
Abstract
Long non-coding RNAs (lncRNAs) play a broad spectrum of distinctive regulatory roles through interactions with proteins. However, only a few plant lncRNAs have been experimentally characterized. We propose GPLPI, a graph representation learning method, to predict plant lncRNA-protein interaction (LPI) from sequence and structural information. GPLPI employs a generative model using long short-term memory (LSTM) with graph attention. Evolutionary features are extracted using frequency chaos game representation (FCGR). Manifold regularization and l2-norm are adopted to obtain discriminant feature representations and mitigate overfitting. The model captures locality preserving and reconstruction constraints that lead to better generalization ability. Finally, potential interactions between lncRNAs and proteins are predicted by integrating catboost and regularized Logistic regression based on L-BFGS optimization algorithm. The method is trained and tested on Arabidopsis thaliana and Zea mays datasets. GPLPI achieves accuracies of 85.76% and 91.97% respectively. The results show that our method consistently outperforms other state-of-the-art methods.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The recent advancement in high-throughput sequencing technology has led to the exponential growth in the repertoire of the genome sequence. Non-coding RNAs (ncRNAs), the largest portion of the eukaryotic genome, are classified based on their genomic origin or mechanism of action. In particular, long non-coding RNAs (lncRNAs) are more enriched in the nucleus and function in various biological processes such as cell growth, differentiation and chromatin modification (Quinn and Chang 2016). Based on the genomic origin lncRNAs can be categorized as intergenic, intronic, sense, and antisense (Qiu et al. 2019). As a key mediator of cellular functions, lncRNAs perform essential regulatory roles in the plant cell nucleus by interacting with proteins. For instance, cold-induced Arabidopsis lncRNAs, COLDAIR and COOLAIR, are transcripts transcribed by Flowering Locus C (FLC), antisense that is regulated by the cis (Yu et al. 2019). So far, many plant lncRNAs have been identified and implicated in flowering time control, biotic and abiotic stress responses, and reproduction. Moreover, emerging evidence shows that plant protection against pathogen attacks have correlation with lncRNA-dependent immune systems (Zaynab et al. 2018). There are two modes of decoding interactions between RNAs and proteins, by recognition of RNA-binding proteins (RBP) direct contact with RNA bases or indirectly by examining RNA structure and thermodynamic aspects (Lam et al. 2019). Computational methods based on quantitative or machine learning models complement experimental methods in uncovering interaction between proteins and RNAs (Cirillo et al. 2017).
New lncRNA-disease association (LDA) and lncRNA-protein interaction (LPI) prediction have received considerable attention. In medicine, uncovering association between lncRNAs and diseases is important for promoting diagnosis and treatment of complex diseases. Studies have found that similar lncRNAs interact with similar diseases (Yu et al. 2017). Based on this theory, several computational methods for LDA have been proposed including LDAP (Lan et al. 2016), BRWLDA (Yu et al. 2017), MFLDA (Fu et al. 2017), and WMFLDA (Yu et al. 2018). Predicting lncRNA-protein interaction is essential for studying molecular mechanisms involving these lncRNAs, understanding the pathogenesis of diseases and deciphering their functions. High-throughput technologies for detecting binding of proteins to RNA include cross-linking immunoprecipitation (CLIP), enhanced CLIP (eCLIP), and in-cell protein-RNA interaction (incPRINT) (Graindorge et al. 2019). Although these wet-lab experimental methods are valuable, they are time-consuming and expensive. Recently, a surge of computational prediction methods for RNA–protein interaction have been proposed. Significant progress has been made via pattern-based, feature-based, and kernel-based computational methods. A web server for predicting mutual binding sites in RNA and protein at the nucleotide and residue level called PRIdictor (Protein-RNA Interaction predictor) was developed (Tuvshinjargal et al. 2016). In 2016, a computational method called RBPPred was proposed (Zhang and Liu 2016). They combined hydrophobicity, polarity, normalized van der Waals volume, polarizability, secondary structure, solvent accessibility, side-chain’s charge and polarity, PSSM profile features and used SVM classifier to distinguish between binding and non-RNA protein binding sites. Recently, a sequence-based generative method for constructing protein binding motifs was proposed (Park and Han 2020). For lncRNA-protein specific interaction prediction, data repositories, models and algorithms have been summarized (Peng et al. 2020). SFPEL-LPI, a sequence-based feature projection ensemble learning framework was proposed to predict LPI(Zhang et al. 2018). A kernel ridge regression model based on fast kernel learning was developed for LPI prediction (Shen et al. 2018). Network-based methods proposed to predict LPI based on the integration of heterogeneous networks include LPIHN, RWR and LPI-NRLMF (Li et al. 2015; Ge et al. 2016; Liu et al. 2017).
