Results
3.1 Evaluation Metrics
In the experiments, precision, recall, accuracy, F1 score, AUC and AUPR are applied to assess the performance of EnANNDeep. For the six measurements, higher values indicate better prediction ability. The experiments are repeatedly implemented for 20 times and the average values on the 20 rounds are selected as the final performance.
3.2 Experimental Settings
We conduct grid search to find the optimal parameters in SFPEL, PMDKN, CatBoost, PLIPCOM, and EnANNDeep when the five LPI prediction approaches obtain the best performance. The details are listed in Table 4. The parameters in LPI-SKF are set to default values provided by Zhou et al. [17].
In addition, to investigate the prediction performance of EnANNDeep for a new lncRNA or protein, three different fivefold CVs are designed.
-
1.
Fivefold CV on lncRNAs ( \(CV_{l}\)): rows in Y are randomly hidden for testing, that is, 80% of lncRNAs are randomly chosen as a training set and the remaining 20% is used as a testing set in each round. \(CV_{l}\) is used to find interacting proteins for a new lncRNA without any associated proteins.
-
2.
Fivefold CV on proteins ( \(CV_{p}\)): columns in Y are randomly hidden for testing, that is, 80% of proteins are randomly chosen as a training set and the remaining 20% is used as a testing set in each round. \(CV_{p}\) is used to identify interacting lncRNAs for a new protein without any associated lncRNAs.
-
3.
Fivefold CV on lncRNA–protein pairs ( \(CV_{lp}\)): lncRNA–protein pairs in Y are randomly hidden for testing, that is, 80% of lncRNA–protein pairs are chosen as a training set and the remaining 20% is used as a testing set in each round. \(CV_{lp}\) is used to uncover interaction information based on known LPIs.
3.3 Comparison with Five State-of-the-Art LPI Prediction Methods
We compare the proposed EnANNDeep method with five representative LPI prediction methods (SFPEL, PMDKN, CatBoost, PLIPCOM, and LPI-SKF) to measure the prediction performance of EnANNDeep. SFPEL is an ensemble learning method for LPI prediction based on sequence feature projection. SFPEL first extracted sequence features for lncRNAs and proteins and then computed lncRNA similarity and protein similarity. Finally, it used a feature projection-based ensemble learning framework to predict LPIs combining the computed similarity matrices.
PMKDN is a neighborhood nonnegative matrix decomposition model applied to possible LPI inference. PMKDN first selected multiple biological features of lncRNAs and proteins. Second, it combined protein GO ontology annotation and sequences, lncRNA sequences, and modified LPI network to calculate lncRNA similarity and protein similarity. Finally, it utilized a projection-based neighborhood nonnegative matrix decomposition algorithm to infer potential LPIs.
CatBoost is a new gradient boosting algorithm. CatBoost implemented two key techniques, that is, ordered boosting which is a permutation-driven alternative to a classification model, and a categorical feature procession strategy. The combination of them promotes CatBoost to outperform the other available boosting techniques. CatBoost has been applied to LPI discovery and obtained better LPI classification ability.
PLIPCOM employed two network features, diffusion features and HeteSim features, and built an LPI prediction model integrating the Gradient Tree Boosting (GTB) algorithm.
LPI-SKF first computed lncRNA similarity based on expression profiles and sequences of lncRNAs and LPI network, and protein similarity based on statistical features and sequences of proteins and LPI network. It then constructed a universal similarity kernel matrix for new LPI identification based on a similarity kernel fusion technique.
We evaluate the performance of our proposed EnANNDeep framework under three different fivefold CVs. During CVs, we randomly select unknown lncRNA–protein pairs as negative samples (non-LPIs). To reduce the overfitting problem produced by data imbalance, we set the ratio of negative LPIs to known LPIs as 1. That is, the number of the screened negative LPIs is the same as one of observed LPIs in the divided training set and test set. The best measurements are represented as bold in each row in Tables 5, 6 and 7.
Table 5 illustrates the prediction results from six LPI identification models in terms of the above six evaluation metrics under \(CV_{l}\). EnANNDeep achieves the highest average precision, recall, accuracy, F1 score, AUC, and AUPR. In particular, compared to SFPEL, PMDKN, CatBoost, PLIPCOM, and LPI-SKF, the average AUC computed by EnANNDeep outperforms 32.92%, 17.29%, 12.76%, 7.99%, and 3.94%, respectively. The average AUPR calculated by EnANNDeep are better 33.33%, 15.85%, 12.78%, 9.76%, and 5.29% than the above five methods. The result suggest that EnANNDeep may be suitable to linkage discovery for a new lncRNA.
