
Abstract
Communication between tumor cells and stromal cells is crucial to tumor development and progression. Fibroblasts and macrophages are the most common stromal cells in the tumor microenvironment. Endothelial cells are another type of stromal cell in the tumor microenvironment required for angiogenesis via interaction with tumor cells. Tumor angiogenesis provides not only oxygen and nutrients for tumor cells but also the necessary anchorage to facilitate tumor metastasis. The present review summarizes studies on the crosstalk between cancer cells and endothelial cells with a focus on implications for tumor progression. The following four categories are discussed in this review: (1) cell–cell communication in tumor microenvironment; (2) induction of metastasis by interaction between cancer cells and endothelial cells; (3) angiogenesis induced by tumor cells; (4) therapeutic strategies targeting adhesion and signaling molecules as well as chemokines. This review provides useful information highlighting the process of cancer aggressiveness affected by the crosstalk between cancer cells and endothelial cells, and suggests therapeutic strategies against tumor progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The tumor microenvironment is complex, consisting of many cell types and factors (O’Malley et al. 2016). Tumor progression is induced by the activation of adjacent stromal cells in the tumor microenvironment (Li et al. 2007; Egeblad et al. 2010). Endothelial cells are a type of stromal cell present in the tumor microenvironment. Tumor cells penetrate the normal epithelium and interact with the surrounding endothelial cells to produce cytokines and growth factors that affect cells in the microenvironment (Brenner et al. 2010).
Metastasis is the main cause of mortality in cancer patients. A major challenge for cancer therapy is defining an appropriate strategy to control or inhibit metastasis. Adhesion to the endothelium of tumor cells affects the formation of metastases in which many adhesion molecules and chemokines are involved (Iiizumi et al. 2007). Cancer cell invasion is crucial for the metastatic spread of locally proliferating tumors and is a step towards the development of a life-threatening disease (Chambers et al. 2002; Keleg et al. 2003).
The metastatic potential of a tumor is determined not only by the cells in the tumor microenvironment but also by the changes in these cancer cells (Gómez-Cuadrado et al. 2017). Cancer cells can invade blood or lymph vessels through the basal membrane in a process called intravasation, a carcinogenic event in which cancer cells begin to move from primary sites (Soon 2007). Epithelial-mesenchymal transition (EMT) cells with migratory phenotypes can degrade the extracellular matrix (ECM) and enable intravasation into tissues and blood or lymphatic vessels (Tsuji et al. 2008). Many cell adhesion molecules and proteinases are involved in the intravasation process of the primary tumor into the blood vessels (Farahani et al. 2014). Using a cell line derived from rat mammary adenocarcinoma, the correlation between intravasation and lung metastasis was revealed (Wyckoff et al. 2000).
Metastasis begins when tumor cells penetrate normal tissues. Transcoelomic metastasis spreads through the peritoneal cavity that connects the organ surface (Tan et al. 2006). In hematogenous metastasis, cancer cells penetrate the blood vessels and extend their reach through blood vessels that are widely spread throughout the body (Sunami et al. 2000). A lymphatic metastasis penetrates the lymph nodes through the lymphatic system and subsequently spreads to other parts of the body (Karaman and Detmar 2014). When cancer cells arrive at a new location, they multiply again, creating a small tumor called a micrometastasis (Rampaul et al. 2001). The process of metastasis ends when the micrometastasis is fully grown.
Cancer cells stimulate endothelial cells to promote tube formation, which leads angiogenesis. Tumor cells induce vascular growth by secreting various growth factors such as basic fibroblast growth factors (bFGF) or vascular endothelial growth factors (VEGF) (Ferrara 2002; Naoyo et al. 2006). Interrupting this crosstalk reduces angiogenesis and the tumor size within the tumor (Vasudev and Reynolds 2014). Therefore, it is important to understand the surrounding microenvironment of cancer and identify biomarkers that may influence cancer progression for the diagnosis and treatment of cancer. The present review summarizes the current collective understanding of the interaction between cancer cells and endothelial cells in the tumor microenvironment. We focus on the key molecules involved in this crosstalk in the tumor microenvironment and on therapeutic strategies targeting these molecules.
Cell–cell communication in tumor microenvironment
The microenvironment includes extracellular matrix as well as components surrounding the tumor cells and vascular endothelial, fibroblast, and bone marrow-derived cells (Tahmasebi and Carloni 2017). The mechanism of cancer development is highly dependent on the interaction between tumors with the components secreted by tumor microenvironment (Witz 2009; Korneev et al. 2017). Cancer cells are surrounded by tumor microenvironment, consisting of fibroblasts, immune and inflammatory cells, blood vascular networks, ECM, and so on (Hanahan and Coussens 2012; Gkretsi et al. 2015). The physiological condition of the tumor microenvironment is related at every stage of tumorigenesis (Wang et al. 2017). A microenvironment in an unhealthy state may cause tumor growth and invasion (Goubran et al. 2014).
The interaction between cancer cells and stromal cells in the microenvironment surrounding tumors plays an important role in the formation and progression of cancer (Bremnes et al. 2011). Major stromal cells include vascular endothelial cells, cancer-associated fibroblasts (CAFs), and tumor-associated macrophages (TAMs) in tumor microenvironment (Junttila and de Sauvage 2013). Endothelial cells are also important determinants of the tumor microenvironment (Chouaib et al. 2010).
CAFs have heterogeneous origins, phenotypes, and functions within the tumor microenvironment (Ishii et al. 2016). CAFs are a heterogeneous group of fibroblasts that are redirected by cancer cells to carcinomas (Lim and Moon 2016). CAFs are fibroblasts similar to activated fibroblasts with stimulation of inflammatory conditions (Augsten 2014). CAFs secrete signaling factors such as VEGF, FGF, and platelet-derived growth factor (PDGF) to support tumor growth (Xing et al. 2010). CAFs also secrete transforming growth factor (TGF)-β associated with EMT (Yu et al. 2014).
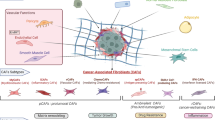
TAMs play a major role in tumor progression by producing cytokines and matrix metalloproteinases (MMPs) (Baay et al. 2011; Quatromoni and Eruslanov 2012). MMPs are involved in ECM composition, and cancer cells play an important role in cell migration during invasion and metastasis (Bodey et al. 2001; Nabeshima et al. 2002). Circulating monocytes in blood are differentiated into M1 macrophages and M2 macrophages which are also called as TAMs (Chanmee et al. 2014; Almatroodi et al. 2016). Infiltrating M1 macrophages present in the early stages of tumorigenesis secrete pro-inflammatory cytokines and inhibit tumor growth (Mantovani et al. 2002). M2 macrophages secrete proteases such as cathepsin, cytokines, and an epidermal growth factor in the later stages of tumorigenesis (Ham and Moon 2013; Rhee 2016; Choi et al. 2017). TAMs secrete cytokines, chemokines, MMPs, and a variety of growth factors, which are associated with angiogenesis, tumor growth, invasion, and metastasis (Baay et al. 2011). TAMs promote the migration and invasion of cancer cells through the cell-ECM (Finkernagel et al. 2016). TAMs can produce proteases such as MMP-2 and MMP-9 which digest the ECM (Yang and Zhang 2017). In addition, an increase in TAM-derived interleukin (IL)-6 has been shown to promote the development of hepatocellular carcinoma (Kong et al. 2016). These results suggest that TAMs play an important role in the occurrence of cancer. Crosstalk between the cancer cells and the endothelial cells (Upreti et al. 2013; Lim and Moon 2016) in the tumor microenvironment was depicted in Fig. 1.

Crosstalk between cancer cells and endothelial cells in tumor microenvironment
Tumor endothelial cells proliferate and migrate more rapidly than normal endothelial cells (Hida et al. 2010). Cancer cells induce changes in endothelial cells by targeting cells through adhesion receptors, gap junctions, and vesicles (Lopes-Bastos et al. 2016). Cancer cells stimulate signaling pathways by activating stromal cells, secreting proteases into the extracellular space, or changing the pH and temperature (Lopes-Bastos et al. 2016). Breast cancer cells regulate lymphatic endothelial cells to promote metastasis (Lee et al. 2014a). Tumor cell-secreted IL-6 induces the phosphorylation of signal transducer and activator of transcription (STAT)-3 in lymphatic endothelial cells (Nilsson et al. 2005). The phosphorylation of STAT3 induces hypoxia-inducible factor (HIF)-1α and VEGF, which activates chemokine (C–C motif) ligand (CCL)-5 expressions in lymphatic endothelial cells (Lee et al. 2014a).
