
Abstract
Alzheimer’s disease (AD) is associated with a very large burden on global healthcare systems. Thus, it is imperative to find effective treatments of the disease. One feature of AD is the accumulation of neurotoxic β-amyloid peptide (Aβ). Aβ induces multiple pathological processes that are deleterious to nerve cells. Despite the development of medications that target the reduction of Aβ to treat AD, none has proven to be effective to date. Non-pharmacological interventions, such as physical exercise, are also being studied. The benefits of exercise on AD are widely recognized. Experimental and clinical studies have been performed to verify the role that exercise plays in reducing Aβ deposition to alleviate AD. This paper reviewed the various mechanisms involved in the exercise-induced reduction of Aβ, including the regulation of amyloid precursor protein cleaved proteases, the glymphatic system, brain-blood transport proteins, degrading enzymes and autophagy, which is beneficial to promote exercise therapy as a means of prevention and treatment of AD and indicates that exercise may provide new therapeutic targets for the treatment of AD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is an irreversible and progressive neurodegenerative disorder [1, 2]. It is the most common form of dementia and accounts for 60 to 70% of total dementia cases and affects about 27 million people worldwide [3, 4]. This creates a large economic burden, as the global healthcare cost for people with dementia is estimated to increase from 818 billion US dollars in 2015 to 2 trillion US dollars by 2030 [5]. The typically clinical features of AD are a gradual loss of cognitive function, episodic memory, and learning ability, followed by a decline in visuospatial skills and language [6, 7]. Patients with AD also may have behavioral problems, such as aggression, apathy, depression, and sleeping problems [8]. These symptoms adversely affect the daily life and social participation of AD patients. Currently, the pharmaceutical treatments for AD include cholinesterase inhibitors, such as galantamine, donepezil and rivastigmine, and memantine, which protects against glutamate-mediated neurotoxicity [9]. However, these medications only offer short-term remission of the development of AD rather than long-term therapeutic effects for the disease; in addition, these drugs are frequently associated with unpleasant side effects, such as weight loss, dizziness, and nausea [10]. Therefore, to clarify the mechanism of AD is a crucial issue for seeking out an effective and novel therapy for AD is urgently required.
Two typically pathophysiological changes, consisting of extracellular deposits of insoluble β-amyloid peptide (Aβ) plaque and neurofibrillary tangles (NFT) of phosphorylated tau (P-tau) within neurons, are the hallmarks of AD [11]. The accumulation of Aβ and P-tau are considered to result in atrophy and death of neurons, leading to cognitive dysfunction [12]. As one of the pathological hallmarks of AD, Aβ is a crucial target to treat AD [13]. The molecular mass of Aβ is 4 kDa and the amino acid sequence exhibits microheterogeneity [14]. According to a widely accepted theory, Aβ originates from the sequentially proteolytic cleavage of amyloid precursor protein (APP) [15]. The main pathways of Aβ removal include clearance by the glymphatic system, transportation across the blood–brain barrier (BBB), proteolytic degradation, and autophagy. An imbalance between the clearance and the production of Aβ causes cerebral dysfunction [16]. Aβ deposition has been proved to play a key role in AD progression, being other pathological events observed (including the NFT formation, endoplasmic reticulum stress, mitochondrial dysfunction, oxidative stress, or neuroinflammation), ultimately followed by neuronal loss [4, 17]. Decreasing the accumulation of Aβ is the main target for the treatment of AD. However, clinical trials using anti-amyloid drugs have repeatedly failed, which might be due to the presence of the blood–brain barrier or other factors. Therefore, we need to explore other types of interventions to alleviate the harmfulness of Aβ [18].