Table 6 describes the six evaluation values under \(CV_{p}\). From Table 6, it can be found that EnANNDeep computes the best average precision, recall, accuracy, AUC and AUPR under \(CV_{p}\). Although EnANNDeep calculates relatively lower F1 score, it greatly boosts the precision, recall, accuracy, AUC, and AUPR performance. For example, compared to SFPEL, PMDKN, CatBoost, PLIPCOM, and LPI-SKF, its AUC boosts 42.76%, 22.23%, 31.74%, 21.79%, and 25.74%, respectively, AUPR improves 36.15%, 14.68%, 31.87%, 23.25%, and 18.82%, respectively. AUC and AUPR can more representatively characterize the performance of classifiers compared to the other four measurements. EnANNDeep distinctly outperforms the other five algorithms in terms of AUC and AUPR. Therefore, it is appropriate to prioritize potential lncRNAs for a new protein.
The experimental results under \(CV_{lp}\) are listed in Table 7. The results illustrate the optimal LPI classification ability of EnANNDeep. Under \(CV_{lp}\), EnANNDeep obtains the best average recall, accuracy, F1 score, AUC, and AUPR. For example, it computes F1 score of 0.8569, which is 9.46%, 30.93%, 8.51%, 3.09%, and 18.09% better than SFPEL, PMDKN, CatBoost, PLIPCOM, and LPI-SKF, respectively. The computed average AUC outperforms 7.07%, 17.53%, 6.55%, 2.43%, and 1.13%, respectively, and AUPR is better 4.67%, 14.98%, 4.19%, 3.74%, and 4.83%, respectively. SFPEL, PMDKN, CatBoost, PLIPCOM, and LPI-SKF are state-of-the-art LPI prediction algorithms. EnANNDeep greatly outperforms the five methods. The comparative results suggest the powerful performance of EnANNDeep under \(CV_{lp}\). That is, EnANNDeep can more accurately mine underlying relationships between lncRNAs and proteins even in the absence of some LPIs.
3.4 Comparison of Different Voting Methods
We conduct several experiments to observe the affect of voting techniques on the classification performance. We consider two voting techniques: soft voting approach and hard voting approach. Given an unobserved lncRNA–protein pair, the hard voting method first obtains label of an lncRNA–protein pair based on classification results from AkNN, DNN, and deep forest, respectively. Hard voting then classifies the sample as positive if its classification results from no less than two basic predictors are positive; otherwise, the pair is labeled as a negative class. The comparison results of two voting approaches under three CVs are shown in Tables 8, 9 and 10. From Tables 8, 9 and 10, we can find that the soft voting-based ensemble learning model can obtain better performance compared to the hard voting method.
3.5 The Effect of Numbers of RNA Secondary Structures on the Performance
Although abundant biological information contributes to improve LPI prediction performance, the biological features exist information robust and increase computational complexity. Therefore, we select the representative features to describe lncRNA secondary structures. Tables 11, 12 and 13 list the performance of EnANNDeep based on 5, 10, 64, and 128 RNA secondary structures with high probability. The results indicate that five features with high probability structures can accurately depict RNA secondary structures. Therefore, we chose five lncRNA secondary structures to reduce computation cost.
3.6 Case Study
In this section, we implement several cases to further evaluate the performance of EnANNDeep. We run the experiments for ten times and compute the average performance from the ten time results.
3.6.1 Finding Interacting Proteins for New lncRNAs
LncRNA Small Nucleolar RNA Host Gene 1 (SNHG1) has close linkage with multiple human diseases. For example, SNHG1 is up-regulated in gastric cancer and may serve as a potential therapeutic target of gastric cancer [64]. It promotes cell proliferation and cell cycle progression carcinoma and inhibits cell apoptosis in hepatocellular carcinoma [65]. It also enhances neuroinflammation in Parkinson’s disease [66]. In addition, nonsmall cell lung cancer has been reported to associate with upregulated SNHG1 [67].
In the three human dataset, SNHG1 interacts with 6, 18, and 4 proteins, respectively. To find interacting proteins with SNHG1, all its interaction information is hidden. The six LPI prediction algorithms are then applied to discover potential proteins for SNHG1. The predicted top 5 proteins are shown in Table 14. It can be found that Q15717, O00425, Q9Y6M1, P35637, and Q9NZI8 are inferred to have the highest interaction probabilities with SNHG1 in dataset 1. Although interactions between the five proteins and SNHG1 are unlabeled in Dataset 1, O00425 and P35637 have been reported to have close relationships with SNHG1 in datasets 3 and 2, respectively. Q15717, Q9Y6M1, and Q9NZI8 have been shown to link with SNHG1 in both datasets 2 and 3. In addition, all the inferred top 5 proteins linking with SNHG1 have higher rankings in SFPEL, PMDKN, CatBoost, PLIPCOM, LPI-SKF, and EnANNDeep. The ranking results again demonstrate the LPI classification ability of EnANNDeep for a new lncRNA.