Induction of metastasis via interaction with endothelial cells
Adhesion molecules mediating the interaction between cancer cells and endothelial cells
The initial contact between cancer cells and the endothelium is mild or transient and is mediated via recognition of carbohydrate–carbohydrate interactions (Dube and Bertozzi 2005; Nakahara and Raz 2008). This contact initiates activation of endothelial and cancer cells via cytokines, free radicals, physiologically active lipids, and growth factors (Orr et al. 2000). These mediators induce the expression of adhesion molecules by endothelial and cancer cells, thereby enhancing or fixing initial adhesion bonds (Tang and Honn 1994; Kannagi 1997). Selectins, integrins, cadherins, immunoglobulins, tetraspanins, and thrombospondin (TSP) are known to regulate the adhesion between cancer cells and the endothelium (Nicolson 1988; Pauli et al. 1990). Adhesion and intravasation of cancer cells by various cytokines and proteins are depicted in Fig. 2.

Adhesion and intravasation of cancer cells by various cytokines and proteins
Selectin is a vascular cell adhesion molecule (VCAM) mediating the interaction between endothelium and leukocytes and platelets in the blood circulation (Ala et al. 2003). E-selectin expressed in activated endothelial cells has been detected in the liver metastatic colonies (Soto et al. 2014). Down-regulation of E-selectin expression has been shown to result in experimental liver metastasis (Brodt et al. 1997; Khatib et al. 1999; Bendas and Borsig 2012). Selectin binds to a variety of molecules, most of which function in vivo (Rinko et al. 2004). The selectin family includes P-, E-, and L-selectin. According to other studies, at least one selectin binds to all human carcinomas that have been tested so far, and selectin mediates contact with the tumor (Faryammanesh et al. 2014).
Expression of P-selectin on the cell surfaces of endothelial cells and platelets contributes to metastasis by inducing nuclear factor-kappa B (NF-κB) activation and further expression of P-selectin (Foreman et al. 1994). The expression of P-selectin on the cell surfaces of endothelial cells and platelets also contributes to metastasis (Reyes–Reyes et al. 2006). The absence of L-selectin induces a significant reduction in metastasis, indicating that L-selectin contributes positively to leukocyte recruitment and metastatic crevice formation (Läubli and Borsig 2010).
E-selectin binds to its receptors and mediates the adhesion of tumor cells (Zen et al. 2008). Inhibition or down-regulation of E-selectin has been shown to attenuate experimental liver metastasis, which was induced by the overexpression of E-selectin (Kang et al. 2016). The cytokines secreted by breast cancer cells stimulate macrophages in order to produce tumor necrosis factor (TNF)-α, the regulatory factor of E-selectin expression, resulting in increased adhesion of endothelial cells (Eichbaum et al. 2011; Reymond et al. 2013). In colorectal cancer cells, in vitro studies using sialyl-Lewis X (an E-selectin ligand)-related carbohydrate determinants showed adhesion of cultured vascular endothelial cells to TNF-α-induced E-selectin (Takada et al. 1991). E-selectin levels were significantly increased in the serum and tissues of breast cancer patients compared to those of the control group (Ragab et al. 2017). The E-selectin gene was found more often in malignant tissues than in control tissues (Ragab et al. 2017), suggesting that E-selectin may be associated with aggressive tumors.
Integrin is a transmembrane receptor that promotes ECM attachment and activates signal transduction pathways that mediate cellular signals such as cancer progression and metastasis (Seguin et al. 2015). Integrin is composed of two chains, that is, α- and β-subunits (Campbell and Humphries 2011). Binding of vascular integrins to ECM components contributes to the invasion of endothelial cells. Tumor-associated blood vessels express αvβ3 and αvβ5 integrins, and the targeting of these vessels has been studied with a promising anti-angiogenic approach. (Brooks et al. 1994; Bendas and Borsig 2012). Antagonists of integrin such as cilengitide, an inhibitor of αvβ3 and αvβ5, have been shown to be promising anti-cancer agents (Desgrosellier and Cheresh 2010).
Tetraspanins, cell surface proteins, are associated with adhesion receptors in the integrin family (Hemler 2005). The expression of tetraspanins correlates with the tumor stage and type (Lazo 2007; Vences-Catalán et al. 2015). Cell surface proteins of the tetraspanin family are present in almost all cell and tissue types and regulate integrin-dependent cell migration (Berditchevski 2001). Several members of the tetraspanin superfamily, including CD9, CD81, and CD151, are located in the tumor cell-endothelial cell contact area (Longo et al. 2001). The interaction between CD9 and TGF-α was decreased through ectodomain shedding to release soluble TGF-α (Imhof et al. 2008). CD9 can interact with transmembrane TGF-α to activate epidermal growth factor receptor (EGFR) (Imhof et al. 2008). Increased TGF-α-EGFR signaling is known to induce cancer progression (Lee et al. 1995; Kenny and Bissell 2007). Expression of CD9 is often markedly reduced in a variety of metastatic cancers, including lung, colon, and pancreatic cancers (Parkes and Jewell 2001). CD81 directly interacts with integrin α4β1 and CD151 directly binds with integrin α3β1 and α6β1 (Serru et al. 1999). Tetraspanin CD151 mediates cell adhesion with integrins (α3β1, α6β1, α6β4, and α7β1) on different types of laminin (Boucheix and Rubinstein 2001). Platelets increase cancer cell adhesion to endothelial cells, which enhances angiogenesis of endothelial colony-forming cells in platelets with integrin α6β1 (Reymond et al. 2013; Huang et al. 2016). The regulation of tetraspanin in tumor cell lines significantly increases cell growth, morphology, invasion, tumor growth, and metastasis (Detchokul et al. 2014; Hemler 2014). Among VCAMs and TSPs, VCAM-1 and thrombospondin (TSP)-1 have been extensively studied in cancer progression and metastasis (Sargiannidou et al. 2001; Lin et al. 2007a).
TSPs, a family of ECM proteins, are involved in cell proliferation and differentiation (Bornstein 2009; Huang et al. 2017). TSP-1 plays an important role in the microenvironment of the tumor and also affects tumor cell adhesion, proliferation, invasion, migration, apoptosis, and tumor immunity (Baenziger et al. 1971; Jeanne et al. 2015). TSP-1 controls inflammation by regulating the activity of other secreted factors (Varma et al. 2008; Lopez-Dee et al. 2011; Stenina-Adognravi 2014). TSP-1 regulates the production and activation of pro-inflammatory cytokine IL-1β by macrophages (Stein et al. 2016). TSP-1 has been shown to exert its CD47-dependent inflammatory effect on the IL-1β pathway (Stein et al. 2016). Other studies have confirmed that CD47 is an important regulator of lymphocyte function-associated antigen (LFA)-1 and very late antigen (VLA)-4 integrin-adherence in the proliferation and recruitment of T cells (Azcutia et al. 2017).
VCAM-1 is involved in this process (Schlesinger and Bendas 2015). VCAM-1 is abnormally expressed in breast cancer cells and has been shown to bind to the natural ligand α4β1 integrin (also known as VLA-4) (Sharma et al. 2017). This binding triggers metastasis of breast cancer cells to the lungs, bones, and brain (Sharma et al. 2017). Intercellular adhesion molecule (ICAM)-1 and VCAM-1 are involved in tumor progression and metastasis (Regidor et al. 1998). Up-regulation of these adhesion molecules promotes endothelial cell adhesion and angiogenesis, and also contributes to changes in the invasive phenotype (Regidor et al. 1998).
Invasion and migration in the interaction between cancer cells and endothelial cells
The ability of malignant tumor cells to induce cell migration and the invasion of cancer has been investigated for years (Clark and Vignjevic 2015; Krakhmal et al. 2015). Metastasis occurs when cancer cells penetrate the basement membrane and wall of the endothelium and travel to distant organs (Valastyan and Weinberg 2011; van Zijl et al. 2011). Stroma and tumor cells exchange signals to modify the ECM and stimulate cell migration (Lomberk 2010). It is known that tumor cells overcome the ECM barrier and spread to surrounding tissues (Krakhmal et al. 2015).
Cells are polarized during this migration process (Friedl and Wolf 2003). This polarity can be reflected through specific regions and molecules on the cell surface (Ridley et al. 2003). Upon polarization, phosphatidylinositol-3, 4, 5-trisphosphate (PIP3), activated Rac, and Cdc42 are found in the same direction (Ridley et al. 2003). In the opposite direction, Rho GTPase and phosphatase and tensin homolog (PTEN) are observed (Parent and Devreotes 1999). After establishment of polarity, actin filament polymerization at the leading edge stimulates lamellipodium (Atilgan et al. 2005). Next, translocation of the cell body occurs due to the formation of adhesive contact (Shieh et al. 2011). The depolymerization of the actin network completes the cell migration assembly process (Carlsson 2010).
Tumor cells attach to endothelial cells and invade connective tissue (Mierke et al. 2008). Vascular endothelial cells promote cancer invasion and metastasis, mostly via Akt and NF-κB pathways (Wang et al. 2013). The integrin-induced signal pathway is involved in cell migration, and the integrins-focal adhesion kinases -Rho GTPases are activated in both endothelial and cancer cells (Feng et al. 2017). Cancer cells secrete growth factors, which significantly increase endothelial cell proliferation, migration, and tube formation (Hwang et al. 2016).