Due to the lack of a disease-modifying treatment for AD, researchers have been investigating the role that exercise, a non-pharmacological intervention, plays in alleviating AD symptoms and progression [19]. A variety of clinical studies demonstrated that aerobic exercise could promote cardiorespiratory fitness, memory, and executive function in patients with mild AD [20,21,22,23,24]. Fifteen weeks of physical activity was demonstrated to improve walking quality and alleviate cognitive decrease in AD patients. Another study showed that exercise can reduce the loss of global cognition in older individuals aged 70–80 with mild-to-moderate AD. The meta-analyses of randomized controlled trials indicate the slight-to-moderate effects of aerobic exercise on cognitive function across the AD spectrum [25, 26]. Due to the limitation of severe motor dysfunction in advanced AD patients, the evidence to investigate the effect of exercise on patients with advanced AD is limited. The increased physical activities caused by technology-aided program promote the positive personal involvement and independent occupation for advanced AD patients [27, 28]. Moreover, regular exercise may alleviate the progress of functional deterioration in mild AD and decrease falls in patients with advanced AD [29].``
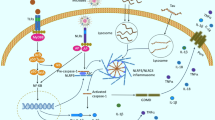
On the other hand, exercise plays a multifactorial role in attenuating various pathophysiologic mechanisms that are associated with cognitive impairment and neurodegeneration in AD, such as the aggregation of tau and Aβ, inflammation, oxidative stress, pyroptosis, endothelial dysfunction, and so on [30,31,32]. Indeed, a series of studies have confirmed the effectiveness of exercise in reducing Aβ accumulation that is one of the most considerable roles in AD [16, 30, 33,34,35,36,37,38,39]. A study that used a mouse model of AD found evidence that voluntary running in the late stage of AD alleviated an increase in the number of Aβ plaques and improved hippocampus neurogenesis and memory [40]. However, up to now, it is still unclear how exercise reduces Aβ deposition. Hence, this review summarized the evidence of the beneficial effects of exercise on Aβ-dependent pathophysiology of AD, including Aβ generation, the glymphatic system, Aβ transport proteins across the BBB, autophagy, degrading enzymes, and other mechanisms to clarify the mechanisms involved and to explore novel and effective interventional targets for the treatment of AD (Fig. 1).

Summary of mechanisms by which exercise reduces Aβ. The beneficial effect of exercise on Aβ, including APP-cleaved proteases, the glymphatic system, Aβ transport proteins across BBB, autophagy, degrading enzymes, and others. Aβ, β-amyloid peptide; ER stress, endoplasmic reticulum stress; PGC1-α, peroxisome proliferator-activated receptor-gamma coactivator 1α; FNDC5, fibronectin type III domain-containing protein 5; BDNF, brain-derived neurotrophic factor; SIRT1, sirtuin-1; AQP4, aquaporin-4: BBB, blood–brain barrier; RAGE, the receptor for advanced glycation end products; LRP1, low-density lipoprotein receptor-related protein 1; IDE, insulin-degrading enzyme; ABCA1, ATP-binding cassette transporter A1; GSK3, glycogen synthase kinase-3
The Mechanisms of the Inhibition of Aβ Deposition by Exercise Within Neurons in AD
The Effect of the Regulation of APP-Cleaved Proteases by Exercise
According to the amyloid cascade hypothesis, Aβ is generated by the protease cleavage of APP, which is predominately expressed in the central nervous system (CNS) [41, 42]. APP can undergo two alternative proteolytic processes through the non-amyloidogenic pathway and the amyloidogenic pathway. In the non-pathogenic pathway, APP is cleaved by α-secretase, such as a disintegrin and metalloprotease-10 (ADAM10), which releases an N-terminal-secreted APP (sAPPα) and C-terminal fragment of 83 amino acids (C83); sAPPα may exert a neuroprotective function [14, 43, 44]. In the amyloidogenic pathway, APP is cleaved by β-secretase, such as beta-site APP-cleaving enzyme 1 (BACE1), and γ-secretase and results in the production of Aβ [45]. Under normal physiological conditions, the two pathways coexist in equilibrium, although the non-amyloidogenic pathway is favored [46]. However, aging, genetic, and environmental factors related to AD may cause a metabolic shift that favors the amyloidogenic pathway of APP [47]. Although medications that target Aβ production to treat AD have been developed, such as γ-secretase modulators and BACE1 inhibitors, they have only recently become available [48]. Therefore, we need to explore novel methods to treat AD. Physical exercise has been proven to have the potential to reduce Aβ production by activating or inhibiting APP-cleaved proteases via multiple pathways (Fig. 2), which are summarized in the following sections.