The key factors that influence the prediction of interaction between genome molecules are the choice of feature extraction method and classification algorithm (Ru et al. 2019). A diverse pool of studies has explored feature extraction and feature selection techniques to study the interaction prediction problem. Feature extraction methods transform raw data into attributes suitable for processing by machine learning algorithms. The feature extraction methods are similarity-based, probabilistic, and likelihood-based methods (Mutlu and Oghaz 2019). The most commonly used matrix factorization methods for feature extraction include principal component analysis, tensor decomposition analysis, and factor analysis (Li et al. 2018c). On the other hand, feature selection is a preprocessing procedure considered a prerequisite for model building. It helps in reducing overfitting, identifying correlation among features to reduce redundancy, increase class relevance in feature subset, and ultimately improve the performance of the learning algorithm. For example, locality preserving projections (LPP) and locality-constrained linear coding (LLC) applies the linearization approach to map between input space and the reduced space (Yu et al. 2016; Xie et al. 2019). Recently, graph feature learning has received attention in the bioinformatics research community (Cho et al. 2016; Yue et al. 2019). It represents learning by encoding to preserve relational information from the graph. The chaos game representation (CGR) is a graphical representation of a sequence derived from a D/RNA or protein sequence. Each point of the plot corresponds to one base of the sequence. CGR explores the evolutionary relationships of genomic sequences based on amino acid or nucleotide properties(Bhoumik and Hughes 2018). Unlike feature selection and dimensionality reduction techniques that alter original representation, feature extraction and aggregation techniques such as serial and parallel feature fusion, combine input features, and select a subset (Saeys et al. 2007). The aim is to obtain discriminative features and reduce computational complexity.
Deep learning (DL) models have gained popularity in myriad domains including bioinformatics, computer vision, and natural language processing. Particularly in bioinformatics, DL provides biological insights due to its ability to capture hidden sequence signals (Li et al. 2019a; Camargo et al. 2020). To date, scalable and cost-efficient computational approaches have been developed to complement and enhance experimental results. For instance, DeepBind is an exemplary DL based method developed by integrating sequence and structure for RBP to infer specificity patterns (Alipanahi et al. 2015). (Li et al. 2019b) proposed RDense, a hybrid of bidirectional LSTM and CNN, to predict protein-RNA interaction. Other protein-RNA binding prediction models based on autoencoder, recurrent neural network, and convolutional networks include Thermonet (Su et al. 2019), IPMiner (Pan et al. 2016), DLPRB (Ben-Bassat et al. 2018), and cDeepBind (Gandhi et al. 2018). Graph representation learning and attention mechanism have been proven effective in enhancing the performance of DL models. The most successful DL methods in graph representation learning are graph convolutional networks(Kipf and Welling 2016) and graph attention networks (Veličković et al. 2017). The key advantage is that graph embedding methods such as random walk captures explicit relations in structured data (Li et al. 2018b; Salehi and Davulcu 2019). Attention mechanism computes representations by dealing with variable sized inputs, focusing on the most relevant parts of the input to make decisions. Moreover, DL models can be combined with other models such as Conditional random field (CRF) and quantization techniques. CRF imposes constraints that enable the model to regenerate the input features given the latent labels accurately. CRFs take into account inter-relation information between labels of neighboring residues (Liu et al. 2018). Quantization is the process of minimizing the number of bits that represent a number. In DL, quantization is achieved by measuring the dynamic range of activations and reducing the size of the floating point for weights (Rastegari et al. 2016). Regularization and activation techniques are implemented in DL models during training to overcome data size limitations inherent to the traditional biological datasets and to improve performance. Dropout and data augmentation are the widely used regularization techniques, while rectifier linear unit (ReLU) and sigmoid are used as activation functions. The main impediments in the ncRNA-protein interaction prediction are in the tradeoff between the feature information and the complexity of the approach used for analysis. Notably, continuous efforts have been dedicated to high-quality computational techniques to study the interactions between RNAs and proteins. Albeit the progress based on the success of DL models in this research area, lncRNA-protein interaction in plants has received little attention.
The development of a computational method for lncRNA-protein interaction prediction is imperative to avert the impending shortage of plant lncRNA functions. This paper introduces GPLPI, a graph-based neural network framework. Frequency chaos game representation (FCGR) is used to extract evolutionary sequence pattern information of the lncRNAs. To fully exploit autoencoder for enhanced feature learning, graph attention is constructed similar to the study by Taheri et al. (2019). Contrary to the standard attention mechanism that guides the model to derive contextual information, graph attention uses attention parameters to guide the learning algorithm to focus on the part of data that optimizes the objective function. The graph attention also improves interpretability by understanding how to assign attention by considering the volume of available data and the structure. Inspired by Schulz et al. (2020), we implement limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) optimization algorithm on logistic regression classifier. Locality-preserving projection is adopted to improve efficiency and extract the most representative information. Our contributions are twofold: (1) multiscale feature generation provide diverse information and locally linear embedding reduce feature redundancy, (2) graph attention mechanism learns arbitrary context distributions for better interpretability.