The invasiveness of cancer cells plays an important role in the cytokines secreted following the interaction with endothelial cells (Kamińska et al. 2015). TGF-β can induce EMT and enhance intravasation (Tsuji et al. 2008). Also, activation of EGFR family members stimulates invadopodia through phosphoinositide 3-kinase (PI3K), neural Wiskott–Aldrich syndrome protein (N-WASP), RhoA, and WASP (Keklikoglou et al. 2012; Chiang et al. 2016). Intratavation and invasion are associated with urokinase-type plasminogen activator (uPA)/uPAR in relation to proteinases, and the role of MMPs is important (Ossowski 1988; Ploug et al. 2001; Shin et al. 2011). Endothelial cells co-cultured with invasive hepatocellular carcinoma cells have been shown to increase the levels of IL-8 as well as growth-regulated oncogene (Gro)-β expression in intravasation (Fig. 2) (Loukinova et al. 2000; Mierke et al. 2008). Expression of two chemokine receptors, chemokine (C-X-C motif) receptor (CXCR) 2, has been shown to be up-regulated in invasive cancer cells (Murdoch et al. 1999; Salcedo et al. 2000). Invasive cancer cells, along with CXCR2 expression, contribute to the destruction of the endothelial barrier (Mierke et al. 2008). Metastasis chips have been developed in which endothelial cells and stromal cells are patterned close to tumor cells (Caballero et al. 2017). These metastasis chips have made it possible to imitate the production of microvascular blood vessels, allowing for the identification of angiogenesis, intravasation, and extravasation (Lee et al. 2014b).
The chemokines that are most important for endothelial progenitor cell migration include CCL2 (MCP-1) and CCL5 (RANTES) (Yu et al. 2016; Phi et al. 2017). Cancer cells secrete chemokines such as CCL2, which secrete inflammatory stimuli that activate endothelial cells and induce the expression of VCAM1 and vascular adhesion protein 1 (VAP1) (Reymond et al. 2013). CCL5 is produced by several tumor cells (Azenshtein et al. 2002). High plasma CCL5 levels are associated with advanced breast cancer, and breast cancer cell-derived CCL5 promotes the progression and invasion of breast cancer (Niwa et al. 2001; Azenshtein et al. 2002). Expression of CCL2 and CCL5 is also increased in prostate cancer (Zhang et al. 2010). CCL2, CCL3, CCL4, and CCL5 are expressed by inflammatory stimuli (Laurence 2006; Bobanga et al. 2013). In mouse models of liver cancer, tumor-associated endothelial cells have been shown to up-regulate the expression of CCL2, CCL3, CCL4, CCL7, and CCL8 (Spring et al. 2005; Ryschich et al. 2006). CCL1 and CCL3 promote the progression of tumor metastasis in leukemia (Ridiandries et al. 2016). Chemokine (C-X-C motif) ligand (CXCL) 1, CXCL2, and CXCL3 are important in the growth of pancreatic cancer, melanoma, lung cancer, and gastric cancer (Bendall 2005). CXCL4 was shown to be involved in the proliferation of cancer as well as overexpressed in many cancers including prostate, breast, ovarian, and lung cancers, as well as melanoma (Müller et al. 2001; Kijima et al. 2002; Darash-Yahana et al. 2004; Sarvaiya et al. 2013).
Angiogenesis induced by tumor cells
Heterologous interaction between tumor cells and endothelial cells plays an important role in the vascularization of neoplastic cells and the pathological angiogenesis of tumors (Longo et al. 2001). Tumor angiogenesis is a complex process in which new blood vessels are formed in response to the interaction between tumor cells and endothelial cells, growth factors, and ECM components (Jung et al. 2002; Khodarev et al. 2003). Angiogenesis is characterized by mitosis of the ECM and endothelial cells (Dudley 2012).
Since endothelial cells form a capillary-like tube structure, they proliferate and migrate in the presence of these growth factors (Prior et al. 2004). Tumors induce vascular growth by secreting various growth factors such as bFGF or VEGF (Ferrara 2002; Naoyo et al. 2006). VEGF, a potent stimulator of angiogenesis, plays an important role in angiogenesis (Bloor 2005; Carmeliet 2005). The secreted VEGF binds to receptors on the surface of vascular endothelial cells, creating new blood vessels that supply oxygen and nutrients to the tumor (Sounni and Noel 2013). VEGF induces a large amount of signal transduction in endothelial cells (Hofer and Schweighofer 2007). VEGF receptors (VEGFRs) are associated with endothelial cell-dependent tumor angiogenesis (Meng et al. 2017). uPA inhibits the uPA dependence of VEGFR1 and VEGFR2 gene transcription by binding to the hematopoietically expressed homeodomain protein or proline-rich homeodomain protein (HHEX/PRH), mediating the angiogenic effect of VEGF and the control of pathological angiogenesis (Stepanova et al. 2016; Song et al. 2018).
PDGF induces mitogenesis with angiogenesis, fibroblasts, osteoblasts, stromal cells, vascular smooth muscle cells, and mesenchymal stem cells (Hollinger et al. 2008). PDGF is one of the many growth factors that regulate cell growth and differentiation (De Donatis et al. 2008). The important roles of PDGF-B and PDGFR-β in angiogenesis have been demonstrated by gene targeting experiments, and their expression has been found to be associated with endothelial vascularization and maturation (Raica and Cimpean 2010). PDGF-B directly induces endothelial cell proliferation, migration, and tube formation, whereas PDGF-A shows no such effect (Gacche and Meshram 2014). PDGF-D regulates VEGF signaling and promotes tumor cell growth in a variety of cancer cell types (Li et al. 2003).
Notch signaling plays an important role in the development and differentiation of various hematopoietic systems (Artavanis-Tsakonas et al. 1999; Milner and Bigas 1999). Notch receptors promote the growth and survival of tumor cells through the interaction between tumor cells and Notch ligands (Jundt et al. 2004). One of the Notch ligands, Jagged (JAG) 1, is overexpressed in many cancer types (Grochowski et al. 2016). JAG1 can indirectly affect tumor microenvironmental components such as the vasculature of the tumor (Li et al. 2014). Blocking the Notch in the tumor vasculature has been shown to inhibit tumor growth (Wu et al. 2010). Tumor vessels use Notch signaling for vascular stability while controlling vascular wall cell function (Kofler et al. 2011). JAG1 expression is induced by TGF-β which induces EMT phenotype in vitro (Camenisch et al. 2002; Zavadil et al. 2004).
Semaphorin (SEMA)-4D, also known as CD100, is a protein belonging to class IV semaphorin that is strongly implicated in tumor progression via interaction with the high affinity receptor Plexin-B1 (Lin et al. 2007b; Okuno et al. 2010). SEMA4D-Plexin-B1 signaling pathway in angiogenesis occurs as SEMA4D binds to Plexin-B1 and induces tumor angiogenesis via two independent downstream pathways (Ch’ng and Kumanogoh 2010). SEMA4D-Plexin-B1 signaling pathway in angiogenesis occurs as SEMA4D binds to Plexin-B1 and induces tumor angiogenesis via two independent downstream pathways (Ch’ng and Kumanogoh 2010). When SEMA4D interacts with Plexin-B1, a Plexin-B1-Met interaction on its binding is possible, resulting in Met activation and tyrosine phosphorylation (Conrotto et al. 2005). Another mechanism involves Plexin-B1 and PDZ-binding motifs in order to activate RhoA (Basile et al. 2007). In addition, cancer cells activate the PI3K/Akt signaling pathway and increase endothelial tube formation as well as survival (Lauring et al. 2013; Massihnia et al. 2016; Zhou et al. 2016). Cancer cells also partially activate the PI3K/Akt signaling pathway and promote endothelial tube formation and survival (Cheng et al. 2017).
Strategies targeting the crosstalk between cancer cells and endothelial cells
Targeted cancer therapy should cause minimal collateral damage to normal cells while targeting cancer cells. Drugs used in chemotherapy work in multiple ways to stop the growth of cancer cells by killing, stopping the division of, or preventing the spread of cells (Shewach and Kuchta 2009). Molecular targets include adhesion molecules, signaling molecules, and chemokines mediating the interaction of cancer cells with endothelial cells (Agemy et al. 2013; Kummar and Doroshow 2013; Farahani et al. 2014). Molecules associated with angiogenesis and regulators of invasion can also be effective targets for anti-cancer strategies (Ferrara and Kerbel 2005; Zhao and Adjei 2015).
