
Abstract
Angiogenesis is a critical process required for tumor progression. Newly formed blood vessels provide nutrition and oxygen to the tumor contributing to its growth and development. However, endothelium also plays other functions that promote tumor metastasis. It is involved in intravasation, which allows invasive cancer cells to translocate into the blood vessel lumen. This phenomenon is an important stage for cancer metastasis. Besides direct association with cancer development, endothelial cells are one of the main sources of cancer-associated fibroblasts (CAFs). The heterogeneous group of CAFs is the main inductor of migration and invasion abilities of cancer cells. Therefore, the endothelium is also indirectly responsible for metastasis. Considering the above, the endothelium is one of the important targets of anticancer therapy. In the chapter, we will present mechanisms regulating endothelial function, dependent on cancer and cancer niche cells. We will focus on possibilities of suppressing pro-metastatic endothelial functions, applied in anti-cancer therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Endothelial cells
- Cancer development
- Tumor endothelial cells
- Cancer microenvironment
- Cancer niche
- Sprouting
- Metastasis
- CAFs
- Microvessels
- Tip cells
- Tumor angiogenesis
- VEGF
- Hypoxia
- Endothelial-mesenchymal transition
- TGF-β
6.1 Introduction
The vascular endothelium is a versatile structure that separates the circulating blood from tissues. Moreover, apart from regulation and maintenance of blood fluidity, it plays multifunctional roles in the delivery of water and nutrient, maintenance of metabolic homeostasis, trafficking of immune cells, activation of innate and acquired immune responses, as well as angiogenesis [30, 73]. The endothelium is a thin monolayer, composed of endothelial cells (ECs) that are able to organize the growth and development of connective tissue cells, forming the surrounding layers of the blood vessel wall. This process is controlled by a paracrine/endocrine network which involves fibrinolytic, pro- and anticoagulants, vasoactive, pro- and anti-inflammatory factors, as well as growth factors produced by ECs [84]. Thus, ECs must be constantly poised to sense and respond to changes within their environment. In tumor and its microenvironment, some agents like hypoxia and chronic growth factor stimulation might lead to endothelial dysfunction. There is more and more evidence showing that these abnormalities contribute to cancer progression.
The tumor has been recently described as an aberrant organ not only composed of cancer cells but also of numerous stromal, inflammatory, and vascular cells. Like other organs, in order to develop, the tumor requires a blood supply to provide nutrients and oxygen and waste removal. Initially, cancer cells might adopt tissue-resident vessels. However, the tumor eventually recruits its own vascular supply through the angiogenesis process [123]. The tumor-associated angiogenesis has been defined as sprouting of new vessels from preexisting vessels, which involves endothelial cells [112]. Tumor modulates its microenvironment by releasing numerous cytokines, chemokines, and growth factors to activate normal, quiescent endothelial cells and adapt them to the angiogenic response. Moreover, surrounding stromal cells might also secrete a plethora of factors and cytokines influencing tumorigenesis and metastasis. Within them, TGF-β is considered one of the main factors modulating interactions between cancer and surrounding cells, located within the tumor niche. Among the TGF-β-dependent effects is regulation of cancer cell proliferation, affecting immune response by suppressing immune cells function, conversion of fibroblasts to myofibroblasts and epithelial-mesenchymal transition (EMT). Furthermore, TGF-β promotes the formation of cancer-associated fibroblasts (CAFs), a specialized group of fibroblasts involved in tumor growth and invasion of cancer cells by modulation of the tumor niche [119]. Until now normal fibroblasts (NFs) have been considered the main source of CAFs, but in the last years, endothelial cells have also become an important origin of CAFs. It has been shown that TGF-β is responsible for such EC conversion in a process called endothelial-mesenchymal transition (EndMT) [56]. During EndMT, endothelial cells lose endothelial markers and gain mesenchymal ones, which is followed by increased expression of transcription factors such as Snail and Slug. The changes are accompanied by defaulting of their cellular function and taking on some characteristics of mesenchymal cells, including loss ability to form capillary tubes and cell-cell junctions, increased cell migration properties, and secretion of extracellular matrix proteins.
In this review, we will focus on the role of endothelial cells in tumor microenvironment particularly on their direct and indirect role in cancer metastasis. While endothelial cells were originally believed to be involved in the direct development of primary tumor due to vascularization, there is more and more evidence suggesting their indirect effect on cancer progression. CAFs are known to play an important role in tumor growth and progression via secretion of various growth factors and chemokines. The contribution of endothelial cells in CAF formation will be discussed. Finally, we will also present current and future therapeutic possibilities targeting at endothelial cells, CAF formation, and chemokines in the context of anti-metastatic treatment.
6.2 Heterogeneity of Normal and Tumor Endothelial Cells
The vascular endothelium is a specific inner cellular lining that separates the circulating blood from the tissues. That thin monolayer plays an important multifunctional property, including the control of vasomotor tone, proliferation/angiogenesis, permeability, hemostasis, humidification, thermoregulation, leukocyte transmigration, sieve function, and scavenging innate and adaptive immunity [2]. This plethora of functions is a consequence of the fact that ECs, being part of the vascular tree, are differentially regulated in space and time. Thus, ECs differ in various organs, but also between distinct segments within or between neighboring of vascular architecture of the same organ. The EC thickness varies across the vascular tree, ranging from less than 0.1 μm in capillaries and veins to 1 μm in the aorta [2]. Endothelial cells are usually flat, but they might be plump or cuboidal occasionally [2]. Endothelium cells in monolayer are held by two main types of junctions: adherent junctions (AJs) and tight junctions (TJs). Their organization varies along the vascular tree [12]. For instance, the large artery is rich in TJs, whereas venules display less organized TJs. Similarly, in the brain, where protection of the nervous system is required, junctions are well developed and rich in TJs [31]. In contrast, post-capillary venules have a poorly organized TJs due to the dynamic trafficking of circulating cells and proteins suspended in plasma [31]. Another feature of endothelium diversity is its continuity. Continuous endothelium might be fenestrated or non-fenestrated. Fenestrated continuous endothelium is found in the places where increased filtration or increased transendothelial transport is needed, like capillaries of exocrine and endocrine glands. Non-fenestrated continuous endothelium is found in capillaries, veins, and arteries. Discontinuous endothelium occurs in some sinusoidal vascular beds, first of all in the liver [2]. It has been proposed that angiogenesis, being one of the main processes engaging endothelial cells, requires at least a few cells of discontinuous. ECs, called tip cells, are directly engaged in vessel sprouting. Highly proliferative stalk cells follow tip cells, and phalanx cells that are involved in improving the perfusion and oxygenation of newly formed blood vessels [51].