3.6.2 Finding Interacting lncRNAs for New Proteins
Q9UKV8 can inhibit translation initiation through interaction with translation initiation factor EIF6 and prevent the recruitment from translation initiation factor EIF4-E. It up-regulates translation under the situation of serum starvation by binding to the AU element. More importantly, it is also interrelated with transcriptional gene silencing [41].
Q9UKV8 interacts with 207, 205, and 222 lncRNAs on three human datasets, respectively. In this section, its association information with lncRNAs is hidden and EnANNDeep is used to reveal its relevant lncRNAs. The found top 5 human lncRNAs interacting with Q9UKV8 are shown in Table 15.
On dataset 1, it can be observed that DANCR, RPI001_1039837 and AL139819.1 are inferred to interact with Q9UKV8. Although the associations between the three lncRNAs and Q9UKV8 are unknown in dataset 1, DANCR has been reported to interact with Q9UKV8 in dataset 2, RPI001_1039837 and AL139819.1 have been validated to interact with Q9UKV8 in dataset 3.
On dataset 2, it can be seen that RMRP, SNORD17, and RPI001_483534 have been predicted to interact with Q9UKV8. Although the relationships between RMRP and SNORD17 and Q9UKV8 are unknown in dataset 2, the two lncRNAs have been shown to link with Q9UKV8 in datasets 1 and 3, respectively. The interaction between RPI001_483534 and Q9UKV8 can not be retrieved on the three datasets. However, it ranks as 5, 2, 478, 183, 9, and 86 by EnANNDeep, SPFEL, PMDKN, PLIPCOM, LPI-CatBoost, and LPI-SKF, respectively. The higher rankings in EnANNDeep, SPFEL, and PLIPCOM suggest that RPI001_483534 may be relative to Q9UKV8 and needs further validation.
On dataset 3, RPI001_84645 and EXOC3 are identified to interact with Q9UKV8. The associations between the two lncRNAs and Q9UKV8 can be searched in datasets 2.
3.6.3 Finding New LPIs Based on Known LPIs
Potential LPIs are subsequently identified by EnANNDeep based on labeled LPIs. The inferred top 50 LPIs with the highest correlation probabilities on the five datasets are illustrated in Figs. 4, 5, 6, 7 and 8. The 50 associations contain known and unknown LPIs.

The predicted top 50 LPIs on dataset 1

The predicted top 50 LPIs on dataset 2

The predicted top 50 LPIs on dataset 3

The predicted top 50 LPIs on dataset 4

The predicted top 50 LPIs on dataset 5
The ranking results show that interactions between SNHG10 and Q15717, VIM-AS1, and Q15717, RPI001_1 01_2148 and ENSP00000385269, AthlncRNA-159 and 22328551, and ZmalncRNA-1314 and B6SP74 are the most possible LPIs among unlabeled lncRNA–protein pairs on datasets 1–5, respectively. They are ranked as 4, 14, 5, 33, and 1972 among 55,165, 74,340, 26,730, 3815, and 71,568 lncRNA–protein pairs, respectively.
lncRNA SNHG10 is a novel driver in the process of development and metastasis in hepatocellular carcinoma [68]. The lncRNA has close linkages with cell proliferation in gastric cancer [69], non-small cell lung cancer [70], and osteosarcoma [71]. Q15717 is an RNA-binding protein [72]. The protein contributes to embryonic stem cell differentiation, and can increase the leptin mRNA’s stability, and mediate the CDKN2A anti-proliferative activity [41]. Both SNHG10 and Q15717 have dense linkages with cell proliferation activity. We infer that SNHG10 may interact with Q15717 and is worthy of further validation.
4 Discussion
Identification of LPI candidates contributes to discover functions and mechanisms of lncRNAs. In this manuscript, an ensemble framework combining AkNN, DNN, and deep forest is developed to find possible interactions between lncRNAs and proteins. Three different CVs are conducted to compare the proposed EnANNDeep model with the other LPI prediction methods. The experimental results indicate that EnANNDeep can be more accurately applied to new LPI discovery.