The most widely known approach inhibiting tumor angiogenesis involves blockade of the VEGF pathway (Kuhnert et al. 2011). VEGF-targeted therapy was initially designed to inhibit neoangiogenesis and starve the tumor of needed oxygen and nutrients (Ellis and Hicklin 2008). In clinical trials, the anti-VEGF approach increased survival rates in metastatic cancer patients (Gyanchandani and Kim 2013). Bevacizumab was the first VEGF inhibitor approved as a cancer treatment (Meadows and Hurwitz 2012). Adding a VEGF-specific antibody, bevacizumab, to chemotherapy improves the overall survival in patients with colorectal cancer and lung cancer (Jain et al. 2006). Sunitinib is a targeted therapy that is a receptor protein-tyrosine kinase inhibitor (Demetri et al. 2006). Multikinase inhibitors that inhibit VEGFR1, 2, 3, PDGFR, and c-Kit include sunitinib, sorafenib, and pazopanib (Keating and Santoro 2009; Sternberg et al. 2013). A number of VEGF inhibitors, including brivanib alaninate, cediranib, and vandetanib, are currently in phase 3 clinical trials or in clinical development (Meadows and Hurwitz 2012). These drugs and their target molecules are summarized in Table 1.
Kangai-1 (KAI1), also known as CD82, is a typical tumor metastasis suppressor (Singh et al. 2016). Inhibition of KAI1 has been shown to negatively regulate VEGF-induced angiogenesis (Nomura et al. 2016). KAI1 is known to block the metastatic process without affecting primary tumor growth (Park et al. 2012; Lee et al. 2017). Imatinib mesylate inhibits the growth of cancer cells by blocking a few of the enzymes needed for cell growth (Table 1) (Dewar et al. 2003; Danchev et al. 2008). Imatinib mesylate regulates metastasis by up-regulating KAI1 gene expression in human breast cancer MCF-7 cell line (Shandiz et al. 2016).
TSP-1 serves as an angiogenesis inhibitor by regulating the bioavailability and activity of VEGF (Lawler 2000). TSP-1 is a multifunctional glycoprotein involved in various biological processes including angiogenesis, apoptosis, and activation of TGF-β1 (Crawford et al. 1998; Murphy-Ullrich and Poczatek 2000). Tumors overexpressing TSP-1 show decreased growth, metastases, and angiogenesis, suggesting TSP-1 as a therapeutic target for cancer (Kazerounian et al. 2008). Both ABT-510 and ABT-898, TSP-1 synthetic analogs mimicking anti-angiogenic activity, have been shown to inhibit the growth of prolactinoma (Recouvreux et al. 2012).
Notch signaling is important in tumor angiogenesis through VEGF-A (Funahashi et al. 2008). Inhibition of Notch signaling in endothelial cells limited VEGF-A-induced tumor growth and caused endothelial dysfunction (Patenaude et al. 2014). Using co-culture and tumor growth assays, Notch-mediated nitric oxide (NO) production in endothelial cells demonstrates the need for VEGF-A signaling (Fukumura et al. 2006). NO, mainly produced by endothelial NO synthase (eNOS), acts as a cardiovascular signal molecule. The eNOS activated by the phosphorylation of the Ser1177 residue was reduced through Notch inhibition, which caused tumor growth and diminished vascular function (Miller et al. 2009). BAY41-2272, a soluble guanylate cyclase activator and vasodilator, can inhibit tumor growth and the vascular function of eNOS (Patenaude et al. 2014).
Conclusions
The tumor microenvironment is composed of complex and diverse elements such as extracellular matrix, growth factors, signaling substances, and cells surrounding cancer cells. Cancer-endothelial cell interactions in the tumor microenvironment secrete adhesion molecules and chemokines, which are critical to tumor growth and metastasis (Buess et al. 2009). Drug resistance and cancer recurrence may be overcome through control of this tumor microenvironment. Studies investigating the anti-cancer mechanisms targeting cytokine secretion by cancer-derived stromal cells or stromal cells in particular provide a new breakthrough in the development of selective chemotherapeutic agents. In the present study, we summarize the current perspective on the interaction between cancer cells and endothelial cells and also suggested anti-cancer strategies based on these interactions.
Intercellular interactions between cancer and other cells in the surrounding tumor microenvironment are critical for tumorigenesis and tumor progression. Understanding the mechanisms of these interactions can lead to the development of new therapies that block tumor progression and metastasis. This review provides useful information underlying cancer aggressiveness affected by the crosstalk between cancer cells and endothelial cells, and suggests therapeutic strategies against tumor progression.
Change history
29 August 2018
Unfortunately, there is an error in the original version of the article. The original published article contains an incomplete ‘Acknowledgements’ section. Please find the full ‘Acknowledgements’ section below.
References
Agemy L, Kotamraju VR, Friedmann-Morvinski D, Sharma S, Sugahara KN, Ruoslahti E (2013) Proapoptotic peptide-mediated cancer therapy targeted to cell surface p32. Mol Ther 21(12):2195–2204
Ala A, Dhillon AP, Hodgson HJ (2003) Role of cell adhesion molecules in leukocyte recruitment in the liver and gut. Int J Exp Pathol 84(1):1–16
Almatroodi SA, McDonald CF, Darby IA, Pouniotis DS (2016) Characterization of M1/M2 tumour-associated macrophages (TAMs) and Th1/Th2 cytokine profiles in patients with NSCLC. Cancer Microenviron 9(1):1–11
Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284(5415):770–776
Atilgan E, Wirtz D, Sun SX (2005) Morphology of the lamellipodium and organization of actin filaments at the leading edge of crawling cells. Biophys J 89(5):3589–3602
Augsten M (2014) Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 4:62
Azcutia V, Bassil R, Herter JM, Engelbertsen D, Newton G, Autio A, Mayadas T, Lichtman AH, Khoury SJ, Parkos CA, Elyaman W, Luscinskas FW (2017) Defects in CD4 + T cell LFA-1 integrin-dependent adhesion and proliferation protect Cd47-/- mice from EAE. J Leukoc Biol 101(2):493–505
Azenshtein E, Luboshits G, Shina S, Neumark E, Shahbazian D, Weil M, Wigler N, Keydar I, Ben-Baruch A (2002) The CC chemokine RANTES in breast carcinoma progression: regulation of expression and potential mechanisms of promalignant activity. Cancer Res 62(4):1093–1102
Baay M, Brouwer A, Pauwels P, Peeters M, Lardon F (2011) Tumor cells and tumor-associated macrophages: secreted proteins as potential targets for therapy. Clin Dev Immunol 2011:565187
Baenziger NL, Brodie GN, Majerus PW (1971) A thrombin-sensitive protein of human platelet membranes. Proc Natl Acad Sci USA 68(1):240–243
Basile JR, Gavard J, Gutkind JS (2007) Plexin-B1 utilizes RhoA and Rho kinase to promote the integrin-dependent activation of Akt and ERK and endothelial cell motility. J Biol Chem 282(48):34888–34895
Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, Degroot J, Gattamaneni R, Cher L, Rosenthal M, Payer F, Jürgensmeier JM, Jain RK, Sorensen AG, Xu J, Liu Q, van den Bent M (2013) Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 31(26):3212–3218
Bendall L (2005) Chemokines and their receptors in disease. Histol Histopathol 20(3):907–926
Bendas G, Borsig L (2012) Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol 2012:676731
Berditchevski F (2001) Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci 114(Pt 23):4143–4151
Bloor CM (2005) Angiogenesis during exercise and training. Angiogenesis 8(3):263–271
Bobanga ID, Petrosiute A, Huang AY (2013) Chemokines as cancer vaccine adjuvants. Vaccines (Basel). 1(4):444–462
Bodey B, Bodey B Jr, Siegel SE, Kaiser HE (2001) Matrix metalloproteinase expression in malignant melanomas: tumor-extracellular matrix interactions in invasion and metastasis. In Vivo 15(1):57–64
Bornstein P (2009) Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal 3(3–4):189–200
Boucheix C, Rubinstein E (2001) Tetraspanins. Cell Mol Life Sci 58(9):1189–1205
Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, Camps C, Marinez I, Busund LT (2011) The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol 6(1):209–217
Brenner W, Beitz S, Schneider E, Benzing F, Unger RE, Roos FC, Thüroff JW, Hampel C (2010) Adhesion of renal carcinoma cells to endothelial cells depends on PKCmu. BMC Cancer 10:183