Molecular mechanisms of the reduction of APP-cleaved proteases by exercise. ADAM10, BACE1, and γ-secretase are the main proteases for APP metabolism. SIRT1, PGC-1α, FNDC5, BDNF, and the ER stress are all involved in the regulation of APP-cleaved proteases by exercise. Green arrows represent promotion; red lines represent inhibition; pink lines represent production. ER stress, endoplasmic reticulum stress; PGC1-α, peroxisome proliferator-activated receptor-gamma coactivator 1α; FNDC5, fibronectin type III domain-containing protein 5; BDNF, brain-derived neurotrophic factor; SIRT1, sirtuin-1; RAR-β, retinoic acid receptor-β; BACE1, beta-site APP-cleaving enzyme 1; ADAM10, a disintegrin and metalloproteinase-10; APP, amyloid precursor protein; Aβ, β-amyloid peptide
The Brain-Derived Neurotrophic Factor
Brain-derived neurotrophic factor (BDNF) is a member of the growth factor family and plays a key role in neuronal growth, differentiation, and survival, as well as synaptic plasticity [49, 50]. Converging studies have suggested that impaired BDNF signalling contributes to the pathological mechanisms of several main disorders and diseases, including AD, Huntington’s disease (HD), and depression. BDNF is one of the elements responsible for synaptic integrity and cognitive function. Patients with AD have decreased expression of BDNF in the brain, and the infusion of BDNF can lessen cognitive dysfunction in elderly primates [51]. Moreover, high levels of peripheral BDNF may provide protection against the occurrence of AD [51, 52]. Importantly, BDNF can reduce Aβ deposition by activating α-secretase, which results in the increased levels of sAPPα, which has neuroprotective effects [53]. Several studies have shown that physical exercise in rats with AD obviously promotes the levels of BDNF and decreases the levels of Aβ [53,54,55]. Lin et al. also reported that 10-week treadmill training of APP/presenilin 1 (PS1) double transgenic (TG) mice elevated the expression of the phosphorylated protein kinase B, tyrosine kinase B receptor, and protein kinase C, which were the BDNF signalling pathway molecules, and prevented AD-related degeneration in the hippocampus and amygdala [56]. Therefore, we can infer that exercise partly reduces Aβ production through the BDNF pathway.
Sirtuin-1 Signalling Pathway
Sirtuin-1 (SIRT1), as an important member of the sirtuin family, plays a crucial role in maintaining cellular homeostasis via influencing insulin sensitivity, mitochondrial biogenesis, glucose metabolism, and neuronal survival [57,58,59,60]. SIRT1 has been verified to be related to numerous neurodegenerative pathophysiological process, such as AD and Parkinson’s disease. Especially, SIRT1 can exert a neuroprotective effect against AD [61, 62]. Downregulation of SIRT1 has been found in AD, causing increased Aβ production, while overexpression of SIRT1 can reverse this pathology, suggesting the profound effect of SIRT1 on Aβ production [63]. Porquet et al. also showed that SIRT1 activation by resveratrol improved neurodegeneration by decreasing oxidative stress and neuroinflammation induced by amyloid accumulation in AD [64]. Revilla et al. have reported that the downregulation of SIRT1 in 3xTG (triple-transgenic) AD mice can be recovered by exercise treatment. Moreover, Koo et al. used a mouse model to show that treadmill exercise promoted the expression level of SIRT1, which subsequently caused the activation of ADAM10 by increasing the retinoic acid receptor-β and inhibiting Rho-associated kinase 1; the SIRT1 signalling pathway eventually activated the non-amyloidogenic pathway. The promotion of SIRT1 also increased the levels of peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) and decreased the levels of BACE1, which suppressed the amyloidogenic pathway [58].
Fibronectin Type III Domain-Containing Protein 5 and PGC-1α
Fibronectin type III domain-containing protein 5 (FNDC5), which is the precursor protein of irisin, has been categorized as a PGC-1α-dependent myokine [65]. FNDC5 is proteolytically cleaved to myokine irisin, which regulates the beneficial role of exercise on human metabolism [66]. In recent years, studies have focused on the role of FNDC5 as a mediator of AD. For instance, one study found that increased levels of FNDC5/irisin in the brains of mice with AD improved impaired memory and synaptic plasticity [67]. Another cell study reported that FNDC5 regulated the β-cleavage of APP through intercommunications with APP, which reduced the levels of Aβ [68]. Choi et al. reported that running exercise enhanced the expression of FNDC5 in the hippocampus of 5xFAD TG mice that coinherited and co-overexpressed familial AD mutant forms of human PS1 and APP transgenes, and their results suggested that exercise attenuated the action of β-secretase through FDNC5 [69].
PGC-1α, an upstream activator of FNDC5, can be stimulated during exercise. In addition, PGC-1α plays a regulatory role in energy metabolism during the early stages of neurological diseases [70]. For instance, the increase of PGC-1α could alleviate the damaged cognition and neuronal injury in TG mice with AD by improving mitochondrial dysfunction and alleviating oxidative stress and insulin resistance [71]. Interestingly, low levels of PGC-1α result in Aβ aggregation in the brains of patients with AD [72]. The reason for this may be that the decreased levels of PGC-1α fail to block the action of BACE1, which increases the production of Aβ [73]. In this regard, both FNDC5 and PGC-1α are involved in the mediation of exercise to Aβ pathology.
