
Abstract
Nucleosome is a central element of eukaryotic chromatin, which composes of histone proteins and DNA molecules. It performs vital roles in many eukaryotic intra-nuclear processes, for instance, chromatin structure and transcriptional regulation formation. Identification of nucleosome positioning via wet lab is difficult; so, the attention is diverted towards the accurate intelligent automated prediction. In this regard, a novel intelligent automated model “iNuc-ext-PseTNC” is developed to identify the nucleosome positioning in genomes accurately. In this predictor, the sequences of DNA are mathematically represented by two different discrete feature extraction techniques, namely pseudo-tri-nucleotide composition (PseTNC) and pseudo-di-nucleotide composition. Several contemporary machine learning algorithms were examined. Further, the predictions of individual classifiers were integrated through an evolutionary genetic algorithm. The success rates of the ensemble model are higher than individual classifiers. After analyzing the prediction results, it is noticed that iNuc-ext-PseTNC model has achieved better performance in combination with PseTNC feature space, which are 94.3%, 93.14%, and 88.60% of accuracies using six-fold cross-validation test for the three benchmark datasets S1, S2, and S3, respectively. The achieved outcomes exposed that the results of iNuc-ext-PseTNC model are prominent compared to the existing methods so far notifiable in the literature. It is ascertained that the proposed model might be more fruitful and a practical tool for rudimentary academia and research.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cell is the rudimentary unit of all living organisms, which may be prokaryotic or eukaryotic. It accomplishes different functions such as reproduction, respiration, transportation of molecules, and identity maintenance. Cell constitutes nucleus, Golgi complex, mitochondria, endoplasmic reticulum, ribosomes, etc. Nucleus is a membrane-enclosed organelle, consisting of genetic material in the form of long DNA molecules (Athey et al. 1990; Mavrich et al. 2008a, c). DNA organizes in a supercoiling structure known as chromatin. Nucleosome is composed of histone proteins and DNA molecules, which is considered the basic unit of eukaryotic chromatin (Thoma et al. 1979). The core histone proteins contain four sub-units, namely H2A, H2B, H3 and H4; however, the linker histone is H1. Chromatin DNA is of two types: one is core DNA, which is a double helical DNA strand about 146 bp, coils around the core histones in a left-handed super-helix form, and the other is linker DNA (Berbenetz et al. 2010; Schwartz et al. 2009). Linker DNA is a short sequence of 20–60 bp through which nucleosomes are attached to each other (Athey et al. 1990; Mavrich et al. 2008a, b). Thus, in nucleosome, the final length of DNA becomes 166–167 bp, which may be two full turns (Thoma et al. 1979) known as chromatosome. The histone octamer around the packaging of DNA performs significant roles in biological processes, namely RNA splicing, DNA replication, repair mechanisms, and transcriptional control (Schwartz et al. 2009; Berbenetz et al. 2010; Yasuda et al. 2005). Various traditional methods such as nuclear magnetic resonance (NMR), filter binding assays, and X-ray crystallography were carried out for the recognition of DNA and proteins (Gabdank et al. 2010; Chen et al. 2014; Xi et al. 2010; Eddy 1996; Segal et al. 2006; Field et al. 2008). Owing to a confined number of genomic and proteomic structure availability, time, and lack of laboratory equipment, the traditional methods remained unsuccessful. Apart from that, a huge number of biological sequences are reported in databases owing to the fast technological advancement in the post-genomic era. However, the identification of these unprocessed data is a challenging job for the researchers in the field of bioinformatics and proteomics. Viewing the implications of traditional approaches, the investigators have diverted their attention towards the computational methods by utilizing contemporary machine learning methods (Field et al. 2008). Nucleosome positioning in genomes is identified by performing various studies (Peckham et al. 2007; Satchwell et al. 1986; Yuan et al. 2005; Goñi et al. 2008; Tahir and Hayat 2016; Yuan and Liu 2008; Tolstorukov et al. 2008; Nikolaou et al. 2010). Hidden Markov model (HMM) was applied to capture the central patterns from the provided data (Stolz and Bishop 2010). Segal et al. introduced a probabilistic model by calculating the probabilities of nucleotides and higher rank dependencies among nucleotides (Thoma et al. 1979). Several k-mer methods were utilized by Kaplan et al. (2009) and Field et al. (2008) for improving the success rates of the developed models (Goñi et al. 2008; Isami et al. 2015). Likewise, Xi et al. introduced a novel duration hidden Markov model (dHMM) by executing the linker DNA length as well as nucleosome positions to collect nucleosome positioning information (Nikolaou et al. 2010). In a sequel, Satchwell et al. introduced di-nucleotide and tri-nucleotide composition for the identification of nucleosome positioning in genome (Awazu 2017). Furthermore, SVM in combination with sequence-based features was used by Peckham et al. to analyze some oligo-nucleotides implicated in nucleosome formation and exclusion (Satchwell et al. 1986; Liu et al. 2015a).