Under \(CV_{p}\), majority of performance achieved from SFPEL, PMDKN, CatBoost, PLIPCOM, and LPI-SKF is much lower than those of \(CV_{l}\) and \(CV_{lp}\). Under \(CV_{p}\), 80% of lncRNAs are used to train the model and the remaining is applied to test the model. On five LPI datasets, each lncRNA may associate with 59, 84, 27, 35, and 42 proteins, respectively. When 20% of proteins are masked their associations, it may shield many LPIs and thus reduce the fitting level of a classification model to the LPI data. Therefore, abundance level of data severely affects the learning capacity of the five models. In comparison, under \(CV_{p}\), the performance obtained from EnANNDeep keeps relatively steady or even outperforms ones in comparing to \(CV_{l}\) and \(CV_{lp}\). The results demonstrate the robustness of the proposed EnANNDeep algorithm under CVs.
More importantly, similar to EnANNDeep, SFPEL, CatBoost, and PLIPCOM are three ensemble learning-based algorithms. The four ensemble learning-based LPI prediction methods integrate sequence information related to lncRNAs and proteins. SFPEL is a feature projection-based technique. CatBoost and PLIPCOM are gradient tree boosting and categorical boosting algorithms, respectively. EnANNDeep outperforms the three ensemble learning models, demonstrating the superior classification ability of basic predictors. That is, AkNN, DNN, and deep forest can be more effectively integrated to find possible LPIs. In addition, a few case analysis further suggest that EnANNDeep can mine useful information for a new lncRNA or protein.
The EnANNDeep framework demonstrates the powerful LPI discovery ability, especially under \(CV_{lp}\). It may be attributed to the following characteristics. First, a deep model composed of DNN and deep forest exhibits the optimal feature representation ability. In particular, deep forest works well even on small-scale data. Second, the proposed AkNN classifier can separately pick the most appropriate k for each query point so that the algorithm can better set the confidence intervals. Third, the ensemble framework from AkNN, DNN, and deep forest can effectively integrate the prediction results from the three predictors and thus improves the classification performance of EnANNDeep. Finally, it integrates multiple biological information related to LPI.
Although EnANNDeep can precisely identify new LPIs, it has one limitation: we select negative LPIs from unlabeled lncRNA–protein pairs. Indeed, unknown lncRNA–protein pairs may contain positive LPIs, thereby affecting the prediction ability of a model.
5 Conclusions
lncRNAs play pivotal roles in regulating many hallmarks of cancer biology. To decipher the lncRNA functions, we focus on new LPI mining. First, five LPI-related datasets are arranged. Second, the lncRNA and protein features are fused to depict each lncRNA–protein pair. Third, an ensemble model, composed of AkNN, DNN, and deep forest, is developed to classify unlabeled lncRNA–protein pairs, respectively. Finally, interaction probabilities of each lncRNA–protein pair from three predictors are integrated based on a soft voting technique to obtain the final classification. The results from comparative experiments and case analyses demonstrate that EnANNDeep can optimize the interplays between lncRNAs and proteins. Case analyses suggest that there probably exists an interaction between SNHG10 and Q15717.
In the future researches, first we will integrate various lncRNA-related datasets from different data sources to investigate the interaction biomolecules for lncRNAs, for example, lncRNA-miRNA interactions [73] and lncRNA-DNA interactions [36]. Second, more biological information from lncRNAs and proteins, for example, secondary structures of lncRNAs, secondary and tertiary structures of proteins, will be fused to represent an lncRNA–protein pair. Finally, we will develop a negative sample selection method based on positive-unlabeled learning to screen reliable negative LPIs.
Availability of data and materials
Source codes and datasets are freely available for download at https://github.com/plhhnu/EnANNDeep.