Materials and methods
Overview of GPLPI
The potential lncRNA-protein interactions are computed using a regularized graph attention neural network model. Transformation methods are used to encode lncRNA sequences from nucleotides {A, T, C, G} and protein sequences from 20 types of amino acids {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y} into numeric vectors. Besides, we include structural features from predicted secondary structures from lncRNA and protein sequences. The proposed method assumes that functionally similar proteins interact with similar lncRNAs. Based on this concept, the target lncRNA-protein partners are predicted. The feature vector of m lncRNAs and n proteins is denoted as L = {l1, l2, …, li, …, lm} and P = {p1, p2, …, pj, …, pn}. The label of interaction between lncRNA li and protein pj denoted as y(li, pj) is assigned 1 for interaction and 0 for non-interaction. Each lncRNA-protein sample is described as a 522-dimensional vector as follows:
where L(lm) is a vector of 175-dimensional feature vector and P(pn) is a 347-dimensional feature vector. The feature vector of lncRNA (L(lm)) is composed of 64-dimension from FCGR, 106-dimension from k-mer (64 from trinucleotide, 32 from gapped k-mer and 10 from reverse complement) and 5 structural features.
The feature vector of protein (P(pn)) is composed of 320 binary profile features from protein sequences and 27 structural features represented as follows:
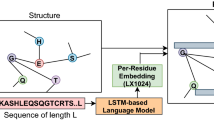
The main procedure followed by the proposed method is summarized as follows: first, selecting positive and negative examples, then, extracting complex features and finally building the model to predict lncRNA-protein interaction pairs effectively. FCGR, k-mer, and RNAFold (for predicting structural features) are used to extract features from lncRNAs. Binary Profile feature (BPF) and SSPro (for predicting secondary structure) are used to extract features from protein sequences. Graph attention LSTM-autoencoder is used to learn high-level abstract representations. In the encoder, LSTM is used to read the input and encode it to a fixed dimensional vector. Another LSTM decodes the output of the vector. Multiple classifiers including random forest, catboost, logistic regression, and extreme gradient boosting, are tested to find the most accurate. To exploit the strength of multiple classifiers, the Logistic regression with Broyden-Fletcher-Goldfarb-Shanno (L-BFGS) algorithm and catboost are combined for prediction. The predictions of the individual models are combined by majority voting, a non-trainable method to output lncRNA-protein interaction matrix Mij. The proposed method is shown in Fig. 1.

Flowchart of the proposed method. Sequence and structural features are extracted from lncRNA and protein sequences and fed into the prediction model to output lncRNA-protein interactions
Data representation
PlncRNADB database (available at https://bis.zju.edu.cn/PlncRNADB is the data resource of the known plant lncRNA-protein interactions data used for prediction in this study. The data includes 22,133 interactions between 1107 lncRNAs and 190 proteins for Zea mays dataset, 948 interactions between 390 lncRNAs and 163 proteins for Arabidopsis thaliana. The non-interactive pairs, 22,133 for Zea mays, and 948 for Arabidopsis thaliana were generated through randomly pairing proteins with lncRNAs and further removing the existing positive pairs (Muppirala et al. 2011). Finally, the Zea mays dataset contains 44,266 and Arabidopsis thaliana contains 1896 lncRNA-protein pairs as shown in Table 1. The data are split into 80% for training and 20% for testing.
The key performance booster for deep learning models is the choice of features. Our deep learning approach utilizes sequence and secondary structure data as inputs. Salient features for lncRNA-protein interaction prediction are obtained using three feature extraction techniques; k-mer, frequency chaos game representation (FCGR), and binary profile features. CGR is an iterative mapping technique proposed by Jeffery for the alignment-free representation of RNA sequences (Jeffrey 1990). It extracts evolutionary information by counting the k-mers i.e. n-tuple or n-gram of nucleic acid or amino acid sequences. k-mer strings are used to identify regions of interest. The k-mer tables are referred to as the frequency chaos game representation (FCGR) (Lichtblau 2019). Unlike other sequence and structure encoding methods such as Fourier Transformation, CGR generates fractals for visual encoding. The four RNA nucleotides are represented by rectangular coordinates (A:-1,1, C:-1,-1, G:1,1 and U:1,-1). The CGR plane is partitioned into a probability matrix of 8 × 8 grids from which the average coordinates of each grid are calculated. The matrix is reshaped to a 64- dimensional feature vector.
The k-mer frequencies model is also used to extract sequence features. For the trinucleotide, given a frequency interval fx where x is an interval, a frequency vector of intervals for a sequence with length L is defined as F = {f1, f2, …, fL-k+1}. The gapped k-mer and reverse complement features are described in Table 2. For the protein sequence, the binary profile feature extraction (BPF) method is used. A binary profile of 20 × b dimension composed of a sequence of length b generated a 320-dimension feature vector, where b = 16. The protein and lncRNA secondary structures are predicted using SSpro (Magnan and Baldi 2014) and RNAfold (Lorenz et al. 2011), respectively. For the lncRNA secondary structure, we extract pairwise probability features.