“iNuc-PseKNC” predictor was developed by Gou et al. for the discrimination of nucleosome positioning in genomes (Peckham et al. 2007). Pseudo k-tuple nucleotide composition utilized six different DNA local structural physicochemical properties for expressing DNA sequences (Peckham et al. 2007).
The notion of pseudo-amino acid (PseAA) composition was broadly implemented in various computational models. It was further extended to DNA representation and introduced several predictors, namely repDNA (Li et al. 2015), Pse-in-One (YongE and GaoShan 2015), and iDNA-KACC (Xiang et al. 2016). Besides, some predictors such as iRSpot-EL (Dong et al. 2016) and iDHS-EL (Xiao et al. 2013) were also established by Liu et al. The concept of PseKNC was successfully implemented and illustrated in RNA/DNA, namely identifying nucleosome (Liu et al. 2015d), predicting splicing site, identifying translation initiation site (Che et al. 2016), predicting recombination spots (Liu et al. 2015d; Luo et al. 2016; Tian et al. 2015), predicting promoters (Liu et al. 2015d), identifying origin of replication (Li et al. 2015), identifying RNA and DNA modification (Yong and GaoShan 2015; Xiang et al. 2016), and others (Dong et al. 2016). According to previous research studies (Guo et al. 2014; Xiao et al. 2013; Chen et al. 2013; Liu et al. 2014a; Qiu et al. 2014; Xu et al. 2013a, b), a precise, reliable, and efficient predictor will be established for a biological system by accomplishing Chou’s 5-steps. They are defined as follows: (1) to choose or design a valid dataset to train and test the model effectively; (2) to mathematically express the samples in such way that can truly represent the motif of target class; (3) to develop or introduce an efficient algorithm for operational engine; (4) to apply a cross-validation test for evaluating the outcome of model; and (5) to develop a web-predictor for the model that can be easily accessible to the public.
Rest of the paper is structured as follows: the next section demonstrates materials and methods, “Results” section presents the performance of supervised algorithms followed by “Discussion” section and finally conclusion is reported at the end of the paper.
Methods
Datasets
In this study, we have targeted three different species such as D. melanogaster, C. elegans, and H. sapiens. The benchmark datasets for these species were selected from Guo et al. 2014. These datasets can be mathematically expressed as
In the above equations, \(S1\), \({S_2}\) and \({S_3}\) represent the benchmark datasets for C. elegans, D. melanogaster, and H. sapiens, respectively. The \(S1\) benchmark dataset contains 4573 samples, of which 2273 belong to \(S_{1}^{+}\) nucleosome-forming samples and 2300 to \(S_{1}^{ - }\) nucleosome-inhabiting samples. The \({S_2}\) benchmark dataset contains 5175 samples, of which 2567 belong to \(S_{2}^{+}\) nucleosome forming and 2608 to \(S_{2}^{ - }\) nucleosome inhabiting. Similarly, \({S_3}\) represents the third benchmark dataset comprised of 5750 samples, of which 2900 belong to \(S_{3}^{+}\) nucleosome-forming and 2850 to \(S_{3}^{ - }\) nucleosome-inhabiting samples. The \(U\) symbol denotes the union of two sets. By removing redundant samples from benchmark datasets, the CD-HIT software was applied, with a cutoff threshold value of 80% (Guo et al. 2014).
Feature extraction techniques
Suppose S is the sequence of DNA with L nucleic acid residues as shown below:
In the above equation, N1 denotes the residue of nucleic acid at the first position in a sequence, N2 denotes the residue of nucleic acid at the second position in a sequence and NL denotes the last residue of the nucleic acid in a DNA sequence at position L (Ioshikhes et al. 1996). These nucleotides are expressed as
where the value of i = 1, 2, …, L.