Brodt P, Fallavollita L, Bresalier RS, Meterissian S, Norton CR, Wolitzky BA (1997) Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer 71(4):612–619
Brooks PC, Clark RA, Cheresh DA (1994) Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264(5158):569–571
Buess M, Rajski M, Vogel-Durrer BM, Herrmann R, Rochlitz C (2009) Tumor-endothelial interaction links the CD44(+)/CD24(-) phenotype with poor prognosis in early-stage breast cancer. Neoplasia. 11(10):987–1002
Caballero D, Kaushik S, Correlo VM, Oliveira JM, Reis RL, Kundu SC (2017) Organ-on-chip models of cancer metastasis for future personalized medicine: from chip to the patient. Biomaterials 149:98–115
Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, Klewer SE (2002) Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol 248(1):170–181
Campbell ID, Humphries MJ (2011) Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol 3(3):a004994
Carlsson AE (2010) Actin dynamics: from nanoscale to microscale. Annu Rev Biophys 39:91–110
Carmeliet P (2005) VEGF as a key mediator of angiogenesis in cancer. Oncology 69(Suppl 3):4–10
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2(8):563–572
Chanmee T, Ontong P, Konno K, Itano N (2014) Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 6(3):1670–1690
Cheng HW, Chen YF, Wong JM, Weng CW, Chen HY, Yu SL, Chen HW, Yuan A, Chen JJ (2017) Cancer cells increase endothelial cell tube formation and survival by activating the PI3 K/Akt signalling pathway. J Exp Clin Cancer Res 36(1):27
Chiang SP, Cabrera RM, Segall JE (2016) Tumor cell intravasation. Am J Physiol Cell Physiol 311(1):C1–C14
Ch’ng ES, Kumanogoh A (2010) Roles of Sema4D and Plexin-B1 in tumor progression. Mol Cancer. 9:251
Choi J, Gyamfi J, Jang H, Koo JS (2017) The role of tumor-associated macrophage in breast cancer biology. Histol Histopathol 6:11916
Chouaib S, Kieda C, Benlalam H, Noman MZ, Mami-Chouaib F, Rüegg C (2010) Endothelial cells as key determinants of the tumor microenvironment: interaction with tumor cells, extracellular matrix and immune killer cells. Crit Rev Immunol 30(6):529–545
Clark AG, Vignjevic DM (2015) Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol 36:13–22
Conrotto P, Valdembri D, Corso S, Serini G, Tamagnone L, Comoglio PM, Bussolino F, Giordano S (2005) Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood 105(11):4321–4329
Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N (1998) Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 93(7):1159–1170
Danchev N, Nikolova I, Momekov G (2008) A new era in anticancer therapy/imatinib—a new era in anticancer therapy. Biotechnol Biotechnol Equip 22(3):769–770
Darash-Yahana M, Pikarsky E, Abramovitch R, Zeira E, Pal B, Karplus R, Beider K, Avniel S, Kasem S, Galun E, Peled A (2004) Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J 18(11):1240–1242
De Donatis A, Comito G, Buricchi F, Vinci MC, Parenti A, Caselli A, Camici G, Manao G, Ramponi G, Cirri P (2008) Proliferation versus migration in platelet-derived growth factor signaling: the key role of endocytosis. J Biol Chem 283(29):19948–19956
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368(9544):1329–1338
Desgrosellier JS, Cheresh DA (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10(1):9–22
Detchokul S, Williams ED, Parker MW, Frauman AG (2014) Tetraspanins as regulators of the tumour microenvironment: implications for metastasis and therapeutic strategies. Br J Pharmacol 171(24):5462–5490
Dewar AL, Domaschenz RM, Doherty KV, Hughes TP, Lyons AB (2003) Imatinib inhibits the in vitro development of the monocyte/macrophage lineage from normal human bone marrow progenitors. Leukemia 17(9):1713–1721
Dube DH, Bertozzi CR (2005) Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev Drug Discov 4(6):477–488
Dudley AC (2012) Tumor endothelial cells. Cold Spring Harb Perspect Med 2(3):a006536
Egeblad M, Nakasone ES, Werb Z (2010) Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 18(6):884–901
Eichbaum C, Meyer AS, Wang N, Bischofs E, Steinborn A, Bruckner T, Brodt P, Sohn C, Eichbaum MH (2011) Breast cancer cell-derived cytokines, macrophages and cell adhesion: implications for metastasis. Anticancer Res 31(10):3219–3227
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8(8):579–591
Farahani E, Patra HK, Jangamreddy JR, Rashedi I, Kawalec M, Rao Pariti RK, Batakis P, Wiechec E (2014) Cell adhesion molecules and their relation to (cancer) cell stemness. Carcinogenesis 35(4):747–759
Faryammanesh R, Lange T, Magbanua E, Haas S, Meyer C, Wicklein D, Schumacher U, Hahn U (2014) SDA, a DNA aptamer inhibiting E- and P-selectin mediated adhesion of cancer and leukemia cells, the first and pivotal step in transendothelial migration during metastasis formation. PLoS ONE 9(4):e93173
Feng T, Yu H, Xia Q, Ma Y, Yin H, Shen Y, Liu X (2017) Cross-talk mechanism between endothelial cells and hepatocellular carcinoma cells via growth factors and integrin pathway promotes tumor angiogenesis and cell migration. Oncotarget. 8(41):69577–69593
Ferrara N (2002) VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2(10):795–803
Ferrara N, Kerbel RS (2005) Angiogenesis as a therapeutic target. Nature 438(7070):967–974
Finkernagel F, Reinartz S, Lieber S, Adhikary T, Wortmann A, Hoffmann N, Bieringer T, Nist A, Stiewe T, Jansen JM, Wagner U, Müller-Brüsselbach S, Müller R (2016) The transcriptional signature of human ovarian carcinoma macrophages is associated with extracellular matrix reorganization. Oncotarget 7(46):75339–75352
Finn RS, Kang YK, Mulcahy M, Polite BN, Lim HY, Walters I, Baudelet C, Manekas D, Park JW (2012) Phase II, open-label study of brivanib as second-line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res 18(7):2090–2098
Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, Eddy SM, Ward PA (1994) C5a-induced expression of P-selectin in endothelial cells. J Clin Invest 94(3):1147–1155
Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3(5):362–374
Fukumura D, Kashiwagi S, Jain RK (2006) The role of nitric oxide in tumour progression. Nat Rev Cancer 6(7):521–534
Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, Sharma A, Kanamaru E, Borisenko V, Desilva DM, Suzuki A, Wang X, Shawber CJ, Kandel JJ, Yamashiro DJ, Kitajewski J (2008) A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res 68(12):4727–4735
Gacche RN, Meshram RJ (2014) Angiogenic factors as potential drug target: efficacy and limitations of anti-angiogenic therapy. Biochim Biophys Acta 1846(1):161–179
Gkretsi V, Stylianou A, Papageorgis P, Polydorou C, Stylianopoulos T (2015) Remodeling components of the tumor microenvironment to enhance cancer therapy. Front Oncol 5:214
Gómez-Cuadrado L, Tracey N, Ma R, Qian B, Brunton VG (2017) Mouse models of metastasis: progress and prospects. Dis Model Mech 10(9):1061–1074
Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T (2014) Regulation of tumor growth and metastasis: the role of tumor microenvironment. Cancer Growth Metastasis 7:9–18
Grochowski CM, Loomes KM, Spinner NB (2016) Jagged1 (JAG1): structure, expression, and disease associations. Gene 576(1 Pt 3):381–384
Gyanchandani R, Kim S (2013) Predictive biomarkers to anti-VEGF therapy: progress toward an elusive goal. Clin Cancer Res 19(4):755–757
Ham MN, Moon A (2013) Inflammatory and microenvironmental factors involved in breast cancer progression. Arch Pharm Res 36(12):1419–1431
Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21(3):309–322
Hantschel O, Rix U, Superti-Furga G (2008) Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk Lymphoma 49(4):615–619
Hemler ME (2005) Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 6(10):801–811
Hemler ME (2014) Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer 14(1):49–60
Hida K, Ohga N, Kurosu T, Totsuka Y, Shindoh M (2010) Crosstalk between blood vessels and tumor microenvironment. Oral Sci Int 7(1):1–10
Hofer E, Schweighofer B (2007) Signal transduction induced in endothelial cells by growth factor receptors involved in angiogenesis. Thromb Haemost 97(3):355–363
Hollinger JO, Hart CE, Hirsch SN, Lynch S, Friedlaender GE (2008) Recombinant human platelet-derived growth factor: biology and clinical applications. J Bone Joint Surg Am 90(Suppl 1):48–54