Graph attention-based autoencoder
Deep neural networks incrementally learn high-level abstract features along with multiple layers. In this study, the LSTM autoencoder with graph attention is implemented (Fig. 2). By stacking layers, the network traverses the kernels length to learn more local spatial information. However, the network complexity increases due to many parameters generated during training, causing the model to overfit and have poor generalization ability. This bottleneck is mitigated by imposing constraints on the network to remove redundant connections and unnecessary neurons through regularization. l2-norm and manifold regularization are implemented to promote sparsity for the neural network model. The l2-norm constraint is a weight-decay regularization imposed on the model parameters. Manifold regularization is imposed on the output of the neural network model through locality preserving constraints. Other regularization mechanisms implemented include dropout and early stopping. The LSTM architecture consists of recurrently connected neurons called the memory cells. A memory block is composed of input, output and forget gate multiplicative units (Zheng et al. 2017). In the LSTM encoder, input from the embedding layer is fed into stacked layers to generate representations that are forwarded to the graph-based attention layer. This representation is then decoded through an LSTM layer to reconstruct the input sequence. A sequence S of length l can be represented as S = {s1, st, st+1, … sl}, where st is the tth nucleotide. The memory block computes a hidden vector ht at a time step t of the input st as follows:
where c is the cell memory. The encoder in our model is multilayered to increase learning capability. The number of layers of the decoder is similar to those of the encoder. The graph attentional layer explicitly assigns different importance to nodes within a neighborhood, thus leveraging self-attentional layers. It integrates graph structure and node-level features by weighting neighbor features with normalization. The setup of the graph attention implemented in this study follows the work of Velickovic et al. (2017). Let a sequence s ϵ S that has been passed through the LSTM layer be the information from neighbors of nodes in the sequence. An attention module A is used to gather local information from the neighbors of s. The graph attention layer represented by Eq. 1 is used to produce the hidden representations.
where x is a d is the dimensional feature vector, Wq, Wk and Wv are the attention weight matrices. Attention weight measures the association of a relation kn to the input qn and output vn. During training, the parameters of the neurons are updated using loss calculated from the difference between the target sequence and the predicted sequence. Given x input and \(\hat{x}\) expected output, the objective of the training is to minimize reconstruction error (L) defined as:

Graph attention neural network architecture. Wi, bi represent LSTM encoder weight and bias parameters; Wa, ba for the attention layer and Wo, bo for the LSTM decoder
The hinge loss is used to minimize the reconstruction error. The loss function penalizes incorrect and less confident predictions, it is defined as follows
where yi are the labels, xi is the input feature vector, hƟ(xi) is the prediction.
Hybrid classifier construction
The intermediate representation of data is done through feature extraction methods to enable classification algorithms to predict outcomes. A feature vector obtained from feature integration provides complementary information that increases accuracy and robustness. Feature fusion mapping is achieved by mathematically combining FCGR, k-mer, binary profile, and structural features. The locally linear embedding (LLE) is adopted to reduce the fusion mapping dimension. The concept of LLE, a linear manifold learning algorithm, is to extract relevant correlation in the feature space, retain variability, and disregard irrelevant features. It extracts intrinsic structure, preserves the neighborhood correlation, and symbolizes a linear estimation of the nonlinear Laplacian eigenmaps (Li et al. 2018a). Let a matrix X of n is the dimension vectors be denoted as X = [x1, x2, …, xn]. Each training sample is denoted as xi where i = 1,2,…,n, seek k nearest neighbors and represent them as a matrix j of n × k dimensions. The selected features enhance classification. Two classifiers, logistic regression (LR) and catboost are incorporated. For LR algorithm, its implementation was depended on L-BFGS optimization algorithm used as the ‘solver’ parameter and other user-defined parameters such as multiclass. For catboost, a gradient boosting algorithm, the implementation was based on parameters such as iterations, depth, learning rate, and loss function. The model’s iteration parameter is used for iterative training of n learners to reduce prediction error. The output from the two classifiers are combined by majority voting. The implementation steps followed by the proposed model are summarized in Algorithm 1.

Implementation and parameter settings
In this work, a deep learning method termed GPLPI is proposed and use Zea mays and Arabidopsis thaliana datasets for evaluation. Sequence and structural features are combined for the prediction task. The high-level abstract features are extracted using DL model and fed as the input for the classifier. The tensorflow library is used for implementation. For the architecture, LSTM is selected for the encoder and decoder. Choosing parameters that seek to find global optima is a significant part of the model training process. The parameters and hyperparameters for our deep learning model are selected after an extensive search for optimal combinations of parameters such as the activation function, the number of hidden layers, and optimizer. In this experiment, ReLU is used as the activation function, Adam as the optimizer and hinge as the cost function. The ReLU activation function maintains a stable convergence speed of the model. Optimization aims at finding parameters for robust training and fast convergence. To minimize loss error, Adam optimizer is selected because it has an improved ability to handle noise by combining root mean square propagation (RMSProp) optimization as a gradient descent and adaptive gradient (Adagrad) algorithms (Wang et al. 2019a). The model learns the weight and bias parameters during training. The list of hyperparameters representing the external configurations, such as the number of hidden layers and activation function for this prediction task are reported in Table 3. The scikit-learn package was used to implement the classification algorithms.
Evaluation
The five-fold cross-validation is used to assess the performance of the proposed method in comparison to other methods. The dataset is arbitrarily divided into five equal subsets, four folds for training and one fold as the test set. We used accuracy (ACC), precision (PRE), recall (REC)/sensitivity (SEN), specificity (SPE) and Mathews correlation coefficient (MCC). The evaluation measures are defined as follows:
where TP, FP, TN and FN represent true positive, false positive, true negative and false negative respectively. In addition, area under the curve (AUC), and area under precision/recall curve (AUPRC) evaluation metrics are also used to show the general performance of the model.