Furthermore, FNDC5 can also regulate the levels of BDNF in hippocampus of mice [74]. One study demonstrated that treadmill exercise potentially reduced Aβ aggregation and improved impaired cognition through the PGC-1α/FNDC5/BDNF pathways in a rat model of AD [75]. Meanwhile, PGC-1α is a substrate of SIRT-1. Thus, it is logical to conclude that the interactions between SIRT1, PGC-1α, FNDC5, and BDNF are related to the exercise-regulated reduction of Aβ production. However, these interactions require further research.
The Unfolded Protein Response Signalling Pathway
The endoplasmic reticulum (ER) is responsible for protein quality control and folding. Multiple environmental and genetic insults can destroy the function of the ER and induce ER stress [76]. The unfolded protein response (UPR) is a complicated adaptive cellular mechanism that is related to ER stress [77]. There are three main signalling branches of the UPR: activating transcription factor 6 (ATF6), inositol-requiring enzyme 1α (IRE1α), and protein kinase RNA-like endoplasmic reticulum kinase (PERK) [78]. The UPR is triggered in multiple neurodegenerative diseases, including AD and Parkinson’s disease, due to the aggregation of misfolded proteins [79]. The accumulation of Aβ disrupted several cellular processes, caused ER stress, and activated the UPR, which aggravated the process of inflammation and apoptosis [80, 81]. Conversely, the overactivated UPR contributes to β-amyloidogenesis. In 5xFAD TG mice that coexpress human APP and PS1 with five familial AD mutations, the activation of PERK in response to ER stress results in elevated eukaryotic initiation factor-2α (eIF2α, the downstream substrate of PERK) phosphorylation, which increases BACE1 translation and Aβ accumulation [82]. In addition, prolonged and excessive ER stress could enhance the expression of PS1 through activating transcription factor 4 (ATF4), which could lead to an increase in Aβ secretion by activating γ-secretase activity [83]. Xia et al. showed that treadmill exercise inhibited the expression of glucose-regulated protein 78 (GRP78) and suppressed activation of ATF4, eIF2α, and PERK, along with the downregulation of the BACE1 in APP/PS1 mice. Therefore, exercise-induced inhibition of ER stress may regulate the amyloidogenic pathway. However, further research is needed to determine if exercise influences the activity of γ-secretase by inhibiting ATF4 [84].
Lipid Rafts
Lipid rafts are special cholesterol-rich membrane microdomains that play a vital role in cell survival and signal transduction [85]. In AD, lipid rafts are closely associated with Aβ deposition. Lipid rafts may obstruct autophagic-lysosomal system, a degenerative pathway of Aβ, thus accelerating AD development. In addition, previous studies indicated that Aβ production was dependent on lipid rafts [86]. The amyloidogenic pathway of APP cleavage primarily occurs in lipid rafts where BACE1 and γ-secretases show their optimum activities [87]. Zhang et al. reported that 12 weeks of treadmill exercise inhibited the formation of lipid rafts in the hippocampus of APP/PS1 TG mice, which hindered the function of BACE1 [88].
Thus, exercise exerts a beneficial role in reducing Aβ levels by mediating ADAM10 and BACE1 through the pathways of BDNF, SIRT-1, PGC-1α, and FNDC5, activating the UPR and decreasing lipid rafts. Besides, these pathways may alleviate cognitive impairment and pathophysiology of AD through an Aβ-independent manner, including increased synaptic plasticity and decreased inflammation and oxidative stress. Relatively little is known about the underlying mechanisms of the association between physical exercise and γ-secretase, which requires further investigation.