Mentioned ECs heterogeneity is provided mainly by one of two distinct mechanisms based on microenvironment pressure or epigenetic modulation [3]. Endothelium is not only a specific inner cellular lining separating the circulating blood from the tissues, but it is exposed to a great variety of factors, secreted by tissue microenvironments. Moreover, to properly perform its functions across the vascular tree, ECs have to detect and respond to environmental stimuli, which is guaranteed by endothelial cells heterogeneity. This mechanism is reversible when ECs are removed from their microenvironment and grow in tissue culture. The second mechanism involved posttranscriptional modification that seemed to be epigenetically programmed and independent of extracellular signals. Although it is widely accepted that microenvironment stimulation is responsible for triggering epigenetic modifications, they may remain during the removal of the signals and be transmitted during mitosis [3].
It should be noted that EC heterogeneity also translates into the heterogeneity of tumor endothelium. In line with Folkman’s hypothesis, tumor growth strictly depends on blood vessels [41]. At the same time, tumor blood vessels are formed by ECs recruited from surrounding tissue transformed to tumor endothelial cells (TECs). The tumor vasculature, in contrast to well-differentiated normal vessels, it is composed of a chaotic mixture of abnormal, disorganized artery–capillary–vein hierarchy vessels [109]. Unlike normal blood vessels, tumor vessels are more dilated and tortuous. They branch irregularly, have chaotic flow patterns, and increased permeability to macromolecules [75]. Due to an imbalance between pro- and antiangiogenic factors and with a predominance of stimulators (angiogenic switch), a classic hierarchical branching pattern system of arterioles, veins, and capillaries is disturbed. The layout of neoplastic capillaries is morphologically immature: chaotic, strongly twisted, with variable vessel diameter and irregular edge [29]. In line to the unsettle tumor vasculature, endothelial cells, forming tumor vessels, are structurally abnormal. TECs have a disturbed redistribution of phospholipids, a discontinuous or absent basement membrane, increased fenestrations and extended intercellular junctions, and a high proliferative rate compared to normal ECs and tend to grow one on top of the other and invade into the vessel lumen [3]. Phenotypic changes, accompanied by changes at the molecular levels, have been identified comparing normal ECs to TECs, isolated from normal and tumor tissues. In 2000, St. Croix et al. performed a comparative analysis of gene expression profiles between tumor endothelial cells and normal endothelial cells and identified the specific genes for TEC called tumor endothelial markers (TEMs) [95]. Since then, several studies have been published on molecular differences between TECs and NECs [15, 66, 77] e.g tumor endothelial markers (TEMs), endoglin (CD105), or endothelial protein-disulfide isomerase EndoPDI [50] has been also demonstrated that TECs can secrete several factors that affect their survival in an autocrine manner [17, 18, 74, 101].
Increased permeability of the walls, hemorrhage, and plasma leakage result from a reduced number of pericytes and increased proteolytic activity within the vessel formation zone. TECs are characterized not only by an increased size, but they also presented aneuploidy, abnormal centrosomes, and high activation of the MAPK pathway, promoting cell survival [5, 43]. TECs exhibit several differences which contribute to their proangiogenic phenotype, including changed responsiveness to growth factors such as EGF, adrenomedullin, and VEGF. VEGF stimulates the migration of TECs and enhances their survival in an autocrine manner, which leads to the antiapoptotic phenotype of TECs [51]. TECs show upregulated aldehyde dehydrogenase (ALDH) expression which is manifested by a formation of increased tube number even under starvation conditions [80].
It is suggested that the persisting hypoxia together with the secretion of cytokines promotes tumor angiogenesis by inducing the mobilization of bone marrow-derived endothelial progenitor cells to cancer [45]. Glioblastoma cells and lymphoma ones are examples of tumor cells that are capable of differentiating into TECs [98, 106]. Interaction between tumor cells and the microenvironment leads to alteration of ECs into TECs that express high levels of biglycan through epigenetic modifications, which stimulates tumor cells to metastasize through activation of different signaling pathways [67]. Furthermore, it was reported that endothelial progenitor cells release microvesicles with gene fragments that can activate endothelial cell angiogenic properties [33]. Due to the mechanisms mentioned above, TECs become cytogenetically abnormal and unstable in the tumor microenvironment.
6.3 Angiogenesis in Tumor Development
Efficient functioning of the circulatory system, responsible for gas exchange, transport of nutrients, and metabolic products, is the basic condition for appropriate development during ontogeny. In embryo development, de novo formation of the vascular plexus from angioblasts (EPCs; endothelial precursor cells) is one of the earliest organogenesis processes, called vasculogenesis [1]. Next, the existing vascular network undergoes proliferation, reorganization, and maturation in the process of angiogenesis (neovascularization) [11]. A new capillary mesh network is created by sprouting of endothelial cells. The last stage is the maturation of the vessel through the migration of pericytes and vascular smooth muscle cells (VSMCs) on a newly formed basal membrane (BM). Under physiological conditions, neovascularization occurs during embryo implantation, the women’s monthly cycle, and wound healing, and in the muscles [16]. In pathological conditions, when the activity between pro-angiogenic factors and antiangiogenic ones is disturbed, it occurs during chronic inflammation and hypoxia and in asthma, rheumatoid arthritis, psoriasis, Crohn’s disease, diabetic retinopathy, as well as endometriosis and obesity. However, angiogenesis plays the most significant role in the process of neoplasia [65].
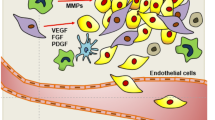
In the initial stage, in order to survive and proliferate, tumor takes oxygen and nutrients by diffusion. The environment in which it develops undergoes hypoxia and acidification as a result of excess metabolic products. When its volume exceeds 1–2 mm3, the tumor must become angiogenic and recruit their vasculature to grow. Cancer cells, together with host/niche cells , stimulate the development of their blood vessels, using various mechanisms of tumor angiogenesis [36]. The most common one and best described is vessel sprouting (Fig. 6.1). In the classical model, the vasodilatation of the mother vessel occurs, which contributes to reduced BM density. It leads to partial degradation of BM and protrusion of endothelial cells in that place. As the ECs do not lose intracellular connection with each other and they migrate parallelly, the polarity of the cells is preserved. At the same time, the new lumen is formed by polarized ECs. They release proteins which rebuild the basal membrane along which pericytes migrate. This phenomenon stabilizes the capillary and contributes to its maturation [83]. The last step of vessels maturation described above is impaired during cancer angiogenesis. Hypoxia-induced factor-1α (HIF-1α) is the main factor that initiates sprouting [69]. It induces secretion by ECs proangiogenic factors such as platelet-derived growth factor, type B (PDGF-B), hepatocyte growth factor (HGF), angiopoietins, epidermal growth factor (EGF), placental growth factor (PlGF) [29, 65] and is the main stimulator of angiogenesis vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) [94]. VEGF, which works in an auto- and paracrine manner, contributes to extravasations of plasma proteins, e.g., fibrinogen, which initiates integrin-dependent migration of ECs, a release of metalloproteinases, and activation of the mitogen-activated protein kinase (MAPK) pathway. Digestion by MMPs of extracellular matrix (ECM) releases the tumor growth factor (TGF-β), basic fibroblast growth factor (FGF-2), and insulin-like growth factor-1 (IGF-1), i.e., antiapoptotic factors, activating the survival signal transduction pathway [27, 47, 52]. The second group consists of tissue-resident cells, including normal tissue epithelial cells, vascular cells (endothelium and pericytes), normal fibroblasts, adipocytes, and leukocytes (mast cells and macrophages) [20, 22, 34, 92]. It has been confirmed that leukocytes as well as ECs are important sources of VEGF-A, which is able to accelerate tumor angiogenesis [35]. But TASCs might increase vascular density in human tumors through secretion of other numerous chemokines and growth factors (Fig. 6.1).