Results
Performance evaluation of GPLPI
The performance of GPLPI is evaluated using two datasets. Figure 3 shows the overall five-fold cross-validation results of GPLPI on the two datasets, Arabidopsis thaliana and Zea mays. GPLPI performed better on Zea mays dataset because the size of the data was more than that of Arabidopsis thaliana. The proposed method obtained 85.76% accuracy, 88.42% precision, 82.41% sensitivity, 88.97% specificity, 71.71% MCC, 91.13% AUC, and 93.41% AUPRC on Arabidopsis thaliana dataset. The method obtained 91.97% accuracy, 92.20% precision, 91.70% sensitivity, 92.24% specificity, 83.94% MCC, 97.76% AUC, and 97.94% AUPRC on Zea mays dataset. The proposed method obtained accuracy with a standard deviation of 2.05 and 0.44, for Arabidopsis thaliana and Zea mays dataset respectively. From the results, the proposed method efficiently extract meaningful information for prediction. This information when used for classification produced good results.

Performance of the proposed method on Zea mays and Arabidopsis thaliana
Ablation study
The proposed model extracts effective sequence and structural features, which are fed as input for the neural network algorithm. To verify the contribution of the feature extraction methods, an ablation study is performed by testing different settings. The baseline classifiers of the GPLPI model are tested on different sets of features. Our aim is to study how the graph-based feature extraction method, frequency chaos game representation (FCGR), k-mer, structural features, and their integration contribute to model effectiveness. Table 4 shows the results of the different feature groups. From the table, the higher value represents a better performance for the evaluation metrics.
From the results in Table 3, the proposed method yields the performance of accuracy (ACC) 91.97%, when structural features are included which is slightly lower than when FCGR and k-mer are used. When FCGR, k-mer, and Secondary Structural features (SS) are combined, the performance improved with an approximately 17% in terms of accuracy and approximately 16% in terms of AUC than when only FCGR is used. There was a slight increment in performance when structural features are added to FCGR and k-mer with approximately 0.8% increase in specificity and MCC while AUC increases by approximately 0.03%. The performance improved in terms of efficiency when the manifold regularization is employed. Overall, the proposed method’s graph attention, loss function, and regularization effectively improve model performance.
Performance comparison of different classifiers
Six classic machine-learning algorithms are tested including logistic regression (LR), catboost, random forest (RF), extreme gradient boosting (XGB), and decision tree (DT). The models were trained on Zea mays dataset. LR and XGB models’ output was observed to be the best performing model in terms of AUC. LR was combined with catboost to construct the proposed model. GPLPI was significantly better than the other methods in all the metrics, as shown in Table 5. The values in the table represent mean (%) and standard deviation obtained and the value in bold denotes the best one yielded on the dataset. The model yielded an average accuracy of approximately 4% better than the other methods. Figure 4 presents the five-fold cross-validation results of GPLPI, LR, catboost, RF, XGB, and DT in the form of boxplots for Zea mays dataset. The better performance is attributed to the ensemble of diverse base classifiers. When the difference between the performances of the individual classifiers is big, majority voting integration is effective. When the difference between the classifiers is small, the classification error degrades, thus, increasing the performance. This indicates that the correlation among classifiers increases the overall performance.

Comparison of the performance in terms of accuracy between GPLPI and LR, catboost, RF, XGB, and DT on Zea mays dataset
Performance comparison of different deep learning methods
In the past decade, many studies have explored the association between RNAs and proteins. In this paper, the proposed model is compared with standard deep learning models to verify its advantage. GPLPI is applied to known plant lncRNA-protein interaction data together with three other methods RPISeq-RF (Muppirala et al. 2011), XRPI (Jain et al. 2018), and RPI-SE (Yi et al. 2020). The three methods are selected for comparison because they can predict non-coding RNA–protein interaction. Five-fold cross-validation was adopted to evaluate their performances. The performances were evaluated by the metrics in terms of the mean (%) and standard deviation as presented in Table 6. In general, the higher values represents a better performance for the evaluation metrics. The ROC curves representing the tradeoffs between true positives and false positives and their associated AUCs of GPLPI, RPISeq-RF, XRPI, and RPI-SE, respectively, are plotted in Fig. 5. For the Arabidopsis thaliana dataset, all the methods were at or above 73% in terms of sensitivity, AUC, and AUPRC. However, accuracy, precision, specificity, and MCC the values range from 26 to 88%. For the Zea mays dataset, all the methods were at or above 80% in terms of accuracy, sensitivity, AUC, and AUPRC. However, precision, specificity, and MCC the values range from 62 to 97%. Notably, our method outperforms other methods. In terms of accuracy and specificity, approximately 2% and 3%, the increase is obtained respectively. As for MCC, a significant performance improvement of approximately 6% enhancement is noted. The results indicate that GPLPI performs significantly better than the other methods in lncRNA-protein interaction prediction. The performance of GPLPI is more outstanding because of the effectiveness of the sequence and structural feature extraction methods that obtained essential information.