Abbreviations
- LPI:
-
Long noncoding RNA–Protein Interaction
- EnANNDeep:
-
Ensemble-based lncRNA–protein interaction prediction framework with adaptive k-nearest neighbor and deep models
- k-NN:
-
k-nearest neighbor
- AkNN:
-
Adaptive k-nearest neighbor
- DNN:
-
Deep neural network
- CVs:
-
Cross validations
References
Chen X, Sun YZ, Guan NN, Qu J, Huang ZA, Zhu ZX, Li JQ (2019) Computational models for LNCRNA function prediction and functional similarity calculation. Brief Funct Genom 18(1):58–82. https://doi.org/10.1093/bfgp/ely031
Wang J, Ma R, Ma W, Chen J, Yang J, Xi Y, Cui Q (2016) Lncdisease: a sequence based bioinformatics tool for predicting lncRNA-disease associations. Nucleic Acids Res 44(9):e90–e90. https://doi.org/10.1093/nar/gkw093
Ching T, Masaki J, Weirather J, Garmire LX (2015) Non-coding yet non-trivial: a review on the computational genomics of lincrnas. BioData Min 8(1):1–12. https://doi.org/10.1186/s13040-015-0075-z
Zhang H, Ming Z, Fan C, Zhao Q, Liu H (2020) A path-based computational model for long non-coding RNA-protein interaction prediction. Genomics 112(2):1754–1760. https://doi.org/10.1016/j.ygeno.2019.09.018
Chen X, Yan CC, Zhang X, You ZH (2017) Long non-coding RNAS and complex diseases: from experimental results to computational models. Brief Bioinform 18(4):558–576. https://doi.org/10.1093/bib/bbw060
Wang W, Dai Q, Li F, Xiong Y, Wei DQ (2020) Mlcdforest: multi-label classification with deep forest in disease prediction for long non-coding rnas. Brief Bioinform. https://doi.org/10.1093/bib/bbaa104
Liu H, Song G, Zhou L, Hu X, Liu M, Nie J, Lu S, Wu X, Cao Y, Tao L et al (2013) Compared analysis of LNCRNA expression profiling in pdk1 gene knockout mice at two time points. Cell Physiol Biochem 32(5):1497–1508. https://doi.org/10.1159/000356586
Vizoso M, Esteller M (2012) The activatory long non-coding RNA dbe-t reveals the epigenetic etiology of facioscapulohumeral muscular dystrophy. Cell Res 22(10):1413–1415. https://doi.org/10.1038/cr.2012.93
De R, Hu T, Moore JH, Gilbert-Diamond D (2015) Characterizing gene-gene interactions in a statistical epistasis network of twelve candidate genes for obesity. BioData Min 8(1):1–16. https://doi.org/10.1186/s13040-015-0077-x
Wang J, Su Z, Lu S, Fu W, Liu Z, Jiang X, Tai S (2018) Lncrna hoxa-as2 and its molecular mechanisms in human cancer. Clin Chim Acta 485:229–233. https://doi.org/10.1016/j.cca.2018.07.004
Tamang S, Acharya V, Roy D, Sharma R, Aryaa A, Sharma U, Khandelwal A, Prakash H, Vasquez KM, Jain A (2019) Snhg12: an lncRNA as a potential therapeutic target and biomarker for human cancer. Front Oncol 9:901. https://doi.org/10.3389/fonc.2019.00901
Liu T, Han Z, Li H, Zhu Y, Sun Z, Zhu A (2018) Lncrna dleu1 contributes to colorectal cancer progression via activation of kpna3. Mol Cancer 17(1):1–13. https://doi.org/10.1186/s12943-018-0873-2
Loewen G, Jayawickramarajah J, Zhuo Y, Shan B (2014) Functions of LNCRNA hotair in lung cancer. J Hematol Oncol 7(1):1–10. https://doi.org/10.1186/s13045-014-0090-4
Mao Z, Li H, Du B, Cui K, Xing Y, Zhao X, Zai S (2017) LncRNA dancr promotes migration and invasion through suppression of lncRNA-let in gastric cancer cells. Biosci Rep. https://doi.org/10.1042/BSR20171070
Zhao Q, Yu H, Ming Z, Hu H, Ren G, Liu H (2018) The bipartite network projection-recommended algorithm for predicting long non-coding RNA-protein interactions. Mol Therapy Nucleic Acids 13:464–471. https://doi.org/10.1016/j.omtn.2018.09.020
Ge M, Li A, Wang M (2016) A bipartite network-based method for prediction of long non-coding RNA-protein interactions. Genom Proteom Bioinform 14(1):62–71. https://doi.org/10.1016/j.gpb.2016.01.004
Zhou YK, Hu J, Shen ZA, Zhang WY, Du PF (2020) Lpi-skf: Predicting lncRNA-protein interactions using similarity kernel fusions. Front Genet 11:1554. https://doi.org/10.3389/fgene.2020.615144
Zheng X, Wang Y, Tian K, Zhou J, Guan J, Luo L, Zhou S (2017) Fusing multiple protein-protein similarity networks to effectively predict lncRNA-protein interactions. BMC Bioinform 18(12):11–18. https://doi.org/10.1186/s12859-017-1819-1