Mechanisms of tumor vascularization. At the point when developing cancer reaches its size 1–2 mm, hypoxia and nutrient deprivation result in release of tumor cell-soluble growth factors, chemokines, and cytokines (VEGF (blue star), PDGF (triangle), FGF (square), angiopoietins (diamond), and SD1a (cross)). The factors induce the sprouting and proliferation of endothelial cells on nearby blood microvessels. The created tumor blood vessels are leaky and tortuous with partially exposed basal lamina where vascular leaks are observed. Additionally, the vascular remodeling is also enhanced by factor secreted by cancer-associated fibroblasts (CAFs) that are recruited to the tumor niche. CAFs cause the rearrangement the profile of extracellular matrix protein and release matrix metalloproteinases (MMPs: MMP-2 (red star), MMP-9 (triangle), and MT1-MMP (square)) that cleave and remodel ECM therefore activating the endogenous angiogenesis inhibitors such as tumstatin and endostatin
Cancer stem cells (CSCs) can differentiate to endothelial cells and, as a consequence, induce new vessels via a phenomenon known as vascular mimicry. However, that ability does not lead to the form of mature and proper blood vessels which would counteract hypoxia. During progression, cancer recruits numerous types of cells to the cancer niche, which can modulate tumor vascularization. The cells located in the tumor microenvironment, called tumor-associated stromal cells (TASCs), can be divided into two main groups. Leukocytes (lymphocytes, neutrophils, monocytes, and macrophages) infiltrating tumor constitute the first group delivered from the bone marrow via systemic circulation. Macrophages that are recruited to the tumor environments, called TAMs (tumor-associated macrophages) , have been described as a source of non-thrombogenic EC-like surfaces, constituting a potential scaffolding for tumor vascularization through mimicry vasculare [89]. However, the mechanism of that process is still unknown.
Tumor cells play a crucial role in initiation and regulation of cancer angiogenesis. It must be noted that, other cells, located in the tumor niche, also secrete numerous signaling molecules and induce pathways that influence the angiogenic response. Apart from sprouting new vessels in response to VEGF stimulation, blood vessels might also originate from cells of the bone marrow or tumor stem cells dedifferentiated to ECs (vascular mimicry). A wide diversity of molecular pathways which are able to induce tumor vascularization can make antiangiogenic therapies ineffective [96].
Tumor endothelial cells may undergo endothelial to mesenchymal transition (EndMT) and become carcinoma-associated fibroblasts, CAFs. It was demonstrated that stromal-derived factor-1 (SDF-1) in CAFs recruits EPCs promoting angiogenesis. Overexpression of MMP-2 by CAFs stimulates epithelial hyperplasia and abnormal branching in the mammary gland. It was shown that high level of MMP-2 production in stromal cells is required to support pathological neoangiogenesis of gliomas. Neovascularization is promoted also by induction of IL-8 secretion by CAFs, isolated from metastatic colon cancer patients [117]. CAFs express a membrane-bound serine protease, called fibroblast activation protein (FAP), which is associated with poor prognosis in several cancer types.
Significant associations were found between tumor angiogenesis and miRNAs in activated endothelial cells. miRNAs have opposing effects on cancer and endothelial cells. Their overexpression inhibits angiogenesis and enhances proliferation of cancer cells. MicroRNA-126 (miR-126) is an endothelial-specific miRNA that regulates angiogenic signaling and vascular integrity as a negative regulator of VEGF-A. However, it was observed that overexpression of miR-126 in endothelial cells enhances VEGF-A activity and promotes vessel formation by repressing the expression of sprouty-related protein-1 (Spred-1) [105]. In oral squamous cell carcinoma, a low miR-126 expression is correlated with tumor progression through the activation of angiogenesis and lymphangiogenesis via VEGF-A pathway [91]. miR-126 is involved in cancer cell–stromal cell crosstalk. CAFs induces downregulation of miR-126 in adjacent human umbilical endothelial cells (HUVEC). The lowered miR-126 confers increased tube formation in the early invasive stage of cervical cancer [53]. VEGFR2 can be targeted by miR-221 and miR-222 [55].
6.4 Intravasation of Cancer Cells
Metastasis is a multi-step process, divided into two main phases: (1) translocation of cancer cells from the primary tumor to distant tissues and (2) colonization of these cancer cells at the secondary site [48]. Here we focused on the role of the endothelium in the first phase. The tumor metastatic potential is dependent on its rapid extravasation into the vascular system [13, 61, 82]. That process is composed of several steps: adhesion of invading cancer cells to ECs, changes in the endothelial barrier and intravasation, dissemination into the bloodstream as migrated and proliferated circulating tumor cells (CTCs) , and finally, after extravasation, colonization of other organs [6, 59, 61, 97]. Transendothelial migration (TEM) of invasive cancer is a critical phenomenon in the intra- and extravasation. During that phenomenon, tumor cells migrate between two endothelial cells [61, 93]. In vitro studies suggest that tumor cells might also pass through individual endothelial cells, in a process called transcellular migration [58, 99]. An interaction between transmigrated cancer cells and ECs induces contraction and disruption of their cell-cell contacts as well as secretion of pro-inflammatory factors by the latter [103]. Metastatic microenvironment is also characterized by platelet aggregation and formed microthrombi which promote ECs activation through induced inflammation [100]. As described above, blood vessels arising during cancer progression [49] are usually immature without proper junctional contact between ECs. The blood vessels are leaky and vulnerable due to abnormal pericyte coverage. Those injuries enable cancer cells to intravasate through the blood barrier [37, 116]. According to a favorable theory, both intravasation and extravasation are active processes, regulated by several factors such as TGF-β [7], VEGF [38, 60, 86], angiopoietin-2 (Angpt2) [88], stromal-derived factor-1α (SDF-1α) [118], or TNF [121]. A notable difference between intravasation and extravasation is found in the fact that intravasation mostly involves abnormal tumor vasculature whereas extravasation targets at normal blood vessels. It has been observed that the interaction between tumor cells and ECs is modulated by VEGF or TNF favor intravasation. The process is modulated when the number of blood microvessels increases and disruption of the blood barrier occurs [Fig. 6.2]. Additionally, presence of macrophages seems to be necessary for this process. However, macrophage-secreted TNF increases endothelial permeability, but its depletion does not reduce intravasation. The authors suggested the importance of other macrophage-secreted factors (probably IL-6) or juxtacrine interactions in induction of intravasation. They also prove the importance of remodeling of the endothelial barrier, induced by tumor-endothelial interaction for translocation of tumor cells via the blood barrier [121].