The Effect of the Regulation of Autophagy by Exercise
Autophagy is a key cellular pathway that is responsible for the disposal of waste components, which allows for cell renewal and ensures cellular bioenergetic homeostasis. As a conservative self-degrading process, autophagy can remove useless and toxic proteins or damaged cytoplasmic constituents and organelles through a lysosomal degradation system [5, 89]. Disorders in the autophagy process accelerate the aggravation of various neurodegenerative diseases that are associated with the aggregation of pathological proteins, such as PD, HD, and AD [90]. Bordi et al. investigated the relationship between autophagy and the pathogenesis of AD and found that lysosomal biogenesis and autophagosome formation were activated by various forms of cellular stress, such as damaged organelles Aβ aggregates, reactive oxygen species, and so on, as an early disease response, and in the late stages of AD, autophagy flux was increasingly hindered due to the inefficient substrate clearance by the lysosomes [91]. Under normal circumstances, Aβ degradation is implemented through the autophagy-lysosomal pathway, which is involved in protein quality control and the clearance of abnormal forms of proteins [92]. In contrast, impaired autophagy results in Aβ deposition. For instance, one study found that the genetic deletion of beclin 1, which was an essential autophagy gene, disrupted autophagy and elevated Aβ accumulation in cultured neurons from APP TG mice; the effect could be reversed by the administration of a beclin 1 viral gene delivery vector [93].
Recently, Li et al. reported that swimming exercise for 12 weeks could promote the autophagy and alleviate the formation of atherosclerosis in the aorta of apolipoprotein E knockout mice [94]. Zhao et al. showed that 12 weeks of treadmill exercise in an APP/PS1 TG mouse model elevated autophagy-lysosomal activity, as evidenced by a reduction in the levels of lysosome-associated membrane protein 1, a lysosomal marker, and p62, an autophagy marker, as well as a decreased Aβ burden [95]. Improvement of abnormal autophagy that is induced by exercise might be achieved by regulating the mTOR signalling pathway, which is a repressor of autophagy; however, the excessive activation of the mTOR signalling pathway could inhibit autophagy and result in the malfunction of Aβ clearance [92, 96, 97]. Thus, exercise has a positive effect on autophagy, which plays an important role in the regulation of Aβ clearance within neurons.
The Mechanisms of Aβ Clearance by Exercise Outside Neurons in AD
The Regulation of Aβ Clearance from the Glymphatic System by Exercise
Recently, a cerebral lymphatic system, known as the glymphatic system, was determined to be responsible for the removal of neuronal extracellular waste protein through a paravascular pathway [98]. In the glymphatic system, the CSF accesses the brain through the periarterial spaces, crosses the interstitium through the astrocytic aquaporin-4 (AQP4) water channels located on the perivascular cells, and drains the interstitial fluid and its solute into the perivenous zones and the deep cervical lymph nodes [99]. The subsequent influx of the CSF into the dense brain parenchyma is implemented by the expression of the AQP4 water channels in significantly polarized astrocytic endfeet that ensheath the cerebral vasculature [100, 101]. The glymphatic system can clear a major percentage of Aβ, tau protein, and other metabolites in the brain parenchyma [99]. However, a damaged glymphatic system is commonly found with senile and neurodegenerative diseases, such as AD [102,103,104]. Moreover, Aβ accumulation in the periarterial space can block perivascular pathway, consequently decreasing glymphatic clearance [105]. As the most affluent water channel in the CNS, AQP4 plays a key role in maintaining brain-water homeostasis and modulating the glymphatic system to accelerate Aβ clearance [106, 107]. Altered localization and expression of AQP4 are related to AD pathology and status [108]. When the glymphatic system is damaged, interstitial clearance decreases by approximately 70%, which leads to a 55–65% inhibition of Aβ clearance in AQP4 knockout mice [100, 101]. Xu et al. demonstrated that AQP4 knockout mice with AD showed more evident spatial memory and learning dysfunction, as well as enhanced amyloid angiopathy, Aβ deposition, atrophy of astrocytes, and synaptic protein loss in both the cortex and hippocampus compared with untreated AD mice [109].
Interestingly, one study showed that after 5 weeks of running on a wheel, the perivascular CSF influx was enhanced in young, awake, and freely behaving mice, which indicated that exercise increased the glymphatic activity and had beneficial effects on brain health through the increased clearance of neurotoxic products from the brain [110]. Moreover, 5 weeks of wheel exercise was shown to improve glymphatic activity, reduce the aggregation of Aβ plaques and neuroinflammation, enhance the level of AQP4, and ultimately prevent spatial cognitive decline and synaptic impairment [111]. Therefore, we propose that exercise may contribute to the clearing function of the glymphatic system via regulating AQP4 expression, which could alleviate the symptoms of AD.