DNA sequence is numerically expressed by computing the frequency of each nucleotide, known as nucleic acid composition (NAC). It can be presented as below:
In the above equation, ƒ(A) indicates the frequency of adenine, ƒ(C) shows the frequency of cytosine and so on in the sequence of DNA; however, the T symbol indicates the transpose operator. Conventional NAC is a simple discrete method, but it does not maintain information regarding sequence order of nucleotides. Consequently, correlation factors among nucleotides are totally ignored. Viewing at the significance of correlation factors and local information, the idea of pseudo-amino acid (PseAA) composition was utilized and took place nearly all the fields of computational proteomics and genomics (Chou 2001a, 2005; Cao et al. 2013; Liu et al. 2014b; Chen and Li 2013). Subsequently, the PseAA composition idea has been extended to handle the sequences of RNA/DNA in the nature of PseKNC.
In this article, we have applied two different discrete feature extraction methods, namely PseDNC and PseTNC to collect variant and prominent numerical descriptors from the sequences of DNA.
Pseudo-di-nucleotide composition
PseDNC expresses a DNA sequence by making a pair of two nucleotides and then calculates the frequency of each pair. Let us suppose, N1N2 is the first pair of di-nucleotide, N2N3 is the second pair of di-nucleotide, and finally, NL−1NL is the last pair of di-nucleotide. Subsequently, 4 × 4 = 16D feature vector is formed. It can be numerically represented as follows:
In the above equations, the T symbol represents the transpose operator, \(f_{1}^{{di}}=f(AA)\) is the frequency of AA pair, \(f_{2}^{{di}}=f(AC)\) is the frequency of AC pair, and \(f_{4}^{{di}}=f(AT)\) is the frequency AT pair in the sequence of DNA and so on.
Pseudo-tri-nucleotide composition
PseTNC expresses the sequence of DNA by combining three nucleotides and then computes the occurrence frequency of three consecutive nucleotide pair. For example, N1N2N3 is the first component of tri-nucleotide, N2N3N4 is the second component of tri-nucleotide, and so on, while the last component of tri-nucleotide is NL−2NL−1NL; accordingly, the corresponding feature vector 4 × 4 × 4 = 64D is generated. The PseTNC is mathematically expressed as
where \(f_{1}^{{3{\text{-tuple}}}}\) = \(f(AAA)\) is the frequency of AAA component, \(f_{4}^{{3{\text{-tuple}}}}\) = \(f(AAG)\) is the frequency of AAG component, while \(f_{{64}}^{{3{\text{-tuple}}}}\) = \(f(TTT)\) is the frequency of TTT component in the sequence of DNA.
Framework of proposed predictor
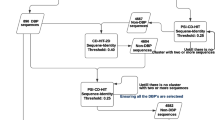
In this research, a novel predictor was introduced, namely iNuc-ext-PseTNC for the discrimination of nucleosome positioning in genomes. Two feature extraction methods: PseDNC and PseTNC are utilized for numerical representation of DNA sequences. Three distant natures of classifiers namely: K-nearest neighbor (KNN), probabilistic neural network (PNN) and support vector machine (SVM) are executed. The predicted outcomes of the individual classifier were then fused to develop an ensemble model “iNuc-ext-PseTNC”. The developed model shows outstanding performance compared to the current state of arts in the literature, so far. The framework of the proposed prediction ensemble model has been shown in Fig. 1.

The framework of iNuc-GA-PseTNC
Classification algorithms
In pattern recognition and machine learning, classification is a supervised learning, in which a novel observation is recognized as already defined target classes on the basis of a training dataset. The process of classification is accomplished in two steps: training and testing. In the training step, the pattern of the pre-defined classes is memorized from the provided data. In the testing step, the new observation is identified on the basis memorized pattern. In this study, we have applied KNN, PNN, and SVM classification algorithms (Guo et al. 2014; Tahir and Hayat 2016; Hayat and Khan 2012; Kabir and Hayat 2016).
Ensemble classification
In the last few decades, researchers have diverted their attention from individual classifier to the concept of ensemble classification to reduce prediction error and broadly utilize for signal peptide prediction (Chou and Shen 2007c), predicting protein subcellular location (Chou and Shen 2007a), for enzyme subfamily prediction (Chou 2005) and predicting subcellular location (Chou and Shen 2007b; Zhang et al. 2015b, 2017; Li et al. 2016). During the classification process, the predicted outcome of each classifier is varied and can yield different errors. However, when the prediction of each classifier is merged, the classification errors are minimized because the error of one classifier is recompensed by another classifier (Hayat and Khan 2012; Zhang et al. 2012, 2015, 2016). The ensemble classification fuses the prediction of various classifiers and tries to minimize the variance instigated in these individual classifiers. In this study, various classifiers, namely KNN, PNN, and SVM are used. First, a classifier is trained and the prediction is noted. The predictions of each classifier are then fused to develop the ensemble model (Kabir and Hayat 2016). It can be mathematically expressed as below:
In the above equation, the ensemble model is represented by EnsC and the symbol \(\oplus\) represents the combination operator.
where C1, C2 and C3 are the individual classifiers; S1 and S2 represent the two classes of nucleosome forming and nucleosome inhabiting.
where
Outcome of the ensemble model adopting GA is generated as
where GAEnsC is the outcome of the ensemble model, Max represents the maximum output, and x1, x2, and x3 are the optimum weight of the individual classifiers.