Molecular pathways that regulate the intravasation. Cancer cells enter the circulation by transmigrating either paracellularly through the endothelial cell (EC) junctions or transcellularly through the EC body. Matrix metalloproteinase 1 (MMP-1) is crucial for paracellular intravasation in regions where protease-activated receptor 1 (PAR1) on ECs mediates the remodeling of endothelial junctions (a). Cancer cells can use Notch receptors to bind to Notch ligands on ECs and thereby transmigrate through the endothelial junctions (a). Alternatively, a vascular endothelial cadherin (VE-cadherin) and angiopoietin-1 receptor (TIE2) are cleavage by metalloproteinase-12 (ADAM12), which leads to disruption of endothelial junctions (b). Cancer cells moving to blood vessels are also promoted by tumor-associated macrophages (b) by secreting epidermal growth factor (EGF). Retraction of endothelial junctions, that facilitate cancer cell transendothelial migration (TEM), might be induced by transforming growth factor β1 (TGFβ1) secreted by cancer cells (c). That process can be stimulated by macrophage-secreted tumor necrosis factor 1α (TNF1α) as well. Transcellular intravasation is observed in sites of cancer cell attachment. There complexes of Ca2+ –calmodulin induce phosphorylation of myosin light chain (MLC) by myosin light chain kinase (MLCK) causing actomyosin contraction. Finally, that pathway results in creation of transitory pore-like structure enable cancer cell to cross the EC barrier
Some data suggested that endothelial-mesenchymal transition , leading to disruption of the cell-cell junction between ECs and disruption of blood barrier, also contributes and facilitates cancer cell intravasation.
6.5 CAF Formation
Endothelial cells forming a single-cell layer lining the inner surface of the blood vessels [55] are characterized by wide plasticity [23]. During cancer progression, the endothelium that undergoes endothelial-mesenchymal transition (EndMT) is becoming, besides normal fibroblasts (NFs), one of the main sources of cancer-associated fibroblasts (CAFs). It has been postulated that about 40% of CAFs are formed from endothelial cells [56].
During EndMT, cells lose cell-cell connections, detach from the cell layer, and elongate. Additionally, their adhesion ability is decreased, and migration properties increased (Fig. 6.3). Those behavioral modulations are accompanied by decreased endothelial marker levels, such as CD31 (platelet endothelial cell adhesion molecule-1 (PECAM-1)) or claudin, and gain of mesenchymal markers, such as fibroblast-specific protein 1 (FSP1; S100A4) or α-smooth muscle actin (αSMA) [9, 10, 76, 85, 120]. CAFs are also characterized by increased expression of contraction proteins like caldesmon and tropomyosin [26, 124].

Endothelial-mesenchymal transition . During the tumor progression, the tumor cells secrete TGF-β (a), which affects EndMT. TGF-β by stimulation the located in cell membrane TGF receptors (activated after phosphorylation (P) TGFβRI (I) and constitutively active TGFβRII (II)) leads to the activation of Smad proteins, which translocate to the nucleus (b). The Smad proteins induce expression of Snail and Slug, TWIST, and ZEB-1 transcription regulators. These transcription modulators cause an increase in the expression of mesenchymal markers and numerous cytoskeletal proteins, leading to the elongation of the endothelial cell and induced its invasive character (as described in the text)
The best-known inductors of EndMT belong to the transforming growth factor superfamily (TGF-β) which includes TGF-β1, TGF-β2, and bone morphogenetic protein (BMP). Activation of receptors for these factors leads to an induction of Smad-dependent and Smad-independent pathways [40, 102, 71]. In the canonical pathway, TGF-β1 or TGF-β2 binds to constitutively activate II TGF-β receptor (TGF-βRII) and then to recruit and activate I TGF-β receptor (TGF-βRI) via its phosphorylation [90]. The last of the receptors binds and phosphorylates Smad2/3 which made a complex with Smad4. The created transcription complex moves to the nucleus and triggers the expression of numerous genes which are specific for EndMT [40, 70] such as NOTCH1, TWIST1, and SNAI1/2 [102]. The in vitro observation was confirmed during in vivo analysis on mouse models. The knockdown and knockout of several TGF-β signaling-related genes, such as SMAD2, SMAD3, and TGFBR2, prevented EndMT [28, 115].
TGF-β signaling might be induced indirectly by caveolin-1 (CAV1), Wnt pathway, and endothelin-1 (ET-1). CAV1, located in caveolae, is involved in the internalization of TGF-β receptors [32]. It has been shown that its expression is upregulated during cancer progression. In vivo studies demonstrated that lack of CAV1 induced spontaneous EndMT in mice model. Additionally, TGF-β might accelerate the process [62]. Wnt proteins are involved in EndMT by Smad-dependent TGF-β signaling. They can modulate the phenomenon through canonical (i.e., involving β-catenin) and non-canonical Wnt signaling pathways [4, 63, 107]. Although numerous studies demonstrated that Notch signaling work together with TGF-β pathway [21, 42, 79, 107], it has been shown they act independently in development of Kaposi’s sarcoma-associated herpes virus [46]. It has been recently revealed that ET-1, which is an endogenous vasoconstrictor polypeptide, might alone or together with TGF-β cause EndMT in human ECs [25, 113, 114].
Cellular elongation and acquisition of migration ability observed during EndMT correlate with cytoskeleton remodeling. The alterations concern to all types of cellular filaments such as microfilaments, microtubules, and intermediate filaments. EndMT is characterized by a gain of vimentin expression, which was described as a marker of mesenchymal cells. The regulation of actin cytoskeleton is controlled by proteins belonging to the Rho GTPase family (RhoA, RhoB, Rac-1, cdc42) whose activity is regulated by TGF-β signaling. Activation of small G-proteins causes incorporation of globular actin proteins (G-actin) into filaments of F-actin. This process is critical to forming the stress fibers and results in an increased contraction ability of CAFs [87]. The G-actin pool is released from cytosolic complexes with MRTFs (MRTF-A and MRTF-B) which in “actin-free stage” translocate to the nucleus. MRTFs are the well-described coactivators of serum response factor (SRF) which regulate expression of cytoskeleton regulators and focal adhesion protein such as FAK, vinculin, and α-SMA, necessary for shaping the mesenchymal and contractile nature of CAFs [81]. In our studies, we found that activation of MRTFs are dependent on RhoA and Rac-1/MMP-9 and finally induce ILK and vinculin expression [26]. That axis regulates generation and maturation of focal adhesion, which is characterized by accelerated cell movement, typical for EndMT.