Studies have found that the glymphatic system is suggested to function almost completely during sleep [112]. In addition, a genetic variation in AQP4 may impair the clearance mechanism and impact the relationship between sleep and the brain Aβ-amyloid burden [113]. These evidences suggest that Aβ removal by the glymphatic system is closely linked to sleep. Unfortunately, sleep disorders occur frequently with AD and affect nearly 45% of patients with AD [114]. Enhanced sleep latency is related to higher brain Aβ burden in older adults with normal cognition [115]. Several clinical trials have suggested that exercise could exert a positive influence on improving sleep quality in patients with AD [116, 117]. Therefore, exercise may be an auxiliary treatment choice for sleep dysfunction in patients with AD because it plays a crucial role in the clearance of Aβ via the glymphatic system, which can delay the progress of AD [118].
The Effect of the Regulation of Neuroinflammation by Exercise
Neuroinflammation is considered as a main feature of AD, which contributes to the pathogenesis of AD [119, 120]. In physiological conditions, astrocytes, microglia, and the other types of innate immune cells in the CNS can deal directly with multiple pathogens, toxins, and tissue damage [21]. As for astrocytes, AD-related neuroinflammation is generally along with reactive astrogliosis and significant changes of morphology and function following CNS damage [121]. Reactive astrocytes can seek, absorb, and degrade Aβ, potentially through its high binding ability to the nicotinic acetylcholine receptors of astrocytes [122]. However, astrocytes may become overloaded with Aβ, resulting in consequent lysis, which in turn contributes to forming Aβ plaque deposition [123]. With respect to microglia, AD-induced polarization of microglia to M1 phenotype causes the release of various pro-inflammatory factors and the failure of removing pathological protein accumulation, thus facilitating Aβ deposition and APP expression [124, 125]. In contrast, the shift of activated microglia from the M1 phenotype to the M2 phenotype inhibits inflammation by expressing various cytokines, thus alleviating the toxic effect of Aβ deposition[126, 127]. Thus, neuroinflammation promotes amyloid pathology, whereas anti-inflammatory strategies potentially hold promise for alleviating AD.
Accumulating evidence from animal experiments and clinical trials proves the general anti-inflammatory effect of exercise on AD. In AD rats, treadmill exercise improved spatial learning memory function by inhibiting neuronal apoptosis and suppressing pro-inflammatory cytokines via inhibiting NF-κB/MAPK signalling pathway [128]. Moreover, resistance exercise alleviated the locomotor hyperactivity associated with AD behavior and elevated microglia recruitment, which might further contribute to the reduction in the volume of Aβ deposition, and reduced the overexpression of cytokines in APP/PS1 TG mice [129]. Furthermore, 12 weeks of treadmill exercise obviously suppressed oxidative stress, elevated the shift of M1 to M2 microglia polarization, and inhibited neuroinflammation in the hippocampus of APP/PS1 TG mice, which were related to significant improvement of cognition ability and decrease of Aβ deposition [130]. In clinical research, the quality of life and psychological states of AD patients were improved following aerobic exercise training, along with inhibition of systemic inflammation, including the decreased levels of interleukin (IL)-6 and tumor necrosis factor (TNF)-α in serum [131]. Moreover, physical training evidently improved the problem-solving ability and judgment function, and reduced the level of reactive oxygen species, catalase activity, and neuron-specific enolase (a sign of neuronal injury) in AD patients [132]. However, Camilla et al. found that physical exercise exerted a mild inflammatory systematic effect on AD patients, which is inconsistent with abovementioned results [133]. Therefore, the further research is required to confirm the effect of exercise on AD and explore detailed molecular mechanisms of exercise in AD-associated inflammation.
The Effect of the Regulation BBB Transport Proteins by Exercise on AD
The BBB is a dynamically protective boundary between the CNS and the peripheral circulation. The BBB is a highly specific chemical barrier with a semipermeable structure that isolates the brain from circulating blood and extracellular fluids [134, 135]. It provides an extremely stable intracerebral environment and prevents foreign materials, such as toxins or microorganisms, from disrupting the brain homeostasis [136].