Metrics for measuring prediction performance
In the statistical prediction model, the fundamental task is the partition of provided data into training and testing subsets. In the literature, cross-validation test is extensively applied for evaluating the quality and effectiveness of the developed model. Sub-sampling or K-fold, self-consistency, independent dataset, and jackknife tests are the types of the cross-validation test. Here, six-fold cross-validation test is applied, in which the data are divided into six-fold, where onefold is used for testing and the rest of folds are utilized for the training process. The same process is repeated six times and finally, the outcome is yielded on the basis of average. The metrics for measuring the prediction performance are mathematically expressed as (Manavalan et al. 2018; Liu et al. 2015c, 2016a, 2017c, 2018; Hayat and Tahir 2015; Ahmad et al. 2017; Ehsan et al. 2018; Feng et al. 2018; Cheng et al. 2017a, b, c, d, 2018; Xiao et al. 2017, 2018)
Equations (15–18) are widely utilized to compute the prediction of classifiers; however, in some cases, these equations are not suitable for biologists, because of the lack of intuitiveness. In this study, we have used the following equations to solve this complication (Schwartz et al. 2009; Xu et al. 2013a, 2014; Chou 2001b; Chen et al. 2013a, 2016, 2017; Lin et al. 2014; Jia et al. 2016; Zhang et al. 2016; Liu et al. 2016c, 2017a,b; Feng et al. 2017):
In the above equations, \({Z^ - }\) denotes the whole number of the true nucleosome-inhibiting sample while \({Z^+}\) signifies the whole number of true nucleosome forming, whereas \(Z_{+}^{ - }\) represents the whole number of nucleosome inhibiting predicted incorrectly while \(Z_{ - }^{+}\) shows the whole number of nucleosome forming predicted incorrectly.
Results
The success rates of two feature spaces are empirically analyzed and performance comparisons have been drawn as well.
Performance comparison of classifiers using PseDNC feature space
Tables 1, 2 and 3 present the experimental results of individual and ensemble classifiers for the three datasets \({S_1}\), \({S_2}\), and \({S_3}\). Among the individual classifiers, PNN has obtained an efficient result for dataset \({S_1}\) on the value of spread = 4.51, whereas SVM has yielded the higher outcomes for dataset \({S_2}\) on the value of cost function (c = 1.33 and gamma (\(g\) = 0.0025)) and again PNN classifier has achieved an efficient result for dataset \({S_3}\) on the value of spread = 2). After that, the individual classifiers or learner hypotheses prediction is combined through optimization technique GA. GA-based ensemble model achieved efficient outcome compared to individuals. Besides, accuracy, specificity, sensitivity, and MCC are employed to illustrate the high strength of GAEnsC. The accuracy of GAEnsC using PseDNC is shown in Figs. 2, 3 and 4.

The performance of GAEnsC PseDNC using S1

The performance of GAEnsC PseDNC using S2

The performance of GAEnsC PseDNC using S3
Performance comparison of classifiers using PseTNC feature space
Tables 1, 2 and 3 show the experimental results of individual and ensemble classifiers for the three datasets \({S_1}\), \({S_2}\), and \({S_3}\) using PseTNC feature spaces. SVM has obtained promising results for all the three datasets \({S_1}\), \({S_2}\), and \({S_3}\) on the value of cost function (c = 1.25 and gamma (g = 0.0035)). The success rate of GA-based ensemble model is quite efficient compared to individual classifiers. The accuracy of GAEnsC using PseTNC feature space is illustrated in Figs. 5, 6 and 7.