Microtubules , the largest cytoskeleton fibers are involved in the translocation of newly expressed mesenchymal markers, one of which is N-cadherin [68]. It has been proposed that the alteration of β-tubulin subunit expression modulates microtubule dynamics [44]. We revealed that upregulation of tubulins β-3 and β-4 levels, during EndMT, is critical for faster CAFs movement [110, 111].
6.6 Perspective: Endothelium as the Therapeutic Agent in Anticancer Therapy
6.6.1 Antiangiogenesis [AA] Therapies
A variety of signaling molecules such as VEGF-VEGFRs , ephrin-Eph receptors, angiopoietin-Tie, and the Delta-Notch play important roles in angiogenesis. These vascular endothelial growth factors and their receptors regulate both vasculogenesis and pathological angiogenesis. The VEGF family members, i.e., VEGF-A/VEGF-B/VEGF-E and PlGF, regulate angiogenesis and vascular permeability by activating receptors VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk1 in mice). VEGF-C/VEGF-D and their receptor VEGFR-3 (Flt-4) are mainly observed in lymphangiogenesis. VEGFR-2 is a major signal transducer for neovascularization by the activation of the MAPK signaling pathway. VEGF-A, which demonstrates a variety of functions, including proangiogenic and vascular permeability activity, is the main player. Due to this fact, the VEGF-VEGFR system is an important target for antiangiogenic therapy in cancer progression [94, 104]. Currently, there are four main approaches targeting at cancer angiogenesis tested in clinical trials and approved for clinical practice: (1) neutralizing monoclonal antibody that binds circulating VEGF; (2) recombinant protein (decoy receptor or VEGF-Trap) that binds more than one proangiogenic growth factor; (3) small-molecule tyrosine kinase inhibitors that block tyrosine kinase activity of VEGFRs; and (4) therapeutic monoclonal antibodies targeting VEGFR-2 [72, 78].
One of the first antiangiogenic therapies was a therapy with a humanized monoclonal antibody, neutralizing circulating VEGF-A, i.e., Bevacizumab (Avastin®, Roche/Genentech). The first phase III trial results showed that Bevacizumab combined with chemotherapy in metastatic colorectal cancer (MCRC) improved progression-free survival (PFS) (10.6 vs. 6.2 months) and overall survival (OS) (23 vs. 15.3 months) compared to chemotherapy [54]. Aflibercept (Zaltrap ®, Sanofi Genzyme) is a human recombinant fusion protein that acts as a decoy receptor of VEGF-A, VEGF-B, and PlGF. Aflibercept treatment was approved in MCRC with infusional fluorouracil, leucovorin, and irinotecan [24]. Tyrosine kinase inhibitors (TKIs) are small-molecular-weight drugs that inhibit the kinase activity of different receptors and their downstream signaling. Sorafenib (Nexavar®, Bayer/Onyx) or Sunitinib (Sutent®, Pfizer) target not only at VEGFR but other kinases such as PDGFR and FGFR [78]. Ramucirumab (Cyramza® Eli Lilly) is a human monoclonal antibody that inhibits angiogenesis by blocking binding VEGF to the extracellular domain of VEGFR2. It is recommended in combination with FOLFIRI (folinic acid, 5′-fluorouracil and irinotecan) in MCRC patients if the disease progresses after therapy with Bevacizumab, oxaliplatin, and fluoropyrimidine [8].
Direct suppression of tumor angiogenesis and vascular normalization results in suppression of tumor growth. However, after a long-term therapy, tumor cells acquire a resistant phenotype as a result of hypoxia and low nutrition stress. Overall, the survival benefits of antiangiogenic (AA) drugs have not been impressive and surprisingly most cancer patients stop responding or do not respond to the AA therapy at all. What is more, recently it was shown that AA drugs cause a switch to vasoinvasion of tumor cells, leading to increased metastasis and shortened life in mice [39]. The tumor resistance to AA agents can partly be a consequence of non-sprouting mechanisms of vessel recruitment. In intussusceptive microvascular growth, new vessels are generated by creating columns from connective tissue within the lumen of existing vessels. Glomeruloid angiogenesis is characterized by tight nests of vessels that resemble renal glomerulus. In vessel co-option, tumor cells incorporate host vessels in the normal surrounding tissue, and vasculogenic mimicry tumor cells directly from perfused channels bind to the host vasculature. In turn, in the case of looping angiogenesis, contractile myofibroblasts pull host vessels into the cancer tissue [78].
Several phase I and II studies targeting at fibroblast activated protein (FAP) with a humanized monoclonal antibody (Sibrotuzumab) failed to produce clinical benefits in the colon and non-small-cell lung cancer alone or in combination with docetaxel. The latest proposed strategy is based on a specific location of FAP which can be used for precise administration of cytotoxic prodrugs. This strategy is expected to enhance efficacy of the drug delivered to the tumor microenvironment [14].
VEGFR-2 is known as a target for Sunitinib which is a receptor tyrosine kinase inhibitor. ECs transfected with miR-221/miR-222 and treated with Sunitinib showed a reduction in total tube length, and enhancement of cellular proliferation was observed. Sunitinib was not able to abolish the effect of miR-221/222 at pharmacologically relevant concentrations. Such resistance to treatment with Sunitinib may develop when the targeted protein is not accessible for the drug binding. In therapeutic implications, inhibition of miR-221 and miR-222 might improve the patient’s survival if administered as an adjuvant therapy in combination with Sunitinib [57].
6.6.2 Inhibition of CAF Formation
Currently, tumor immunomodulation is the main focus of anti-cancer therapies [64]. CAFs, being an important element, regulate cancer invasiveness and characterize by a wide range of cross- talk with other cells located in tumor microenvironments, are a target of anti-cancer therapies. In contrast to preinvasive stages of cancers, the cross-talk processes are mainly observed in invasive cancer stages [64]. That interaction seems to be the main source of chemoresistance. CAFs highly express chemoresistance receptors like retinoic acid receptor β, which improves therapeutic responses of the cells. Cell surface molecules CD10 and GPR77 expressed on CAFs, also contribute to chemoresistance through supporting cancer stemness. Hence, the effectiveness of anticancer therapies in preinvasive stages may not be disturbed but the treatment may additionally intensify tumor growth in invasive stages. Therefore, complexed therapies should be applied.