Huang Z, Miao X, Patarroyo M, Nilsson GP, Pernow J, Li N (2016) Tetraspanin CD151 and integrin α6β1 mediate platelet-enhanced endothelial colony forming cell angiogenesis. J Thromb Haemost 14(3):606–618
Huang T, Sun L, Yuan X, Qiu H (2017) Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget. https://doi.org/10.18632/oncotarget.19165
Hwang SH, Lee BH, Choi SH, Kim HJ, Won KJ, Lee HM, Rhim H, Kim HC, Nah SY (2016) Effects of gintonin on the proliferation, migration, and tube formation of human umbilical-vein endothelial cells: involvement of lysophosphatidic-acid receptors and vascular-endothelial-growth-factor signaling. J Ginseng Res 40(4):325–333
Iiizumi M, Mohinta S, Bandyopadhyay S, Watabe K (2007) Tumor-endothelial cell interactions: therapeutic potential. Microvasc Res 74(2–3):114–120
Imhof I, Gasper WJ, Derynck R (2008) Association of tetraspanin CD9 with transmembrane TGF{alpha} confers alterations in cell-surface presentation of TGF{alpha} and cytoskeletal organization. J Cell Sci 121(Pt 13):2265–2274
Ishii G, Ochiai A, Neri S (2016) Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev 99:186–196
Jain RK, Duda DG, Clark JW, Loeffler JS (2006) Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 3(1):24–40
Jeanne A, Schneider C, Martiny L, Dedieu S (2015) Original insights on thrombospondin-1-related antireceptor strategies in cancer. Front Pharmacol 6:252
Jundt F, Pröbsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, Bargou RC, Manz R, Stein H, Dörken B (2004) Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood 103(9):3511–3515
Jung SP, Siegrist B, Hornick CA, Wang YZ, Wade MR, Anthony CT, Woltering EA (2002) Effect of human recombinant endostatin protein on human angiogenesis. Angiogenesis 5(1–2):111–118
Junttila MR, de Sauvage FJ (2013) Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501(7467):346–354
Kamińska K, Szczylik C, Bielecka ZF, Bartnik E, Porta C, Lian F, Czarnecka AM (2015) The role of the cell-cell interactions in cancer progression. J Cell Mol Med 19(2):283–296
Kang SA, Blache CA, Bajana S, Hasan N, Kamal M, Morita Y, Gupta V, Tsolmon B, Suh KS, Gorenstein DG, Razaq W, Rui H, Tanaka T (2016) The effect of soluble E-selectin on tumor progression and metastasis. BMC Cancer. 16:331
Kannagi R (1997) Carbohydrate-mediated cell adhesion involved in hematogenous metastasis of cancer. Glycoconj J 14(5):577–584
Karaman S, Detmar M (2014) Mechanisms of lymphatic metastasis. J Clin Invest 124(3):922–928
Kazerounian S, Yee KO, Lawler J (2008) Thrombospondins in cancer. Cell Mol Life Sci 65(5):700–712
Keating GM, Santoro A (2009) Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs 69(2):223–240
Keklikoglou I, Koerner C, Schmidt C, Zhang JD, Heckmann D, Shavinskaya A, Allgayer H, Gückel B, Fehm T, Schneeweiss A, Sahin O, Wiemann S, Tschulena U (2012) MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-κB and TGF-β signaling pathways. Oncogene 31(37):4150–4163
Keleg S, Büchler P, Ludwig R, Büchler MW, Friess H (2003) Invasion and metastasis in pancreatic cancer. Mol Cancer 2:14
Kenny PA, Bissell MJ (2007) Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest 117(2):337–345
Khatib AM, Kontogiannea M, Fallavollita L, Jamison B, Meterissian S, Brodt P (1999) Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Res 59(6):1356–1361
Khodarev NN, Yu J, Labay E, Darga T, Brown CK, Mauceri HJ, Yassari R, Gupta N, Weichselbaum RR (2003) Tumour-endothelium interactions in co-culture: coordinated changes of gene expression profiles and phenotypic properties of endothelial cells. J Cell Sci 116(Pt 6):1013–1022
Kijima T, Maulik G, Ma PC, Tibaldi EV, Turner RE, Rollins B, Sattler M, Johnson BE, Salgia R (2002) Regulation of cellular proliferation, cytoskeletal function, and signal transduction through CXCR4 and c-Kit in small cell lung cancer cells. Cancer Res 62(21):6304–6311
Kofler NM, Shawber CJ, Kangsamaksin T, Reed HO, Galatioto J, Kitajewski J (2011) Notch signaling in developmental and tumor angiogenesis. Genes Cancer 2(12):1106–1116
Kong L, Zhou Y, Bu H, Lv T, Shi Y, Yang J (2016) Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J Exp Clin Cancer Res 35(1):131
Korneev KV, Atretkhany KN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA (2017) TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine 89:127–135
Krakhmal NV, Zavyalova MV, Denisov EV, Vtorushin SV, Perelmuter VM (2015) Cancer invasion: patterns and mechanisms. Acta Nat 7(2):17–28
Kuhnert F, Kirshner JR, Thurston G (2011) Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc Cell 3(1):20
Kummar S, Doroshow JH (2013) Molecular targets in cancer therapy. Expert Rev Anticancer Ther 13(3):267–269
Läubli H, Borsig L (2010) Selectins promote tumor metastasis. Semin Cancer Biol 20(3):169–177
Laurence AD (2006) Location, movement and survival: the role of chemokines in haematopoiesis and malignancy. Br J Haematol 132(3):255–267
Lauring J, Park BH, Wolff AC (2013) The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw 11(6):670–678
Lawler J (2000) The functions of thrombospondin-1 and-2. Curr Opin Cell Biol 12(5):634–640
Lazo PA (2007) Functional implications of tetraspanin proteins in cancer biology. Cancer Sci 98(11):1666–1677
Lee DC, Fenton SE, Berkowitz EA, Hissong MA (1995) Transforming growth factor alpha: expression, regulation, and biological activities. Pharmacol Rev 47(1):51–85
Lee E, Fertig EJ, Jin K, Sukumar S, Pandey NB, Popel AS (2014a) Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat Commun 5:4715
Lee H, Park W, Ryu H, Jeon NL (2014b) A microfluidic platform for quantitative analysis of cancer angiogenesis and intravasation. Biomicrofluidics 8(5):054102
Lee J, Byun HJ, Lee MS, Jin YJ, Jeoung D, Kim YM, Lee H (2017) The metastasis suppressor CD82/KAI1 inhibits fibronectin adhesion-induced epithelial-to-mesenchymal transition in prostate cancer cells by repressing the associated integrin signaling. Oncotarget 8(1):1641–1654
Li H, Fredriksson L, Li X, Eriksson U (2003) PDGF-D is a potent transforming and angiogenic growth factor. Oncogene 22(10):1501–1510
Li H, Fan X, Houghton J (2007) Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 101(4):805–815
Li D, Masiero M, Banham AH, Harris AL (2014) The notch ligand JAGGED1 as a target for anti-tumor therapy. Front Oncol 4:254
Lim H, Moon A (2016) Inflammatory fibroblasts in cancer. Arch Pharm Res 39(8):1021–1031
Lin KY, Lu D, Hung CF, Peng S, Huang L, Jie C, Murillo F, Rowley J, Tsai YC, He L, Kim DJ, Jaffee E, Pardoll D, Wu TC (2007a) Ectopic expression of vascular cell adhesion molecule-1 as a new mechanism for tumor immune evasion. Cancer Res 67(4):1832–1841
Lin X, Ogiya M, Takahara M, Yamaguchi W, Furuyama T, Tanaka H, Tohyama M, Inagaki S (2007b) Sema4D-plexin-B1 implicated in regulation of dendritic spine density through RhoA/ROCK pathway. Neurosci Lett 428(1):1–6
Lomberk G (2010) The extracellular matrix and cell migration. Pancreatology 10(1):4–5
Longo N, Yáñez-Mó M, Mittelbrunn M, de la Rosa G, Muñoz ML, Sánchez-Madrid F, Sánchez-Mateos P (2001) Regulatory role of tetraspanin CD9 in tumor-endothelial cell interaction during transendothelial invasion of melanoma cells. Blood 98(13):3717–3726
Lopes-Bastos BM, Jiang WG, Cai J (2016) Tumour-endothelial cell communications: important and indispensable mediators of tumour angiogenesis. Anticancer Res 36(3):1119–1126
Lopez-Dee Z, Pidcock K, Gutierrez LS (2011) Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm 2011:296069
Los M, Roodhart JM, Voest EE (2007) Target practice: lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist. 12(4):443–450
Loukinova E, Dong G, Enamorado-Ayalya I, Thomas GR, Chen Z, Schreiber H, Van Waes C (2000) Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene 19(31):3477–3486
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23(11):549–555
Massihnia D, Galvano A, Fanale D, Perez A, Castiglia M, Incorvaia L, Listì A, Rizzo S, Cicero G, Bazan V, Castorina S, Russo A (2016) Triple negative breast cancer: shedding light onto the role of PI3K/AKT/MTOR pathway. Oncotarget 7(37):60712–60722
McGary EC, Onn A, Mills L, Heimberger A, Eton O, Thomas GW, Shtivelband M, Bar-Eli M (2004) Imatinib mesylate inhibits platelet-derived growth factor receptor phosphorylation of melanoma cells but does not affect tumorigenicity in vivo. J Invest Dermatol 122(2):400–405