Liu H, Ren G, Hu H, Zhang L, Ai H, Zhang W, Zhao Q (2017) Lpi-nrlmf: lncrna-protein interaction prediction by neighborhood regularized logistic matrix factorization. Oncotarget. https://doi.org/10.18632/oncotarget.21934
Zhang T, Wang M, Xi J, Li A (2018) Lpgnmf: predicting long non-coding RNA and protein interaction using graph regularized nonnegative matrix factorization. IEEE/ACM Trans Comput Biol Bioinform 17(1):189–197. https://doi.org/10.1109/TCBB.2018.2861009
Ma Y, He T, Jiang X (2019) Projection-based neighborhood non-negative matrix factorization for lncRNA-protein interaction prediction. Front Genet 10:1148. https://doi.org/10.3389/fgene.2019.01148
Hu H, Zhang L, Ai H, Zhang H, Fan Y, Zhao Q, Liu H (2018) HLPI-ensemble: prediction of human LNCRNA-protein interactions based on ensemble strategy. RNA Biol 15(6):797–806. https://doi.org/10.1080/15476286.2018.1457935
Zhang W, Yue X, Tang G, Wu W, Huang F, Zhang X (2018) Sfpel-lpi: sequence-based feature projection ensemble learning for predicting lncrna-protein interactions. PLoS Comput Biol 14(12):e1006616. https://doi.org/10.1371/journal.pcbi.1006616
Deng L, Wang J, Xiao Y, Wang Z, Liu H (2018) Accurate prediction of protein-LNCRNA interactions by diffusion and hetesim features across heterogeneous network. BMC Bioinform 19(1):1–11. https://doi.org/10.1186/s12859-018-2390-0
Fan XN, Zhang SW (2019) LPI-BLS: predicting LNCRNA-protein interactions with a broad learning system-based stacked ensemble classifier. Neurocomputing 370:88–93. https://doi.org/10.1016/j.neucom.2019.08.084
Wekesa JS, Meng J, Luan Y (2020) Multi-feature fusion for deep learning to predict plant lncRNA-protein interaction. Genomics 112(5):2928–2936. https://doi.org/10.1016/j.ygeno.2020.05.005
Yi HC, You ZH, Wang MN, Guo ZH, Wang YB, Zhou JR (2020) Rpi-se: a stacking ensemble learning framework for ncrna-protein interactions prediction using sequence information. BMC Bioinform 21(1):1–10. https://doi.org/10.1186/s12859-020-3406-0
LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521(7553):436–444. https://doi.org/10.4258/hir.2016.22.4.351
Chu Y, Kaushik AC, Wang X, Wang W, Zhang Y, Shan X, Salahub DR, Xiong Y, Wei DQ (2019) DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features. Brief Bioinform. https://doi.org/10.1093/bib/bbz152
Kaushik AC, Wang YJ, Wang X, Kumar A, Singh SP, Pan CT, Shiue YL, Wei DQ (2019) Evaluation of anti-EGFR-IRGD recombinant protein with gold nanoparticles: synergistic effect on antitumor efficiency using optimized deep neural networks. RSC Adv 9(34):19261–19270. https://doi.org/10.1039/C9RA01975H
Gainza P, Sverrisson F, Monti F, Rodola E, Boscaini D, Bronstein M, Correia B (2020) Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat Methods 17(2):184–192. https://doi.org/10.1038/s41592-019-0666-6
Deng L, Wu H, Liu H (2019) D2vcb: A hybrid deep neural network for the prediction of in-vivo protein-DNA binding from combined DNA sequence. In: 2019 IEEE International Conference on bioinformatics and biomedicine (BIBM). IEEE, pp 74–77. https://doi.org/10.1109/BIBM47256.2019.8983051
Zhang Y, Jia C, Kwoh CK (2020) Predicting the interaction biomolecule types for lncRNA: an ensemble deep learning approach. Brief Bioinform. https://doi.org/10.1093/bib/bbaa228
Wekesa JS, Meng J, Luan Y (2020) A deep learning model for plant lncRNA-protein interaction prediction with graph attention. Mol Genet Genom 295(5):1091–1102. https://doi.org/10.1007/s00438-020-01682-w
Wei H, Liao Q, Liu B (2020) ilncrnadis-fb: identify lncRNA-disease associations by fusing biological feature blocks through deep neural network. IEEE/ACM Trans Comput Biol Bioinform. https://doi.org/10.1109/TCBB.2020.2964221
Zhao T, Hu Y, Peng J, Cheng L (2020) Deeplgp: a novel deep learning method for prioritizing lncrna target genes. Bioinformatics 36(16):4466–4472. https://doi.org/10.1093/bioinformatics/btaa428