TGF-βs are the main EndMT inductors contributing to formation of CAFs. Thus, the inhibition of TGF-β pathway seems to be the most promising strategy to decrease the population of CAFs. Theoretically, three levels of inhibition are possessed: (i) ligand inhibition which prevents TGF-β synthesis, (ii) ligand-receptor interaction blocking, and (iii) restriction of signal transduction. Despite the numerous tested inhibitors, studies on their functions mainly focused on modulation of cancer cells. Only one study demonstrated the role of the inhibitor on the cancer niche. TGFβ-activated microenvironment increases the metastasis ability of colon cancer cell into lungs and liver. Zhang et al. [122] revealed that LY2109761 significantly reduces liver metastases and prolongs survival (by about 25%) in a mouse model. Galunisertib treatment resulted in a blocked formation of subcutaneous tumors by primary colorectal cancer stem cells [19]. Results of these studies demonstrated that STAT3 signaling enhances liver and lung metastasis through TGF-β and IL-11-dependent pathways. The authors prove that targeting at TGF-β signaling can alter cancer cells via cells located in the tumor microenvironment.
Finding effective therapies which would inhibit CAF formation or block their effect on cancer progression appears to be quite difficult.
As described above, CAFs are a heterogeneous group of cells, formed from several sources under the influence of different immunomodulators. Therefore, inhibition of only one pathway is not strong enough to counteract an occurrence of CAFs. Secondly, particular CAF subpopulations demonstrated different functions. It has been recently shown that depending on the location in the cancer niche, CAFs can regulate cancer cells contraction (CAFs located in the close area or within the tumor) or affect the tumor through secreted immunomodulators. Additionally, it has been suggested that different levels of the α-SMA marker, demonstrated by CAFs, depend on the origin of these fibroblasts [70]. Expression of particular markers is another problem that should be considered while searching for anti-CAF therapy. Numerous studies revealed that their expression is dependent on the CAF source. Difficulty is that the presence of particular markers is not specific to CAFs, but it can be observed in other cells of the tumor niche, especially macrophages or lymphocytes [108].
6.7 Conclusion
The endothelium plays a critical role in cancer progression. Inhibition of cancer vascularization, intravasation of cancer cells, and CAF formation are the main reasons for creating effective anti-cancer therapies and, above all, inhibition of metastasis.
Nevertheless, many strategies limiting the growth of blood vessels proved to be ineffective. The reasons for that should be explained by diversified vasculature of the types of recruited inducing cells or those localized in the tumor area as well as possibilities of their interaction. It should be emphasized that the above-mentioned processes can be induced by tumor cells and tumor niche cells in response to the applied treatment. These elements should be included in the search for new effective therapies that inhibit tumor vascularization. Intravasation of cancer cells could be limited in three ways: by preventing cancer vascularization; by blocking an invasion of tumor cells into the vessel, i.e., by maintaining cell-cell connections; and finally, by inhibiting tumor cells invasiveness. Unfortunately, the last two possibilities are practically not used in anti-cancer therapies. CAFs may serve as the last mechanism that may constitute a source of anti-cancer strategies. It is known that these cells significantly contribute to a development of the tumor, either by induction of proliferation in preinvasive cancer stages or through the acceleration of EMT and metastatic capacity in invasive tumor stages. EndMT inhibition, which accounts for approximately 40% of CAFs, could prevent these adverse effects of CAF formations. However, so far, the role of mechanisms conditioning the transformation and functions of individual CAF subpopulations have not been clearly clarified.
References
Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8:464–478
Aird CW (2007) Phenotypic heterogeneity of the endothelium. I. Structure, function, and mechanisms. Circ Res 100:158–173
Aird CW (2012) Endothelial cell heterogeneity. Cold Spring Harb Perspect Med 2:a006429
Aisagbonhi O, Rai M, Ryzhov S et al (2011) Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech 4:469–483
Akino T, Hida K, Hida Y et al (2009) Cytogenetic abnormalities of tumour-associated endothelial cells in human malignant tumours. Am J Pathol 175:2657–2667
Al-Mehdi AB, Tozawa K, Fisher AB, Shientag L et al (2000) Intravascular origin of metastasis from the proliferation of endothelium-attached tumour cells: a new model for metastasis. Nat Med 6:100–102
Anderberg C, Cunha SI, Zhai Z et al (2013) Deficiency for endoglin in tumour vasculature weakens the endothelial barrier to metastatic dissemination. J Exp Med 210:563–579
Aprile G, Rijavec E, Fontanella C et al (2014) Ramucirumab: preclinical research and clinical development. Onco Targets Ther 7:1997–2006
Arciniegas E, Frid MG, Douglas IS, Stenmark KR (2007) Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 293:L1–L8
Armstrong EJ, Bischoff J (2004) Heart valve development: endothelial cell signaling and differentiation. Circ Res 95:459–470
Ausprunk DH, Folkman J (1977) Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumour angiogenesis. Microvasc Res 14:53–65
Bazzoni G, Dejana E (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84:869–901
Bos D, Zhang XH, Nadal C et al (2009) Massague Genes that mediate breast cancer metastasis to the brain. Nature 459:1005–1009
Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR (2012) Targeting carcinoma-associated fibroblasts within the tumour stroma with a fibroblast activation protein-activated prodrug. J Natl Cancer Inst 104:1320–1334
Buckanovich RJ, Sasaroli D, O’Brien-Jenkins A et al (2007) Tumour vascular proteins as biomarkers in ovarian cancer. J Clin Oncol 25:852–861
Burri PH, Hlushchuk R, Djonov V (2004) Intussusceptive angiogenesis: its emergence, its characteristics, and its significance. Dev Dyn 31:474–488
Bussolati B, Deambrosis I, Russo S et al (2003) Altered angiogenesis and survival in human tumour-derived endothelial cells. FASEB J 17:1159–1161
Bussolati B, Assenzio B, Deregibus MC, Camussi G (2006) The proangiogenic phenotype of human tumour-derived endothelial cells depends on thrombospondin-1 downregulation via phosphatidylinositol 3-kinase/Akt pathway. J Mol Med 84:852–863
Calon E, Espinet S, Palomo-Ponce DV et al (2012) Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 22:571–584
Cerasuolo M, Paris D, Iannotti FA et al (2015) Neuroendocrine transdifferentiation in human prostate cancer cells: an integrated approach. Cancer Res 75:2975–2986
Chang ACY, Fu Y, Garside VC et al (2011) Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev Cell 21:288–300
Chen HF, Huang CH, Liu CJ et al (2014) Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat Commun 22:4697