ROC curves of the comparison between the performances of the four methods for aArabidopsis thaliana and bZea mays
Discussion
Identification of lncRNAs in the plant genome has received more research interest than functions and mechanisms. Several machine-learning algorithms for plant lncRNA identification have been proposed (Singh et al. 2017; Negri et al. 2018; Zhao et al. 2018). The available databases and methods for lnRNA-protein interaction have a preference for collecting animal data, and thus, insufficient lncRNA-protein interaction is a major problem in plants. Therefore, it is prudent to develop a computational method for the accurate identification of plant lncRNA-protein interaction. Graph embedding can aid in predicting lncRNA-protein associations, extract biological information and enhance the quality of high-throughput sequencing data analysis. However, most existing methods do not involve topological information. Therefore, the relationship between lncRNAs and proteins is not directly considered. This paper proposed a predictive model for inferring plant lncRNA-protein interaction using a recurrent autoencoder algorithm with graph attention in combination with FCGR, k-mer and BPF sequence coding methods. The efficiency of the proposed GPLPI in addressing lncRNA-protein interaction problem in plant species is supported by the experimental results presented in the results section.
Similar to network-based methods, the proposed graph-based deep learning framework works based on the assumption that potential interactions exist among lncRNAs sharing common interactive partners. However, unlike network-based method that explores neighborhood topology structure, the proposed method is not limited to this exploration process. The feature extraction methods extract evolutionary and structural information for better interaction recognition. The features distinguish the different genome molecules and make different contributions for plant lncRNA-protein interaction. This work demonstrated that feature integration and ensemble learning help provide more accurate measure of lncRNA-protein association. Besides, directly learning the mapping from lncRNA sequence to a 2D space without imposing restriction on the nucleotide sequence length is an appealing attribute of FCGR. Excellent experimental results indicate that GPLPI performed well in predicting association between lncRNAs and RNA-binding proteins with the support of graph attention based algorithm and sequence-structural information. The success of GPLPI may be due to its generalization ability to learn hidden interaction features.
The performance of GPLPI relies on the multiscale feature aggregation, feature reduction through locally linear embedding (LLE) and fusion of multiple ensemble models. We obtained diverse information and achieved best results by combining classification algorithms with higher accuracies. As proven by the experimental results, the FCGR, a graph-based feature-mapping method, enables GPLPI to achieve good performance. The graph attention mechanism learns arbitrary context distributions for better optimization of the training loss and interpretability. These attributes distinguish the proposed method from existing methods. Despite the good performance, GPLPI can be improved in several ways. For instance, combining several deep learning models such as graph convolution neural network, dilated convolution and normalization to boost performance further. Moreover, integrating other models such as matrix factorization can also be advantageous. This can be backed by evidence from closely related studies conducted in pioneering research (Gandhi et al. 2018; Li et al. 2019b; Wang et al. 2019b; Xuan et al. 2019). In this work, we only employ lncRNA-protein interaction data, integrating more biological data such as protein–protein interaction may also lead to better performance. In conclusion, graph attention is proposed to learn context distribution and enhance the discriminative ability. In the feature-learning phase, manifold regularization yields feature learning efficiency. Moreover, l2-norm and the loss function mitigate overfitting. Two classifiers are integrated to demonstrate the effectiveness of the proposed method. Experiments on Zea mays and Arabidopsis thaliana datasets indicate that GPLPI performed well in lncRNA-protein interaction prediction compared to the state-of-the-art methods. GPLPI is applicable to other plant species and is useful in functional analysis.
Availability of data and material
The source code associated with this study is available at https://github.com/Mjwl/GPLPI..
References
Alipanahi B, Delong A, Weirauch MT, Frey BJ (2015) Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat Biotechnol 33:831–838
Ben-Bassat I, Chor B, Orenstein Y (2018) A deep neural network approach for learning intrinsic protein-RNA binding preferences. Bioinformatics 34:i638–i646
Bhoumik P, Hughes AL (2018) Chaos game representation: an alignment-free technique for exploring evolutionary relationships of protein sequences. BioRxiv:276915