The performance of GAEnsC PseTNC using S1

The performance of GAEnsC PseTNC using S2

The performance of GAEnsC PseTNC using S3
Performance comparison with other methods
Our proposed predictor is also compared with other existing methods: 3LS (Awazu 2017), iNuc-STNC (Tahir and Hayat 2016), and iNuc-PseKNC (Guo et al. 2014) on the same benchmark datasets. Table 4 demonstrates that our proposed iNuc-ext-PseTNC model has obtained efficient outcomes compared to existing methods. The experimental outcomes proved that the success rates of GA-based ensemble model are more efficient. This success has been ascribed with optimization-based ensemble classification and high variant features of PseTNC.
Discussion
In this article, a predictor “iNuc-ext-PseTNC” is proposed for the identification of nucleosome positioning. The patterns are collected using PseDNC and PseTNC from protein sequences. Contemporary machine learning algorithms are applied to correctly identify nucleosome positioning in genomes. The empirical results explored that the pair of two nucleotides (PseDNC) did not clearly discern the pattern of nucleosome positioning compared to the pair of three nucleotides (PseTNC). It means that the sequence order information has more significance in identifying the motif of nucleosome positioning in genomes. Despite the substantial results of SVM in the combination of PseTNC feature space, the desired outcomes are not achieved. To obtain the desired outcomes, the notion of ensemble classification is introduced. The ensemble process is carried out through bio-inspired evolutionary approach genetic algorithm (GA). After combining the predicted outcome of each learner through GA, consequently, outstanding results have been obtained, which are not only higher than individual learners but also from existing models in the works of literature, so far.
Conclusion
In this study, iNuc-ext-PseTNC predictor is proposed for the prediction of nucleosome positioning in genomes. In this predictor, two discrete feature extraction methods namely: PseDNC and PseTNC are used for the formulation of DNA sequences. The extracted feature spaces are provided to different classifiers such as KNN, SVM, and PNN to comprehend the pattern of nucleosome positioning in genomes. After analyzing the success rates of the individual prediction model, the result of the single classifiers is fused through the GA optimization approach. GA-based ensemble predictor has achieved efficient outcomes than that of the individual classifiers. This significant success has been achieved on account of highly discriminated features of PseTNC and GA-based optimization method. It is discovered that “iNuc-ext-PseTNC” model might be helpful in drug-related applications. Several recent papers demonstrated that (Guo et al. 2014; Liu et al. 2015a, c; Lin et al. 2014; Levitsky 2004; Chen et al. 2015) user-friendly and publicly accessible web servers show future direction for constructing practically more useful models. Therefore, we shall make efforts in our future work to provide a web server for the computational method presented in this paper since doing so will significantly enhance its impact as revealed in two comprehensive review papers (Chou 2015, 2017).
References
Ahmad J, Javed F, Hayat M (2017) Intelligent computational model for classification of sub-Golgi protein using oversampling and fisher feature selection methods. Artif Intell Med 78:14–22
Athey BD, Smith MF, Rankert DA, Williams SP, Langmore JP (1990) The diameters of frozen-hydrated chromatin fibers increase with DNA linker length: evidence in support of variable diameter models for chromatin. J Cell Biol 111:795–806
Awazu A (2017) Prediction of nucleosome positioning by the incorporation of frequencies and distributions of three different nucleotide segment lengths into a general pseudo k-tuple nucleotide composition. Bioinformatics 33:42–48
Berbenetz NM, Nislow C, Brown GW (2010) Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet 6:e1001092
Cao D-S, Xu Q-S, Liang Y-Z (2013) Propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics 29:960–962
Che Y, Ju Y, Xuan P, Long R, Xing F (2016) Identification of multi-functional enzyme with multi-label classifier. PLoS One 11:e0153503
Chen Y-K, Li K-B (2013) Predicting membrane protein types by incorporating protein topology, domains, signal peptides, and physicochemical properties into the general form of Chou’s pseudo amino acid composition. J Theor Biol 318:1–12