Chi JT, Chang HY, Haraldsen G et al (2003) Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A 100:10623–10628
Ciombor KK, Berlin J (2014) Aflibercept – a decoy VEGF receptor. Curr Oncol Rep 16:368
Cipriani P, Di Benedetto P, Ruscitti P et al (2015) The endothelial mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by Macitentan, a dual endothelin-1 receptor antagonist. J Rheumatol 42:1808–1816
Ciszewski WM, Sobierajska K, Wawro ME et al (2017) The ILK-MMP9-MRTF axis is crucial for EndMT differentiation of endothelial cells in a tumour microenvironment. Biochim Biophys Acta 1864:2283–2296
Conway EM, Collen D, Carmeliet P (2001) Molecular mechanisms of blood vessel growth. Cardiovasc Res 49:507–521
Cooley BC, Nevado J, Mellad J et al (2014) TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med 6:227ra34
Crinò L, Metro G (2014) Therapeutic options targeting angiogenesis in nonsmall cell lung cancer. Eur Respir Rev 23:79–91
Cugno M (2012) Inflammation, coagulation, vascular permeability and thrombosis. Curr Vasc Pharmacol 10:631
Dejana E, Orsenigo F (2013) Endothelial adherens junctions at a glance. J Cell Sci 126:2545–2549
Del Galdo F, Lisanti MP, Jimenez SA (2008) Caveolin-1, transforming growth factor-β receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol 20:713–719
Deregibus MC, Cantaluppi V, Calogero R et al (2007) Endothelial progenitor cell-derived microvesicles activate an angiogenic program in endothelial cells by an horizontal transfer of mRNA. Blood 110:2440–2448
DeRuiter MC, Poelmann RE, VanMunsteren JC et al (1997) Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro. Circ Res 80:444–451
Detmar M, Brown LF, Schon MP et al (1998) Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J Invest Dermatol 111:1
Döme B, Hendrix MJ, Paku S et al (2007) Alternative vascularization mechanisms in cancer. Pathology and therapeutic implications. Am J Pathol 170:1–15
Dudley AC (2012) Tumour endothelial cells. Cold Spring Harb Perspect Med 2:a006536
Dvorak HF, Brown LF, Detmar M, Dvorak AM (1995) Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 146:1029–1039
Ebos JM, Kerbel RS (2011) Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 8:210–221
Evrard SM, Lecce L, Michelis KC et al (2016) Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun 7:11853
Folkman J (1971) Tumour angiogenesis: therapeutics implication. N Engl J Med 285:1182–1186
Fu Y, Chang A, Chang L et al (2009) Differential regulation of transforming growth factor β signaling pathways by Notch in human endothelial cells. J Biol Chem 284:19452–19462
Furuya M, Nishiyama M, Kasuya Y et al (2005) Pathophysiology of tumour neovascularization. Vasc Health Risk Manag 1:277–290
Ganguly H, Yang RS et al (2012) The role of microtubules and their dynamic in cell migration. J Biol Chem 287:43359–43369
Gao D, Nolan D, McDonnell K et al (2009) Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumour growth and metastatic progression. Biochim Biophys Acta 1796:33–40
Gasperini P, Espigol-Frigole G, McCormick PJ et al (2012) Kaposi sarcoma herpes virus promotes endothelial-to-mesenchymal transition through notch-dependent signaling. Cancer Res 72(5):1157–1169
Ghajar CM, George SC, Putnam AJ (2008) Matrix metalloproteinase control of capillary morphogenesis. Crit Rev Eukaryot Gene Expr 18:251–278
Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127:679–695
Hanahan D, Weinberg R (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hida K, Hida Y, Shindoh M (2008) Understanding tumour endothelial cell abnormalities to develop ideal anti-angiogenic therapies. Cancer Sci 99:459–466
Hida K, Maishi N, Annan DA, Hida Y (2018) Contribution of tumour endothelial cells in cancer progression. Int J Mol Sci 19. pii: E1272.
Hillen F, Griffioen AW (2007) Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev 26:489–502
Huang TH, Chu TY (2014) Repression of miR-126 and upregulation of adrenomedullin in the stromal endothelium by cancer-stromal cross talks confers angiogenesis of cervical cancer. Oncogene 33:3636–3647
Hurwitz H, Fehrenbacher L, Novotny W et al (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342
Junqueira LC, Carneiro J (2005) Basic histology: text and Atlas, 10th edn. McGraw-Hill Medical, New York-Burr Ridge-San Francisco, p 215
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Khella HWZ, Butz H, Ding Q et al (2015) miR-221/222 are involved in response to Sunitinib treatment in metastatic renal cell carcinoma. Mol Ther 23:1748–1758
Khuon S, Liang L, Dettman L et al (2010) Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: a three-dimensional FRET study. J Cell Sci 123:431–440
Kienast Y, von Baumgarten L, Fuhrmann M et al (2010) Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16:116–122
Lee T-H, Avraham HK, Jiang S, Avraham S (2003) Vascular endothelial growth factor modulates the transendothelial migration of MDA-MB-231 breast cancer cells through regulation of brain microvascular endothelial cell permeability. J Biol Chem 278:5277–5384
Leong HS, Robertson AE, Stoletov K et al (2014) Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep 8:1558–1570
Li Z, Wermuth PJ, Benn BS et al (2013) Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am J Pathol 182:325–331
Li L, Chen L, Zang J et al (2015) C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism 64:597–610
Locy H, de Mey S, de Mey W et al (2018) Immunomodulation of the tumour microenvironment: turn foe into friend. Front Immunol 9:2909
Loizzi V, Del Vecchio V, Gargano G et al (2017) Biological pathways involved in tumour angiogenesis and bevacizumab based anti-angiogenic therapy with special references to ovarian cancer. Int J Mol Sci 18:1967
Lu C, Bonome T, Li Y et al (2007) Gene alterations identified by expression profiling in tumour-associated endothelial cells from invasive ovarian carcinoma. Cancer Res 67:1757–1768
Maishi N, Ohba Y, Akiyama K et al (2016) Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci Rep 6:28039
Mary S, Charrasse S, Meriane M et al (2002) Biogenesis of N-cadherin-dependent cell-cell contacts in living fibroblasts is a microtubule-dependent kinesin-driven mechanism. Mol Biol Cell 13:285–301
Masoud GN, Li W (2015) HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 5:378–389
Medici D, Kalluri R (2012) Endothelial mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol 144:724–732
Medici D, Potenta S, Kalluri R (2011) Transforming growth factor-β2 promotes Snail-mediated endothelial–mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J 437:515–520
Medinger M, Mross K (2010) Clinical trials with anti-angiogenic agents in hematological malignancies. J Angiogenes Res 2:10
Michiels C (2003) Endothelial cell functions. J Cell Physiol 196:430–443
Muraki C, Ohga N, Hida Y et al (2011) Cyclooxygenase-2 inhibition causes antiangiogenic effects on tumour endothelial and vascular progenitor cells. Int J Cancer 130:59–70
Nagy JA, Chang SH, Shih SC et al (2010) Heterogeneity of the tumour vasculature. Semin Thromb Hemost 36:321–331
Nakajima Y, Yamagishi T, Hokari S, Nakamura H (2000) Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor-beta and bone morphogenetic protein. Anat Rec 258:119–127