Meadows KL, Hurwitz HI (2012) Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med 2(10):a006577
Meng J, Liu Y, Han J, Tan Q, Chen S, Qiao K, Zhou H, Sun T, Yang C (2017) Hsp90β promoted endothelial cell- dependent tumor angiogenesis in hepatocellular carcinoma. Mol Cancer 16(1):72
Mierke CT, Zitterbart DP, Kollmannsberger P, Raupach C, Schlötzer-Schrehardt U, Goecke TW, Behrens J, Fabry B (2008) Breakdown of the endothelial barrier function in tumor cell transmigration. Biophys J 94(7):2832–2846
Miller TW, Isenberg JS, Roberts DD (2009) Molecular regulation of tumor angiogenesis and perfusion via redox signaling. Chem Rev 109(7):3099–3124
Milner LA, Bigas A (1999) Notch as a mediator of cell fate determination in hematopoiesis: evidence and speculation. Blood 93(8):2431–2448
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410(6824):50–56
Murdoch C, Monk PN, Finn A (1999) Cxc chemokine receptor expression on human endothelial cells. Cytokine 11(9):704–712
Murphy-Ullrich JE, Poczatek M (2000) Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev 11(1–2):59–69
Nabeshima K, Inoue T, Shimao Y, Sameshima T (2002) Matrix metalloproteinases in tumor invasion: role for cell migration. Pathol Int 52(4):255–264
Nakahara S, Raz A (2008) Biological modulation by lectins and their ligands in tumor progression and metastasis. Anticancer Agents Med Chem 8(1):22–36
Naoyo N, Hirohisa Y, Takashi N, Toshiharu K, Masamichi K (2006) Angiogenesis in cancer. Vasc Health Risk Manag 2(3):213–219
Nicolson GL (1988) Cancer metastasis: tumor cell and host organ properties important in metastasis to specific secondary sites. Biochim Biophys Acta 948(2):175–224
Nilsson MB, Langley RR, Fidler IJ (2005) Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res 65(23):10794–10800
Niwa Y, Akamatsu H, Niwa H, Sumi H, Ozaki Y, Abe A (2001) Correlation of tissue and plasma RANTES levels with disease course in patients with breast or cervical cancer. Clin Cancer Res 7(2):285–289
Nomura S, Iwata S, Hatano R, Komiya E, Dang NH, Iwao N, Ohnuma K, Morimoto C (2016) Inhibition of VEGF-dependent angiogenesis by the anti-CD82 monoclonal antibody 4F9 through regulation of lipid raft microdomains. Biochem Biophys Res Commun 474(1):111–117
Okuno T, Nakatsuji Y, Moriya M, Takamatsu H, Nojima S, Takegahara N, Toyofuku T, Nakagawa Y, Kang S, Friedel RH, Sakoda S, Kikutani H, Kumanogoh A (2010) Roles of Sema4D-plexin-B1 interactions in the central nervous system for pathogenesis of experimental autoimmune encephalomyelitis. J Immunol 184(3):1499–1506
O’Malley G, Heijltjes M, Houston AM, Rani S, Ritter T, Egan LJ, Ryan AE (2016) Mesenchymal stromal cells (MSCs) and colorectal cancer: a troublesome twosome for the anti-tumour immune response? Oncotarget 7(37):60752–60774
Orr FW, Wang HH, Lafrenie RM, Scherbarth S, Nance DM (2000) Interactions between cancer cells and the endothelium in metastasis. J Pathol 190(3):310–329
Ossowski L (1988) Plasminogen activator dependent pathways in the dissemination of human tumor cells in the chick embryo. Cell 52(3):321–328
Parent CA, Devreotes PN (1999) A cell’s sense of direction. Science 284(5415):765–770
Park JW, Finn RS, Kim JS, Karwal M, Li RK, Ismail F, Thomas M, Harris R, Baudelet C, Walters I, Raoul JL (2011) Phase II, open-label study of brivanib as first-line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res 17(7):1973–1983
Park JJ, Jin YB, Lee YJ, Lee JS, Lee YS, Ko YG, Lee M (2012) KAI1 suppresses HIF-1α and VEGF expression by blocking CDCP1-enhanced Src activation in prostate cancer. BMC Cancer 12:81
Parkes M, Jewell D (2001) Ulcerative colitis and Crohns disease: molecular genetics and clinical implications. Expert Rev Mol Med 2001:1–18
Patenaude A, Fuller M, Chang L, Wong F, Paliouras G, Shaw R, Kyle AH, Umlandt P, Baker JH, Diaz E, Tong J, Minchinton AI, Karsan A (2014) Endothelial-specific Notch blockade inhibits vascular function and tumor growth through an eNOS-dependent mechanism. Cancer Res 74(9):2402–2411
Pauli BU, Augustin-Voss HG, el-Sabban ME, Johnson RC, Hammer DA (1990) Organ-preference of metastasis. Cancer Metastasis Rev 9(3):175–189
Phi JH, Suzuki N, Moon YJ, Park AK, Wang KC, Lee JY, Choi SA, Chong S, Shirane R, Kim SK (2017) Chemokine ligand 5 (CCL5) derived from endothelial colony-forming cells (ECFCs) mediates recruitment of smooth muscle progenitor cells (SPCs) toward critical vascular locations in moyamoya disease. PLoS ONE 12(1):e0169714
Pick AM, Nystrom KK (2012) Pazopanib for the treatment of metastatic renal cell carcinoma. Clin Ther 34(3):511–520
Ploug M, Østergaard S, Gårdsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Danø K (2001) Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry 40(40):12157–12168
Prior BM, Yang HT, Terjung RL (2004) What makes vessels grow with exercise training? J Appl Physiol (1985) 97(3):1119–1128
Quatromoni JG, Eruslanov E (2012) Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res 4(4):376–389
Ragab HM, Afufy M, Samy N, El Maksoud NA, Shaaban HM (2017) Evaluation of serum soluble E-selectin in breast cancer. J App Pharm Sci 7(3):57–61
Raica M, Cimpean AM (2010) Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals (Basel). 3(3):572–599
Rampaul RS, Miremadi A, Pinder SE, Lee A, Ellis IO (2001) Pathological validation and significance of micrometastasis in sentinel nodes in primary breast cancer. Breast Cancer Res 3(2):113–116
Recouvreux MV, Camilletti MA, Rifkin DB, Becu-Villalobos D, Díaz-Torga G (2012) Thrombospondin-1 (TSP-1) analogs ABT-510 and ABT-898 inhibit prolactinoma growth and recover active pituitary transforming growth factor-β1 (TGF-β1). Endocrinology 153(8):3861–3871
Regidor PA, Callies R, Regidor M, Schindler AE (1998) Expression of the cell adhesion molecules ICAM-1 and VCAM-1 in the cytosol of breast cancer tissue, benign breast tissue and corresponding sera. Eur J Gynaecol Oncol 19(4):377–383
Reyes-Reyes ME, George MD, Roberts JD, Akiyama SK (2006) P-selectin activates integrin-mediated colon carcinoma cell adhesion to fibronectin. Exp Cell Res 312(20):4056–4069
Reymond N, d’Água BB, Ridley AJ (2013) Crossing the endothelial barrier during metastasis. Nat Rev Cancer 13(12):858–870
Rhee I (2016) Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res 39(11):1588–1596
Ridiandries A, Tan JT, Bursill CA (2016) The role of CC-chemokines in the regulation of angiogenesis. Int J Mol Sci 17(11):1856
Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR (2003) Cell migration: integrating signals from front to back. Science 302(5651):1704–1709
Rinko LJ, Lawrence MB, Guilford WH (2004) The molecular mechanics of P- and L-selectin lectin domains binding to PSGL-1. Biophys J 89(1 Pt 1):544–554
Ryschich E, Lizdenis P, Ittrich C, Benner A, Stahl S, Hamann A, Schmidt J, Knolle P, Arnold B, Hämmerling GJ, Ganss R (2006) Molecular fingerprinting and autocrine growth regulation of endothelial cells in a murine model of hepatocellular carcinoma. Cancer Res 66(1):198–211
Salcedo R, Resau JH, Halverson D, Hudson EA, Dambach M, Powell D, Wasserman K, Oppenheim JJ (2000) Differential expression and responsiveness of chemokine receptors (CXCR1-3) by human microvascular endothelial cells and umbilical vein endothelial cells. FASEB J 14(13):2055–2064
Sargiannidou I, Zhou J, Tuszynski GP (2001) The role of thrombospondin-1 in tumor progression. Exp Biol Med (Maywood) 226(8):726–733
Sarvaiya PJ, Guo D, Ulasov I, Gabikian P, Lesniak MS (2013) Chemokines in tumor progression and metastasis. Oncotarget 4(12):2171–2185
Schlesinger M, Bendas G (2015) Vascular cell adhesion molecule-1 (VCAM-1)–an increasing insight into its role in tumorigenicity and metastasis. Int J Cancer 136(11):2504–2514
Seguin L, Desgrosellier JS, Weis SM, Cheresh DA (2015) Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol 25(4):234–240
Serru V, Le Naour F, Billard M, Azorsa DO, Lanza F, Boucheix C, Rubinstein E (1999) Selective tetraspan-integrin complexes (CD81/alpha4beta1, CD151/alpha3beta1, CD151/alpha6beta1) under conditions disrupting tetraspan interactions. Biochem J 340(Pt1):103–111