Chen W, Feng P-M, Lin H, Chou K-C (2013a) iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res 41(6):e68
Chen W, Feng P, Lin H, Chou K (2013b) iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res gks1450
Chen W, Lei T-Y, Jin D-C, Lin H, Chou K-C (2014) PseKNC: a flexible web server for generating pseudo K-tuple nucleotide composition. Anal Biochem 456:53–60
Chen W, Zhang X, Brooker J, Lin H, Zhang L, Chou K-C (2015) PseKNC-general: a cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 31:119–120
Chen W, Ding H, Feng P, Lin H, Chou K-C (2016) iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget 7:16895
Chen W, Feng P, Yang H, Ding H, Lin H, Chou K-C (2017) iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget 8:4208
Cheng X, Xiao X, Chou K-C (2017a) pLoc-mGneg: predict subcellular localization of Gram-negative bacterial proteins by deep gene ontology learning via general PseAAC. Genomics 110:231–239
Cheng X, Xiao X, Chou K-C (2017b) pLoc-mHum: predict subcellular localization of multi-location human proteins via general PseAAC to winnow out the crucial GO information. Bioinformatics 34:1448–1456
Cheng X, Xiao X, Chou K-C (2017c) pLoc-mPlant: predict subcellular localization of multi-location plant proteins by incorporating the optimal GO information into general PseAAC. Mol Biosyst 13:1722–1727
Cheng X, Xiao X, Chou K-C (2017d) pLoc-mVirus: predict subcellular localization of multi-location virus proteins via incorporating the optimal GO information into general PseAAC. Gene 628:315–321
Cheng X, Xiao X, Chou K-C (2018) pLoc-mEuk: predict subcellular localization of multi-label eukaryotic proteins by extracting the key GO information into general PseAAC. Genomics 110:50–58
Chou KC (2001a) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct Funct Bioinform 43:246–255
Chou K-C (2001b) Prediction of signal peptides using scaled window. Peptides 22:1973–1979
Chou K-C (2005) Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 21:10–19
Chou K-C (2015) Impacts of bioinformatics to medicinal chemistry. Med Chem 11:218–234
Chou K-C (2017) An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr Top Med Chem 17:2337–2358
Chou K-C, Shen H-B (2007a) Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J Proteome Res 6:1728–1734
Chou K-C, Shen H-B (2007b) Recent progress in protein subcellular location prediction. Anal Biochem 370:1–16
Chou K-C, Shen H-B (2007c) Signal-CF: a subsite-coupled and window-fusing approach for predicting signal peptides. Biochem Biophys Res Commun 357:633–640
Dong C, Yuan Y-Z, Zhang F-Z, Hua H-L, Ye Y-N, Labena AA, Lin H, Chen W, Guo F-B (2016) Combining pseudo dinucleotide composition with the Z curve method to improve the accuracy of predicting DNA elements: a case study in recombination spots. Mol BioSyst 12:2893–2900
Eddy SR (1996) Hidden markov models. Curr Opin Struct Biol 6:361–365
Ehsan A, Mahmood K, Khan YD, Khan SA, Chou K-C (2018) A novel modeling in mathematical biology for classification of signal peptides. Sci Rep 8:1039
Feng P, Ding H, Yang H, Chen W, Lin H, Chou K-C (2017) iRNA-PseColl: identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Mol Ther Nucleic Acids 7:155–163
Feng P, Yang H, Ding H, Lin H, Chen W, Chou K-C (2018) iDNA6mA-PseKNC: identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics. https://doi.org/10.1016/j.ygeno.2018.01.005
Field Y, Kaplan N, Fondufe-Mittendorf Y, Moore IK, Sharon E, Lubling Y, Widom J, Segal E (2008) Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol 4:e1000216
Gabdank I, Barash D, Trifonov EN (2010) Single-base resolution nucleosome mapping on DNA sequences. J Biomol Struct Dyn 28:107–121
Goñi JR, Fenollosa C, Pérez A, Torrents D, Orozco M (2008) DNAlive: a tool for the physical analysis of DNA at the genomic scale. Bioinformatics 24:1731–1732
Guo S-H, Deng E-Z, Xu L-Q, Ding H, Lin H, Chen W, Chou K-C (2014) iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 30(11):1522–1529
Hayat M, Khan A (2012) Discriminating outer membrane proteins with fuzzy K-nearest neighbor algorithms based on the general form of Chou’s PseAAC. Protein Pept Lett 19:411–421
Hayat M, Tahir M (2015) PSOFuzzySVM-TMH: identification of transmembrane helix segments using ensemble feature space by incorporated fuzzy support vector machine. Mol BioSyst 11:2255–2262