Nanda A, St Croix B (2004) Tumour endothelial markers: new targets for cancer therapy. Curr Opin Oncol 16:44–49
Nasir A (2019) Angiogenic signaling pathways and anti-angiogenic therapies in human cancer: applications in precision medicine. Predictive Biomarkers Oncol:243–262
Noseda M, McLean G, Niessen K et al (2004) Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res 94:910–917
Ohmura-Kakutani H, Akiyama K, Maishi N et al (2014) Identification of tumour endothelial cells with high aldehyde dehydrogenase activity and a highly angiogenic phenotype. PLoS One 9:e113910
Olson EN, Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11:353–365
Padua D, Zhang XZ, Wang Q et al (2008) TGFbeta primes breast tumours for lung metastasis seeding through angiopoietin-like 4. Cell 133:66–77
Paku S, Paweletz N (1991) First steps of tumour-related angiogenesis. Lab Investig 65:334–346
Pantsulaia I, Ciszewski WM, Niewiarowska J (2016) Senescent endothelial cells: potential modulators of immunosenescence and ageing. Ageing Res Rev 29:13–25
Potts JD, Runyan RB (1989) Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor-beta. Dev Biol 134:392–401
Prager GW, Lackner E-M, Krauth M-T et al (2010) Targeting of VEGF-dependent transendothelial migration of cancer cells by bevacizumab. Mol Oncol 4:150–160
Raffaghello L, Vacca A, Pistoia V, Ribatti D (2015) Cancer associated fibroblasts in hematological malignancies. Oncotarget 6:2589–2603
Rigamonti N, De Palma M (2013) A role for angiopoietin-2 in organ-specific metastasis. Cell Rep 4:621–623
Rong X, Huang B, Qiu S et al (2016) Tumour-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget 7:83976–83986
Sanchez-Duffhues G, Orlova V, ten Dijke P (2016) In brief: endothelial-to-mesenchymal transition. J Pathol 238(3):378380
Sasahira T, Kurihara M, Bhawal UK et al (2012) Downregulation of miR-126 induces angiogenesis and lymphangiogenesis by activation of VEGF-A in oral cancer. Br J Cancer 107:700–706
Schully S, Francescone R, Faibish M et al (2012) Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J Neurosci 32:12950–12960
Schumacher D, Strilic B, Sivaraj KK et al (2013) Platelet-derived nucleotides promote tumour-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 24:130–137
Shibuya M (2011) Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2(12):1097–1105
St Croix B, Rago C, Velculescu V et al (2000) Genes expressed in human tumour endothelium. Science 289:1197–1202
Stockmann C, Schadendorf D, Klose R, Helfrich I (2014) The impact of the immune system on tumour: angiogenesis and vascular remodeling. Front Oncol 4:69
Stoletov K, Kato H, Zardouzian E et al (2010) Visualizing extravasation dynamics of metastatic tumour cells. J Cell Sci 123:2332–2341
Streubel B, Chott A, Huber D et al (2004) Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N Engl J Med 351:250–259
Tremblay PL, Huot J, Auger FA (2008) Mechanisms by which E-selectin regulates diapedesis of colon cancer cells under flow conditions. Cancer Res 68:5167–5176
Tsai JH, Yang J (2013) Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 27:2192–2206
Tsuchiya K, Hida K, Hida Y et al (2010) Adrenomedullin antagonist suppresses tumour formation in renal cell carcinoma through inhibitory effects on tumour endothelial cells and endothelial progenitor mobilization. Int J Oncol 36:1379–1386
van Meeteren LA, ten Dijke P (2011) Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res 347:177–186
van Zijl F, Krupitza G, Mikulits W (2011) Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res 728:23–34
Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K (2000) Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res 60:203–212
Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 15:261–271
Wang R, Chadalavada K, Wilshire J et al (2010) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468:829–833
Wang S-H, Chang JS, Hsiao J-R et al (2016) Tumour cell-derived WNT5B modulates in vitro lymphangiogenesis via induction of partial endothelial mesenchymal transition of lymphatic endothelial cells. Oncogene 36:1–13
Wang M, Zhao J, Zhang L et al (2017) Role of tumour microenvironment in tumourigenesis. J Cancer 8:761–773
Warren B (1979) The vascular morphology of tumours. In: Peterson H-I (ed) Tumour blood circulation: angiogenesis, vascular morphology and blood flow of experimental and human tumours. CRC Press, Boca Raton, pp 1–47
Wawro ME, Sobierajska K, Ciszewski WM et al (2017) Tubulin beta 3 and 4 are involved in the generation of early fibrotic stages. Cell Signals 38:26–38
Wawro ME, Chojnacka K, Wieczorek-Szukała K et al (2019) Invasive colon cancer cells induce transdifferentiation of endothelium to cancer-associated fibroblasts through microtubules enriched in tubulin-β3. Int J Mol Sci 20:53
Weis SM, Cheresh DA (2011) Tumour angiogenesis: molecular pathways and therapeutic targets. Nat Med 17:1359–1370
Wermuth PJ, Li Z, Mendoza FA, Jimenez SA (2016) Stimulation of transforming growth factor-β1-induced endothelial-to-mesenchymal transition and tissue fibrosis by endothelin-1 (ET-1): a novel profibrotic effect of ET-1. PLoS One 11:e0161988
Widyantoro B, Emoto N, Nakayama K et al (2010) Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 121:2407–2418
Xavier S, Vasko R, Matsumoto K et al (2015) Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol 26:817–829
Xian X, Håkansson J, Ståhlberg A et al (2006) Pericytes limit tumour cell metastasis. J Clin Invest 116:642–651
Xing F, Saidou J, Watabe K (2010) Cancer associated fibroblasts (CAFs) in tumour microenvironment. Front Biosci 15:166–179
Yagi H, Tan W, Dillenburg-Pilla P, Armando S et al (2011) A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci Signal 4:ra60
Zeisberg M, Kalluri R (2013) Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 304(3):C216–C225
Zeisberg EM, Potenta S, Xie L et al (2007) Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67:10123–10128
Zervantonakis IK, Hughes-Alford SK, Charest JL et al (2012) Three-dimensional microfluidic model for tumour cell intravasation and endothelial barrier function. Proc Natl Acad Sci U S A 109:13515–13520
Zhang B, Halder SK, Zhang S, Datta PK (2009) Targeting transforming growth factor-beta signaling in liver metastasis of colon cancer. Cancer Lett 277:114–120
Ziyad S, Iruela-Arispe L (2011) Molecular mechanisms of tumour angiogenesis. Genes Cancer 2:1085–1096
Wawro ME, Sobierajska K, Ciszewski WM, Niewiarowska J (2019) Nonsteroidal Anti-Inflammatory Drugs Prevent Vincristine-Dependent Cancer-Associated Fibroblasts Formation. International Journal of Molecular Sciences 20(8):1941
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sobierajska, K., Ciszewski, W.M., Sacewicz-Hofman, I., Niewiarowska, J. (2020). Endothelial Cells in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1234. Springer, Cham. https://doi.org/10.1007/978-3-030-37184-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-37184-5_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-37183-8
Online ISBN: 978-3-030-37184-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)