Shandiz SAS, Khosravani M, Mohammadi S, Noorbazargan H, Mirzaie A, Inanlou DN, Jalali MD, Jouzaghkar H, Baghbani-Arani F, Keshavarz-Pakseresht B (2016) Evaluation of imatinib mesylate (Gleevec) on KAI1/CD82 gene expression in breast cancer MCF-7 cells using quantitative real-time PCR. Asian Pac J Trop Biomed 6(2):159–163
Sharma R, Sharma R, Khaket TP, Dutta C, Chakraborty B, Mukherjee TK (2017) Breast cancer metastasis: putative therapeutic role of vascular cell adhesion molecule-1. Cell Oncol (Dordr). 40(3):199–208
Shewach DS, Kuchta RD (2009) Introduction to cancer chemotherapeutics. Chem Rev 109(7):2859–2861
Shieh JC, Schaar BT, Srinivasan K, Brodsky FM, McConnell SK (2011) Endocytosis regulates cell soma translocation and the distribution of adhesion proteins in migrating neurons. PLoS ONE 6(3):e17802
Shin MK, Kim SK, Jung H (2011) Integration of intra- and extravasation in one cell-based microfluidic chip for the study of cancer metastasis. Lab Chip 11(22):3880–3887
Singh R, Bhatt ML, Singh SP, Kumar V, Goel MM, Mishra DP, Srivastava K, Kumar R (2016) Expression levels of tetraspanin KAI1/CD82 in breast cancers in North Indian females. Asian Pac J Cancer Prev 17(7):3431–3436
Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M (2009) CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene 28(1):85–94
Song P, Hai Y, Wang X, Zhao L, Chen B, Cui P, Xie Q, Yu L, Li Y, Wu Z, Li H (2018) Realgar transforming solution suppresses angiogenesis and tumor growth by inhibiting VEGF receptor 2 signaling in vein endothelial cells. Arch Pharm Res 41(4):467–480
Soon LL (2007) A discourse on cancer cell chemotaxis: where to from here? IUBMB Life 59(2):60–67
Soto MS, Serres S, Anthony DC, Sibson NR (2014) Functional role of endothelial adhesion molecules in the early stages of brain metastasis. Neuro Oncol 16(4):540–551
Sounni NE, Noel A (2013) Targeting the tumor microenvironment for cancer therapy. Clin Chem 59(1):85–93
Spring H, Schüler T, Arnold B, Hämmerling GJ, Ganss R (2005) Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci USA 102(50):18111–18116
Stein EV, Miller TW, Ivins-O’Keefe K, Kaur S, Roberts DD (2016) Secreted thrombospondin-1 regulates macrophage interleukin-1β production and activation through CD47. Sci Rep 6:19684
Stenina-Adognravi O (2014) Invoking the power of thrombospondins: regulation of thrombospondins expression. Matrix Biol 37:69–82
Stepanova V, Jayaraman PS, Zaitsev SV, Lebedeva T, Bdeir K, Kershaw R, Holman KR, Parfyonova YV, Semina EV, Beloglazova IB, Tkachuk VA, Cines DB (2016) Urokinase-type plasminogen activator (uPA) promotes angiogenesis by attenuating prolince-rich homeodomain protein (PRH) transcription factor activity and de-repressing vascular endothelial growth factor (VEGF) receptor expression. J Biol Chem 291(29):15029–15045
Sternberg CN, Hawkins RE, Wagstaff J, Salman P, Mardiak J, Barrios CH, Zarba JJ, Gladkov OA, Lee E, Szczylik C, McCann L, Rubin SD, Chen M, Davis ID (2013) A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: final overall survival results and safety update. Eur J Cancer 49(6):1287–1296
Sunami E, Tsuno N, Osada T, Saito S, Kitayama J, Tomozawa S, Tsuruo T, Shibata Y, Muto T, Nagawa H (2000) MMP-1 is a prognostic marker for hematogenous metastasis of colorectal cancer. Oncologist 5(2):108–114
Tahmasebi MB, Carloni V (2017) Tumor microenvironment, a paradigm in hepatocellular carcinoma progression and therapy. Int J Mol Sci 18(2):405
Takada A, Ohmori K, Takahashi N, Tsuyuoka K, Yago A, Zenita K, Hasegawa A, Kannagi R (1991) Adhesion of human cancer cells to vascular endothelium mediated by a carbohydrate antigen, sialyl Lewis A. Biochem Biophys Res Commun 179(2):713–719
Tan DS, Agarwal R, Kaye SB (2006) Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol 7(11):925–934
Tang DG, Honn KV (1994) 12-Lipoxygenase, 12(S)-HETE, and cancer metastasis. Ann N Y Acad Sci 744:19–215
Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, Hu GF (2008) Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res 68(24):10377–10386
Upreti M, Jyoti A, Sethi P (2013) Tumor microenvironment and nanotherapeutics. Transl Cancer Res 2(4):309–319
Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147(2):275–292
van Zijl F, Krupitza G, Mikulits W (2011) Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res 728(1–2):23–34
Varma V, Yao-Borengasser A, Bodles AM, Rasouli N, Phanavanh B, Nolen GT, Kern EM, Nagarajan R, Spencer HJ 3rd, Lee MJ, Fried SK, McGehee RE Jr, Peterson CA, Kern PA (2008) Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance. Diabetes 57(2):432–439
Vasudev NS, Reynolds AR (2014) Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis 17(3):471–494
Vences-Catalán F, Rajapaksa R, Srivastava MK, Marabelle A, Kuo CC, Levy R, Levy S (2015) Tetraspanin CD81 promotes tumor growth and metastasis by modulating the functions of T regulatory and myeloid-derived suppressor cells. Cancer Res 75(21):4517–4526
Verweij J, Sleijfer S (2013) Pazopanib, a new therapy for metastatic soft tissue sarcoma. Expert Opin Pharmacother 14(7):929–935
Wang YH, Dong YY, Wang WM, Xie XY, Wang ZM, Chen RX, Chen J, Gao DM, Cui JF, Ren ZG (2013) Vascular endothelial cells facilitated HCC invasion and metastasis through the Akt and NF-κB pathways induced by paracrine cytokines. J Exp Clin Cancer Res 32(1):51
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C (2017) Role of tumor microenvironment in tumorigenesis. J Cancer 8(5):761–773
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M (2008) Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 7(10):3129–3140
Witz IP (2009) The tumor microenvironment: the making of a paradigm. Cancer Microenviron 1:9–17
Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan CA, Hymowitz SG, Siebel CW (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464(7291):1052–1057
Wyckoff JB, Jones JG, Condeelis JS, Segall JE (2000) A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res 60(9):2504–2511
Xing F, Saidou J, Watabe K (2010) Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 15:166–179
Yang L, Zhang Y (2017) Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol 10(1):58
Yoshikawa D, Ojima H, Kokubu A, Ochiya T, Kasai S, Hirohashi S, Shibata T (2009) Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as a novel molecular-targeted therapy against cholangiocarcinoma. Br J Cancer 100(8):1257–1266
Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM (2014) Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer 110(3):724–732
Yu B, Wong MM, Potter CM, Simpson RM, Karamariti E, Zhang Z, Zeng L, Warren D, Hu Y, Wang W, Xu Q (2016) Vascular stem/progenitor cell migration induced by smooth muscle cell-derived chemokine (C-C motif) ligand 2 and chemokine (C-X-C motif) ligand 1 contributes to neointima formation. Stem Cells 34(9):2368–2380
Zavadil J, Cermak L, Soto-Nieves N, Böttinger EP (2004) Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J 23(5):1155–1165
Zen K, Liu DQ, Guo YL, Wang C, Shan J, Fang M, Zhang CY, Liu Y (2008) CD44v4 is a major E-selectin ligand that mediates breast cancer cell transendothelial migration. PLoS ONE 3(3):e1826
Zhang J, Lu Y, Pienta KJ (2010) Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst 102(8):522–528
Zhang X, Qin Y, Li H, Bai C, Zhu T, Xu J, Wu C, Wu M, Wang C, Song H, Wei L, He J (2011) Efficacy and safety of vandetanib, a dual VEGFR and EGFR inhibitor, in advanced non-small-cell lung cancer: a systematic review and meta-analysis. Asian Pac J Cancer Prev 12(11):2857–2863
Zhao Y, Adjei AA (2015) Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist 20(6):660–673
Zhou H, Wu J, Wang T, Zhang X, Liu D (2016) CXCL10/CXCL3 axis promotes the invasion of gastric cancer via PI3 K/AKT pathwath-dependent MMPs production. Biomed Pharmacother 82:479–488
Zivi A, Cerbone L, Recine F, Sternberg CN (2012) Safety and tolerability of pazopanib in the treatment of renal cell carcinoma. Expert Opin Drug Saf 11(5):851–859
Acknowledgements
This study was supported by the Duksung Women’s University Research Grant 2017.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Choi, H., Moon, A. Crosstalk between cancer cells and endothelial cells: implications for tumor progression and intervention. Arch. Pharm. Res. 41, 711–724 (2018). https://doi.org/10.1007/s12272-018-1051-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-018-1051-1