Ioshikhes I, Bolshoy A, Derenshteyn K, Borodovsky M, Trifonov EN (1996) Nucleosome DNA sequence pattern revealed by multiple alignment of experimentally mapped sequences. J Mol Biol 262:129–139
Isami S, Sakamoto N, Nishimori H, Awazu A (2015) Simple elastic network models for exhaustive analysis of long double-stranded DNA dynamics with sequence geometry dependence. PLoS One 10:e0143760
Jia J, Liu Z, Xiao X, Liu B, Chou K-C (2016) pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol 394:223–230
Kabir M, Hayat M (2016) iRSpot-GAEnsC: identifying recombination spots via ensemble classifier and extending the concept of Chou’s PseAAC to formulate DNA samples. Mol Genet Genom 291:285–296
Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J (2009) The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458:362–366
Levitsky VG (2004) RECON: a program for prediction of nucleosome formation potential. Nucleic Acids Res 32:W346–W349
Li W-C, Deng E-Z, Ding H, Chen W, Lin H (2015) iORI-PseKNC: a predictor for identifying origin of replication with pseudo k-tuple nucleotide composition. Chemom Intell Lab Syst 141:100–106
Li D, Luo L, Zhang W, Liu F, Luo F (2016) A genetic algorithm-based weighted ensemble method for predicting transposon-derived piRNAs. BMC Bioinform 17:329
Lin H, Deng E-Z, Ding H, Chen W, Chou K-C (2014) iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res 42:12961–12972
Liu B, Zhang D, Xu R, Xu J, Wang X, Chen Q, Dong Q, Chou K-C (2014a) Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 30:472–479
Liu B, Xu J, Lan X, Xu R, Zhou J, Wang X, Chou K-C (2014b) iDNA-Prot| dis: identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS One 9:e106691
Liu B, Liu F, Fang L, Wang X, Chou K-C (2015a) repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics 31:1307–1309
Liu Z, Xiao X, Qiu W-R, Chou K-C (2015c) iDNA-Methyl: identifying DNA methylation sites via pseudo trinucleotide composition. Anal Biochem 474:69–77
Liu B, Fang L, Liu F, Wang X, Chen J, Chou K-C (2015d) Identification of real microRNA precursors with a pseudo structure status composition approach. PLoS One 10:e0121501
Liu G-H, Shen H-B, Yu D-J (2016a) Prediction of protein–protein interaction sites with machine-learning-based data-cleaning and post-filtering procedures. J Membr Biol 249:141–153
Liu B, Long R, Chou K-C (2016b) iDHS-EL: identifying DNase I hypersensitive sites by fusing three different modes of pseudo nucleotide composition into an ensemble learning framework. Bioinformatics 32(16):2411–2418
Liu B, Wang S, Long R, Chou K-C (2016c) iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics 33:35–41
Liu B, Yang F, Huang D-S, Chou K-C (2017a) iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 34:33–40
Liu B, Yang F, Chou K-C (2017b) 2L-piRNA: a two-layer ensemble classifier for identifying Piwi-interacting RNAs and their function. Mol Ther Nucleic Acids 7:267–277
Liu B, Wu H, Zhang D, Wang X, Chou K-C (2017c) Pse-Analysis: a python package for DNA/RNA and protein/peptide sequence analysis based on pseudo components and kernel methods. Oncotarget 8:13338
Liu B, Li K, Huang D-S, Chou K-C (2018) iEnhancer-EL: identifying enhancers and their strength with ensemble learning approach. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty458
Luo L, Li D, Zhang W, Tu S, Zhu X, Tian G (2016) Accurate prediction of transposon-derived piRNAs by integrating various sequential and physicochemical features. PLoS One 11:e0153268
Manavalan B, Shin TH, Lee G (2018) PVP-SVM: sequence-based prediction of phage virion proteins using a support vector machine. Front Microbiol 9:476
Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC (2008a) Nucleosome organization in the Drosophila genome. Nature 453:358–362
Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF (2008b) A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18:1073–1083
Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF (2008c) A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res
Nikolaou C, Althammer S, Beato M, Guigó R (2010) Structural constraints revealed in consistent nucleosome positions in the genome of S. cerevisiae. Epigenetics Chromatin 3:20
Peckham HE, Thurman RE, Fu Y, Stamatoyannopoulos JA, Noble WS, Struhl K, Weng Z (2007) Nucleosome positioning signals in genomic DNA. Genome Res 17:1170–1177
Qiu W-R, Xiao X, Chou K-C (2014) iRSpot-TNCPseAAC: identify recombination spots with trinucleotide composition and pseudo amino acid components. Int J Mol Sci 15:1746–1766