Shaw D, Chen H, Xie M, Jiang T (2021) Deeplpi: a multimodal deep learning method for predicting the interactions between lncrnas and protein isoforms. BMC Bioinform 22(1):1–22. https://doi.org/10.1186/s12859-020-03914-7
Li A, Ge M, Zhang Y, Peng C, Wang M (2015) Predicting long noncoding RNA and protein interactions using heterogeneous network model. BioMed Res Int. https://doi.org/10.1155/2015/671950
Yuan J, Wu W, Xie C, Zhao G, Zhao Y, Chen R (2014) Npinter v2. 0: an updated database of ncrna interactions. Nucleic Acids Res 42(D1):D104–D108. https://doi.org/10.1093/nar/gkt1057
Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, Zhu W, Wu W, Chen R, Zhao Y (2014) Noncodev4: exploring the world of long non-coding RNA genes. Nucleic Acids Res 42(D1):D98–D103. https://doi.org/10.1093/nar/gkt1222
Consortium U (2019) Uniprot: a worldwide hub of protein knowledge. Nucleic Acids Res 47(D1):D506–D515. https://doi.org/10.1093/nar/gky1049
Zhang W, Qu Q, Zhang Y, Wang W (2018) The linear neighborhood propagation method for predicting long non-coding RNA-protein interactions. Neurocomputing 273:526–534. https://doi.org/10.1016/j.neucom.2017.07.065
Bai Y, Dai X, Ye T, Zhang P, Yan X, Gong X, Liang S, Chen M (2019) PLNCRNADB: a repository of plant LNCRNAS and LNCRNA-RBP protein interactions. Curr Bioinform 14(7):621–627. https://doi.org/10.2174/1574893614666190131161002
Shrikumar A, Prakash E, Kundaje A (2019) Gkmexplain: fast and accurate interpretation of nonlinear gapped k-mer svms. Bioinformatics 35(14):i173–i182. https://doi.org/10.1093/bioinformatics/btz322
Tahir M, Hayat M, Khan SA (2019) inuc-ext-psetnc: an efficient ensemble model for identification of nucleosome positioning by extending the concept of chou’s pseaac to pseudo-tri-nucleotide composition. Mol Genet Genom 294(1):199–210. https://doi.org/10.1007/s00438-018-1498-2
Liu B, Liu F, Fang L, Wang X, Chou KC (2015) REPDNA: a python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics 31(8):1307–1309. https://doi.org/10.1093/bioinformatics/btu820
Su Y, Luo Y, Zhao X, Liu Y, Peng J (2019) Integrating thermodynamic and sequence contexts improves protein-RNA binding prediction. PLoS Comput Biol 15(9):e1007283. https://doi.org/10.1371/journal.pcbi.1007283
Magnan CN, Baldi P (2014) Sspro/accpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 30(18):2592–2597. https://doi.org/10.1093/bioinformatics/btu352
Peterson LE (2009) K-nearest neighbor. Scholarpedia 4(2):1883. https://doi.org/10.4249/scholarpedia.1883
Du Q, Faber V, Gunzburger M (1999) Centroidal voronoi tessellations: applications and algorithms. SIAM Rev 41(4):637–676. https://doi.org/10.1137/S0036144599352836
Balsubramani A, Dasgupta S, Freund Y, Moran S (2019) An adaptive nearest neighbor rule for classification. In: NeurIPS, pp 7577–7586. https://par.nsf.gov/biblio/10168808
Zhao T, Hu Y, Valsdottir LR, Zang T, Peng J (2020) Identifying drug-target interactions based on graph convolutional network and deep neural network. Brief Bioinform. https://doi.org/10.1093/bib/bbaa044
Wang L, You ZH, Huang YA, Huang DS, Chan KC (2020) An efficient approach based on multi-sources information to predict circrna-disease associations using deep convolutional neural network. Bioinformatics 36(13):4038–4046. https://doi.org/10.1093/bioinformatics/btz825
Kingma DP, Ba J (2014) Adam: A method for stochastic optimization. arXiv:1412.6980
Cai L, Lu C, Xu J, Meng Y, Wang P, Fu X, Zeng X, Su Y (2021) Drug repositioning based on the heterogeneous information fusion graph convolutional network. Brief Bioinform. https://doi.org/10.1093/bib/bbab319
Zhou ZH, Feng J (2019) Deep forest. National Sci Rev 6(1):74–86. https://doi.org/10.1093/nsr/nwy108
Zhou ZH, Feng J (2017) Deep forest[J]. arXiv preprint arXiv:1702.08835
Qi Y (2012) Random forest for bioinformatics. In: Ensemble machine learning. Springer, New York, pp 307–323. https://doi.org/10.1007/978-1-4419-9326-7_11
Biau G, Scornet E (2016) A random forest guided tour. Test 25(2):197–227. https://doi.org/10.1007/s11749-016-0481-7