Camargo AP, Sourkov V, Pereira Gonçalo AG, Carazzolle Marcelo F (2020) RNAsamba: neural network-based assessment of the protein-coding potential of RNA sequences. NAR Genom Bioinform 2:Iqz024
Chen Z, Zhao P, Li F, Marquez-Lago TT, Leier A, Revote J, Zhu Y, Powell DR, Akutsu T, Webb GI, Chou KC, Smith AI, Daly RJ, Li J, Song J (2019) iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA. Brief Bioinform, RNA and protein sequence data. https://doi.org/10.1093/bib/bbz041
Cho H, Berger B, Peng J (2016) Compact integration of multi-network topology for functional analysis of genes. Cell Syst 3:540–548.e545
Cirillo D, Blanco M, Armaos A, Buness A, Avner P, Guttman M, Cerase A, Tartaglia GG (2017) Quantitative predictions of protein interactions with long noncoding RNAs. Nat Methods 14:5–6
Fu G, Wang J, Domeniconi C, Yu G (2017) Matrix factorization-based data fusion for the prediction of lncRNA–disease associations. Bioinformatics 34:1529–1537
Gandhi S, Lee LJ, Delong A, Duvenaud D, Frey B (2018) cDeepbind: a context sensitive deep learning model of RNA-protein binding. bioRxiv:345140
Ge M, Li A, Wang M (2016) A bipartite network-based method for prediction of long non-coding RNA–protein interactions. Genom Proteom Bioinform 14:62–71
Graindorge A, Pinheiro I, Nawrocka A, Mallory AC, Tsvetkov P, Gil N, Carolis C, Buchholz F, Ulitsky I, Heard E, Taipale M, Shkumatava A (2019) In-cell identification and measurement of RNA-protein interactions. Nat Commun 10:5317
Jain DS, Gupte SR, Aduri R (2018) A data driven model for predicting RNA-protein interactions based on gradient boosting machine. Sci Rep 8:9552
Jeffrey HJ (1990) Chaos game representation of gene structure. Nucleic Acids Res 18:2163–2170
Kipf TN, Welling M (2016) Semi-supervised classification with graph convolutional networks.arXiv:1609.02907 arXiv:1609.02907
Lam JH, Li Y, Zhu L, Umarov R, Jiang H, Héliou A, Sheong FK, Liu T, Long Y, Li Y, Fang L, Altman RB, Chen W, Huang X, Gao X (2019) A deep learning framework to predict binding preference of RNA constituents on protein surface. Nat Commun 10:4941
Lan W, Li M, Zhao K, Liu J, Wu F-X, Pan Y, Wang J (2016) LDAP: a web server for lncRNA-disease association prediction. Bioinformatics 33:458–460
Li A, Ge M, Zhang Y, Peng C, Wang M (2015) Predicting long noncoding RNA and protein interactions using heterogeneous network model. BioMed Res Int 2015:671950
Li HG, Song RQ, Liu JW (2018a) Low-dimensional feature fusion strategy for overlapping neuron spike sorting. Neurocomputing 281:152–159
Li J, Chen L, Wang S, Zhang Y, Kong X, Huang T, Cai Y-D (2018b) A computational method using the random walk with restart algorithm for identifying novel epigenetic factors. Mol Genet Genom 293:293–301
Li Y, Wu F-X, Ngom A (2018c) A review on machine learning principles for multi-view biological data integration. Brief Bioinform 19:325–340
Li Y, Huang C, Ding L, Li Z, Pan Y, Gao X (2019a) Deep learning in bioinformatics: introduction, application, and perspective in the big data era. Methods 166:4–21
Li Z, Zhu J, Xu X, Yao Y (2019b) RDense: a protein-RNA binding prediction model based on bidirectional recurrent neural network and densely connected convolutional networks. IEEE Access 8:14588–14605
Lichtblau D (2019) Alignment-free genomic sequence comparison using FCGR and signal processing. BMC Bioinform 20:742
Liu H, Ren G, Hu H, Zhang L, Ai H, Zhang W, Zhao Q (2017) LPI-NRLMF: lncRNA-protein interaction prediction by neighborhood regularized logistic matrix factorization. Oncotarget 8:103975
Liu Y, Wang X, Liu B (2018) IDP-CRF: intrinsically disordered protein/region identification based on conditional random fields. Int J Mol Sci 19:2483
Lorenz R, Bernhart S, Zu Siederdissen CH, Tafer H, Flamm C, Stadler P (2011) ViennaRNA Package 2.0. Algorithm Mol Biol 6:26
Magnan CN, Baldi P (2014) SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 30:2592–2597
Muppirala UK, Honavar VG, Dobbs D (2011) Predicting RNA-protein interactions using only sequence information. BMC Bioinform 12:489
Mutlu EC, Oghaz TA (2019) Review on graph feature learning and feature extraction techniques for link prediction. arXiv:1901.03425
Negri TdC, Alves WAL, Bugatti PH, Saito PTM, Domingues DS, Paschoal AR (2018) Pattern recognition analysis on long noncoding RNAs: a tool for prediction in plants. Brief Bioinform 20:682–689
Pan X, Fan Y-X, Yan J, Shen H-B (2016) IPMiner: hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genom 17:582
Park B, Han K (2020) Discovering protein-binding RNA motifs with a generative model of RNA sequences. Comput Biol Chem 84:107171
Peng L, Liu F, Yang J, Liu X, Meng Y, Deng X, Peng C, Tian G, Zhou L (2020) Probing lncRNA–protein interactions: data repositories, models, and algorithms. Front Genet 10:1346
Qiu C-W, Zhao J, Chen Q, Wu F (2019) Genome-wide characterization of drought stress responsive long non-coding RNAs in Tibetan wild barley. Environ Exp Bot 164:124–134
Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17:47–62