Satchwell SC, Drew HR, Travers AA (1986) Sequence periodicities in chicken nucleosome core DNA. J Mol Biol 191:659–675
Schwartz S, Meshorer E, Ast G (2009) Chromatin organization marks exon–intron structure. Nat Struct Mol Biol 16:990
Segal E, Fondufe-Mittendorf Y, Chen L, Thåström A, Field Y, Moore IK, Wang J-PZ, Widom J (2006) A genomic code for nucleosome positioning. Nature 442:772–778
Stolz RC, Bishop TC (2010) ICM Web: the interactive chromatin modeling web server. Nucleic Acids Res 38:W254–W261
Tahir M, Hayat M (2016) iNuc-STNC: a sequence-based predictor for identification of nucleosome positioning in genomes by extending the concept of SAAC and Chou’s PseAAC. Mol BioSyst 12:2587–2593
Thoma F, Koller T, Klug A (1979) Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol 83:403–427
Tian K, Yang X, Kong Q, Yin C, He RL, Yau SS-T (2015) Two dimensional Yau-hausdorff distance with applications on comparison of DNA and protein sequences. PLoS One 10:e0136577
Tolstorukov MY, Choudhary V, Olson WK, Zhurkin VB, Park PJ (2008) nuScore: a web-interface for nucleosome positioning predictions. Bioinformatics 24:1456–1458
Xi L, Fondufe-Mittendorf Y, Xia L, Flatow J, Widom J, Wang J-P (2010) Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinform 11:1
Xiang S, Liu K, Yan Z, Zhang Y, Sun Z (2016) RNAMethPre: a web server for the prediction and query of mRNA m 6 A sites. PLoS One 11:e0162707
Xiao X, Wang P, Lin W-Z, Jia J-H, Chou K-C (2013) iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal Biochem 436:168–177
Xiao X, Cheng X, Su S, Mao Q, Chou K-C (2017) pLoc-mGpos: incorporate key gene ontology information into general PseAAC for predicting subcellular localization of Gram-positive bacterial proteins. Nat Sci 9:330
Xiao X, Cheng X, Chen G, Mao Q, Chou K-C (2018) pLoc-mGpos: predict subcellular localization of Gram-positive bacterial proteins by quasi-balancing training dataset and PseAAC. Genomics. https://doi.org/10.1016/j.ygeno.2018.05.017
Xu Y, Shao X-J, Wu L-Y, Deng N-Y, Chou K-C (2013a) iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ 1:e171
Xu Y, Ding J, Wu L-Y, Chou K-C (2013b) iSNO-PseAAC: predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One 8:e55844
Xu Y, Wen X, Wen L-S, Wu L-Y, Deng N-Y, Chou K-C (2014) iNitro-Tyr: prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS One 9:e105018
Yasuda T, Sugasawa K, Shimizu Y, Iwai S, Shiomi T, Hanaoka F (2005) Nucleosomal structure of undamaged DNA regions suppresses the non-specific DNA binding of the XPC complex. DNA Repair 4:389–395
YongE F, GaoShan K (2015) Identify beta-hairpin motifs with quadratic discriminant algorithm based on the chemical shifts. PLoS One 10:e0139280
Yuan G-C, Liu JS (2008) Genomic sequence is highly predictive of local nucleosome depletion. PLoS Comput Biol 4:e13
Yuan G-C, Liu Y-J, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ (2005) Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309:626–630
Zhang W, Niu Y, Xiong Y, Zhao M, Yu R, Liu J (2012) Computational prediction of conformational B-cell epitopes from antigen primary structures by ensemble learning. PLoS One 7:e43575
Zhang W, Liu F, Luo L, Zhang J (2015a) Predicting drug side effects by multi-label learning and ensemble learning. BMC Bioinform 16:365
Zhang W, Niu Y, Zou H, Luo L, Liu Q, Wu W (2015b) Accurate prediction of immunogenic T-cell epitopes from epitope sequences using the genetic algorithm-based ensemble learning. PLoS one 10:e0128194
Zhang W, Zou H, Luo L, Liu Q, Wu W, Xiao W (2016a) Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing 173:979–987
Zhang C-J, Tang H, Li W-C, Lin H, Chen W, Chou K-C (2016b) iOri-Human: identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 7:69783
Zhang W, Shi J, Tang G, Wu W, Yue X, Li D (2017) Predicting small RNAs in bacteria via sequence learning ensemble method. In: Bioinformatics and biomedicine (BIBM), 2017 IEEE international conference on, IEEE, pp 643–647
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Communicated by S. Hohmann.
Rights and permissions
About this article

Cite this article
Tahir, M., Hayat, M. & Khan, S.A. iNuc-ext-PseTNC: an efficient ensemble model for identification of nucleosome positioning by extending the concept of Chou’s PseAAC to pseudo-tri-nucleotide composition. Mol Genet Genomics 294, 199–210 (2019). https://doi.org/10.1007/s00438-018-1498-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-018-1498-2