The Receptor for Advanced Glycation End Products and Low-Density Lipoprotein Receptor-Related Protein 1
There is a tight balance between Aβ influx and efflux across the BBB, which is disturbed in AD and results in aggregation of toxic protein. This balance partly depends on the BBB receptors; receptor for advanced glycation end products (RAGE) and low-density lipoprotein receptor-related protein 1 (LRP1) control the transport of Aβ into and out of the brain, respectively [137]. RAGE, a multiligand receptor, belongs to the immunoglobulin superfamily. Its ligands consist of Aβ, advanced glycation end products, amphoterin, and S100/calgranulin family members [138]. The increased expression of RAGE in the astroglial and neural cells was reported in animal models of AD and closely related to cognitive impairment and AD pathogenesis [139, 140]. LRP-1 is a lipoprotein receptor in the cytomembrane that induces endocytosis or the cellular internalization of diverse ligands; the ligands for LRP1, which include secreted APP, α2-macroglobulin (α2M), apolipoprotein E, and Aβ, are involved in the pathogenesis of AD [138, 141]. Various studies have reported that LRP1 plays an essential role in the three-step mechanism that regulates Aβ clearance from the brain and body [142]. A recent study showed that the upregulation of LRP1 by vitamin D was responsible for the Aβ clearance in models of AD [143].
Studies have confirmed that physical exercise improves several pathological mechanisms of AD, such as neurovascular unit dysfunction or cognitive deficits, through differential modulation of RAGE and LRP1 to reduce the amyloid plaque load in the brain [144,145,146]. However, in 2018, Zhang et al. found no obvious alteration in LRP1, although they did find a decrease in RAGE in the hippocampus after long-term treadmill exercise, which resulted in the reduction of Aβ deposition mainly through the suppression of the amyloidogenic pathway of APP cleavage [146].
LRP1 in the liver induces rapid peripheral Aβ clearance [147]. In healthy human or mice, plasma soluble LRP1 (sLRP1) binds more than 70% of circulating Aβ in order to prevent it from accessing the brain; it also takes part in maintaining brain Aβ homeostasis [148, 149]. In rats treated with Aβ1–42, researchers found that 4 weeks of treadmill exercise elevated the sLRP1 levels in plasma and the LRP1 protein and mRNA levels in the liver and reduced the levels of circulating sAβ1–42 [145]. Other studies found that soluble RAGE (sRAGE) could restrict the binding of RAGE to its ligands as a decoy in plasma, thus inhibiting a variety of pathological processes of AD, such as oxidative stress and inflammation [150,151,152,153]. One study reported that physical activity for 8 months caused a significant elevation in plasma sRAGE levels in 98 participants [154]. These results highlighted the protective effect of exercise on cardiovascular disease or other diseases, such as AD, that were partially mediated by an increase in inflammatory conditions. In conclusion, RAGE and LRP1 have beneficial effects on Aβ clearance in the brain and in the blood. Exercise training can regulate these two factors and remove generated Aβ to alleviate AD.
The Glucose Transporter 1
Glucose transporter 1 (GLUT1), which is mainly expressed at the BBB, is the major transporter of glucose to mediate glucose enter into the brain [155]. The supply of glucose is vital for maintaining brain energy metabolism homeostasis and supporting the activated nerve cells to function properly [156]. Early decrease in glucose transport related to reduced levels of GLUT1 at the BBB is one of the characterized features of AD [157]. Diminished expression of GLUT1 and GLUT3 (the glucose transporter 3) caused the impairment of brain glucose uptake process in the cerebral cortex of AD patients, resulting in hyperphosphorylation of tau protein [158]. The reduction of GLUT1 at BBB aggravated cognitive dysfunction, cerebrovascular degenerative changes, and neuropathology of AD [159]. In addition, GLUT1 deficiency resulted in decrease of Aβ clearance and facilitated Aβ pathology by decreasing the expression of LRP-1, indicating that GLUT1 reduction could promote the disease process via amplifying vascular injury and Aβ deposition [159]. The regular exercise training increased GLUT1 and GLUT3 expression levels in the central nervous system (CNS) of AD model mice, playing an important role in the process of energy metabolic adaptation [156]. In summary, exercise can reduce Aβ by elevating GLUT1/LRP1 expression levels at BBB in AD. However, the exact mechanism among exercise, GLUT1, LRP1 still needs further exploration.
The Effect of the Regulation of Aβ-Degrading Enzymes by Exercise on AD
In the brain, Aβ clearance can also be enzymic [160]. For example, neprilysin (CD10) and insulin-degrading enzyme (IDE) are two key enzymes that are involved in the clearance of Aβ, especially the proteolytic degradation to monomeric Aβ. These enzymes are expressed in multiple cellular constituents of the brain, including the BBB endothelium [161]. Studies have shown that CD10 and IDE are upregulated in mice with AD after a period of exercise [18, 145]. However, other studies have yielded different results. For instance, Adlard et al. found that exercise induced a reduction in the extracellular Aβ plaques that was independent of CD10 and IDE [162]. The researchers concluded that the reduction in Aβ might be related to neuronal metabolic alterations that were known to be modulated by exercise and to influence APP processing [162]. Zhang et al. discovered that APP/PS1 TG mice that were subjected to 5 months of treadmill exercise had decreased CD10 and IDE expressions, which indicated an inhibited degradation pathway [40]. One possible explanation for these discrepancies is that these studies involved different types and durations of exercises or animal models [138].