Rastegari M, Ordonez V, Redmon J, Farhadi A (2016) XNOR-Net: ImageNet classification using binary convolutional neural networks. In: Proceedings of the European conference on computer vision. Springer, Berlin, pp 525–542
Ru X, Cao P, Li L, Zou Q (2019) Selecting essential microRNAs using a novel voting method. Mol Ther Nucl Acids 18:16–23
Saeys Y, Inza I, Larrañaga P (2007) A review of feature selection techniques in bioinformatics. Bioinformatics 23:2507–2517
Salehi A, Davulcu H (2019) Graph attention auto-encoders. arXiv:1905.10715
Schulz F, Roux S, Paez-Espino D, Jungbluth S, Walsh DA, Denef VJ, McMahon KD, Konstantinidis KT, Eloe-Fadrosh EA, Kyrpides NC, Woyke T (2020) Giant virus diversity and host interactions through global metagenomics. Nature 578:432–436
Shen C, Ding Y, Tang J, Guo F (2018) Multivariate information fusion with fast kernel learning to kernel ridge regression in predicting LncRNA-protein interactions. Front Genet 9:716
Shrikumar A, Prakash E, Kundaje A (2019) GkmExplain: fast and accurate interpretation of nonlinear gapped k-mer SVMs. Bioinformatics 35:i173–i182
Singh U, Khemka N, Rajkumar MS, Garg R, Jain M (2017) PLncPRO for prediction of long non-coding RNAs (lncRNAs) in plants and its application for discovery of abiotic stress-responsive lncRNAs in rice and chickpea. Nucleic Acids Res 45:e183
Su Y, Luo Y, Zhao X, Liu Y, Peng J (2019) Integrating thermodynamic and sequence contexts improves protein-RNA binding prediction. PLoS Comput Biol 15:e1007283
Taheri A, Gimpel K, Berger-Wolf T (2019) Sequence-to-sequence modeling for graph representation learning. Appl Netw Sci 4:68
Tuvshinjargal N, Lee W, Park B, Han K (2016) PRIdictor: protein–RNA interaction predictor. Biosystems 139:17–22
Veličković P, Cucurull G, Casanova A, Romero A, Lio P, Bengio Y (2017) Graph attention networks. arXiv:1710.10903
Wang X, Wu Y, Wang R, Wei Y, Gui Y (2019a) A novel matrix of sequence descriptors for predicting protein-protein interactions from amino acid sequences. PLoS ONE 14:e0217312
Wang Y, Yu G, Domeniconi C, Wang J, Zhang X, Guo M (2019b) Selective matrix factorization for multi-relational data fusion. International conference on database systems for advanced applications. Springer, Chiang Mai, pp 313–329
Xie G, Huang S, Luo Y, Ma L, Lin Z, Sun Y (2019) LLCLPLDA: a novel model for predicting lncRNA–disease associations. Mol Genet Genom 294:1477–1486
Xuan P, Sheng N, Zhang T, Liu Y, Guo Y (2019) CNNDLP: a method based on convolutional autoencoder and convolutional neural network with adjacent edge attention for predicting lncRNA–disease associations. Int J Mol Sci 20:4260
Yi H-C, You Z-H, Wang M-N, Guo Z-H, Wang Y-B, Zhou J-R (2020) RPI-SE: a stacking ensemble learning framework for ncRNA-protein interactions prediction using sequence information. BMC Bioinform 21:60
Yu Q, Wang R, Li BN, Yang X, Yao M (2016) Robust locality preserving projections with cosine-based dissimilarity for linear dimensionality reduction. IEEE Access 5:2676–2684
Yu G, Fu G, Lu C, Ren Y, Wang J (2017) BRWLDA: bi-random walks for predicting lncRNA-disease associations. Oncotarget 8:60429–60446
Yu G, Wang Y, Wang J, Fu G, Guo M, Domeniconi C (2018) Weighted matrix factorization based data fusion for predicting lncRNA-disease associations. 2018 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE, Madrid, pp 572–577
Yu Y, Zhang Y, Chen X, Chen Y (2019) Plant noncoding RNAs: hidden players in development and stress responses. Annu Rev Cell Dev Bi 35:407–431
Yue X, Wang Z, Huang J, Parthasarathy S, Moosavinasab S, Huang Y, Lin SM, Zhang W, Zhang P, Sun H (2019) Graph embedding on biomedical networks: methods, applications and evaluations. Bioinformatics 36:1241–1251
Zaynab M, Fatima M, Abbas S, Umair M, Sharif Y, Raza MA (2018) Long non-coding RNAs as molecular players in plant defense against pathogens. Microb Pathogenes 121:277–282
Zhang X, Liu S (2016) RBPPred: predicting RNA-binding proteins from sequence using SVM. Bioinformatics 33:854–862
Zhang W, Yue X, Tang G, Wu W, Huang F, Zhang X (2018) SFPEL-LPI: sequence-based feature projection ensemble learning for predicting LncRNA-protein interactions. PLoS Comput Biol 14:e1006616
Zhao X, Li J, Lian B, Gu H, Li Y, Qi Y (2018) Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat Commun 9:5056
Zheng S, Hao Y, Lu D, Bao H, Xu J, Hao H, Xu B (2017) Joint entity and relation extraction based on a hybrid neural network. Neurocomputing 257:59–66
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 61872055, 31872116).
Author information
Authors and Affiliations
Contributions
JSW and JM designed the methods and conceived the study. JSW and JM implemented the entire method and performed the experiments. JM and YSL analyzed the prediction results. JSW and JM wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wekesa, J.S., Meng, J. & Luan, Y. A deep learning model for plant lncRNA-protein interaction prediction with graph attention. Mol Genet Genomics 295, 1091–1102 (2020). https://doi.org/10.1007/s00438-020-01682-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-020-01682-w