Geurts P, Ernst D, Wehenkel L (2006) Extremely randomized trees. Mach Learn 63(1):3–42. https://doi.org/10.1007/s10994-006-6226-1
Cao Y, Geddes TA, Yang JYH, Yang P (2020) Ensemble deep learning in bioinformatics. Nat Mach Intell 2(9):500–508. https://doi.org/10.1038/s42256-020-0217-y
Chen X, Zhu CC, Yin J (2019) Ensemble of decision tree reveals potential mirna-disease associations. PLoS Comput Biol 15(7):e1007209. https://doi.org/10.1371/journal.pcbi.1007209
Chen X, Xie D, Zhao Q, You ZH (2019) Micrornas and complex diseases: from experimental results to computational models. Brief Bioinform 20(2):515–539. https://doi.org/10.1093/bib/bbx130
Hu Y, Ma Z, He Y, Liu W, Su Y, Tang Z (2017) LNCRNA-SNHG1 contributes to gastric cancer cell proliferation by regulating DNMT1. Biochem Biophys Res Commun 491(4):926–931. https://doi.org/10.1016/j.bbrc.2017.07.137
Zhang M, Wang W, Li T, Yu X, Zhu Y, Ding F, Li D, Yang T (2016) Long noncoding RNA snhg1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomed Pharmacother 80:73–79. https://doi.org/10.1016/j.biopha.2016.02.036
Cao B, Wang T, Qu Q, Kang T, Yang Q (2018) Long noncoding rna snhg1 promotes neuroinflammation in parkinson’s disease via regulating mir-7/nlrp3 pathway. Neuroscience 388:118–127. https://doi.org/10.1016/j.neuroscience.2018.07.019
Cui Y, Zhang F, Zhu C, Geng L, Tian T, Liu H (2017) Upregulated LNCRNA SNHG1 contributes to progression of non-small cell lung cancer through inhibition of MIR-101-3p and activation of wnt/ \(\beta\)-catenin signaling pathway. Oncotarget 8(11):17785. https://doi.org/10.18632/oncotarget.14854
Lan T, Yuan K, Yan X, Xu L, Liao H, Hao X, Wang J, Liu H, Chen X, Xie K et al (2019) LNCRNA SNHG10 facilitates hepatocarcinogenesis and metastasis by modulating its homolog scarna13 via a positive feedback loop. Can Res 79(13):3220–3234. https://doi.org/10.1158/0008-5472
Yuan X, Yang T, Xu Y, Ou S, Shi P, Cao M, Zuo X, Liu Q, Yao J (2020) Snhg10 promotes cell proliferation and migration in gastric cancer by targeting mir-495-3p/ctnnb1 axis. Dig Dis Sci:1–10. https://doi.org/10.1007/s10620-020-06576-w
Liang M, Wang L, Cao C, Song S, Wu F (2020) LNCRNA SNHG10 is downregulated in non-small cell lung cancer and predicts poor survival. BMC Pulm Med 20(1):1–6. https://doi.org/10.1186/s12890-020-01281-w
Zhu S, Liu Y, Wang X, Wang J, Xi G (2020) Lncrna snhg10 promotes the proliferation and invasion of osteosarcoma via wnt/ \(\beta\)-catenin signaling. Mol Therapy Nucleic Acids 22:957–970. https://doi.org/10.1016/j.omtn.2020.10.010
Li J, Sun W (2018) Exploration of radiosensitivity-related LNCRNAS in esophageal cancer stem cell. Int J Radiat Oncol Biol Phys 102(3):e33. https://doi.org/10.1016/j.ijrobp.2018.07.524
Chen X, Wang L, Qu J, Guan NN, Li JQ (2018) Predicting mirna-disease association based on inductive matrix completion. Bioinformatics 34(24):4256–4265. https://doi.org/10.1093/bioinformatics/bty503
Acknowledgements
We would like to thank anonymous reviewers and all authors of the cited references.
Funding
This research was funded by the National Natural Science Foundation of China (Grant 62072172, 61803151).
Author information
Authors and Affiliations
Contributions
Conceptualization: L-HP, J-WT and L-QZ; funding acquisition: L-HP, L-QZ; investigation: L-HP and J-WT; methodology: L-HP and J-WT; project administration: L-HP, L-QZ; software: J-WT; validation: J-WT, X-FT; writing—original draft: L-HP; writing—review and editing: L-HP and J-WT.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Source codes are freely available for download at https://github.com/plhhnu/EnANNDeep.
Rights and permissions
About this article

Cite this article
Peng, L., Tan, J., Tian, X. et al. EnANNDeep: An Ensemble-based lncRNA–protein Interaction Prediction Framework with Adaptive k-Nearest Neighbor Classifier and Deep Models. Interdiscip Sci Comput Life Sci 14, 209–232 (2022). https://doi.org/10.1007/s12539-021-00483-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-021-00483-y