Other Mechanisms Involved in the Therapeutic Effect of Exercise on AD
It has been well established that tau phosphorylation is a key pathophysiological change in AD patients. The resistance training inhibited tau pathology and neuroinflammation, and improved synaptic plasticity in the hippocampus and frontal cortex of AD mice [163]. The treadmill exercise exerted inhibitory effects on tau phosphorylation, neuronal apoptosis, and mitochondrial dysfunction, as well as improved hippocampal-dependent cognitive function in the streptozotocin-induced rat model of AD after 4 weeks of treadmill exercise [30]. In addition, voluntary running exercise diminished the loss of neurons and spatial memory, reduced the levels of tau phosphorylation and Aβ accumulation, and increased hippocampal neurogenesis in TG mice of AD [164].
Glycogen synthase kinase-3 (GSK3) that is a main kinase in AD, which can inhibit hippocampal neurogenesis and stimulate neuroinflammation, is a regulator of tau hyperphosphorylation [165, 166]. Its role in APP phosphorylation may be involved in aberrant APP processing and the pathological aggregation of Aβ. A previous study reported that 5 months of treadmill exercise led to a strong reduction in Aβ accumulation and tau phosphorylation, along with a significantly decreased PS1 expression and APP phosphorylation by inhibiting the GSK3-dependent signalling pathway in APP/PS1 mice [167]. Moreover, exercise training could promote the phosphorylation of PI3K/AKT, the upstream precursors of GSK-3, and then suppress the kinase activity of GSK-3 via phosphorylation, thus mitigating the pathological changes of AD [168].
ATP-binding cassette transporter A1 (ABCA1) is a key transmembrane protein that promotes the extracellular transport of cholesterol; it is mainly regulated by retinoid X receptor (RXR) and liver X receptor (LXR) [169]. It has been shown that LXR and RXR accelerate the intracellular cholesterol efflux by modulating ABCA1, which participates in the deposition and transport of Aβ. However, the exact mechanism by which ABCA1 affects APP processing is not clear. One study found that long-term exercise changed cholesterol transportation and reduced soluble Aβ by increasing ABCA1 expression and influencing the levels of RXR and LXR [146].
Developmental Origins of Health and Disease (DOHaD) is a rising field that aims to delay the rapid growth of non-communicable chronic diseases [170]. The environmental exposures during important periods of development may result in subtle alternations in certain biological functions, although almost invisible, and can raise the risk of dysfunction and disease later in life [129]. DOHaD research lays emphasis on how environmental conditions sustained by the developing fetus affect the subsequent development of health or disease in adulthood. Consistently, there is evidence for beneficial effects of physical or cognitive activity at early stage of ontogenesis [171]. Clinical trials showed that maternal exercise during pregnancy positively affected the fetal health and the cognitive functioning of offspring until childhood [172, 173]. Additionally, Herring et al. studied female transgenic (TG) CRND8 mice with the human APP 695 transgene and reported that wheel running during pregnancy alleviated amyloidogenic APP processing and provided long-lasting protection from neurodegeneration in their unstimulated progeny, indicating that maternal exercise interferes with the AD-like pathology of offspring [174]. Therefore, we can conclude that intrauterine milieu mediated by exercise in the period of pregnancy can provide long-lasting benefits to the health of offspring and some resistance against chronic diseases in adult stage. In summary, exercise could exert neuroprotective effects on AD via different types of mechanisms.
The Related Mechanisms of Exercise on Other Neurodegenerative Diseases
Except for AD, exercise also could exert benefits on various neurodegenerative diseases, such as Parkinson’s disease (PD) and Huntington’s disease (HD). PD is characterized by the deficiency of dopaminergic neurons and the existence of Lewy bodies within the substantia nigra [175, 176]. Exercise could markedly improve motor dysfunctions, such as gait and balance, and non-motor disorders, such as cognitive function and quality of life, in PD patient [177]. Moreover, aerobic exercise can exert neuroprotective and neurorestorative effects in PD via modulating neurotrophic factors to promote angiogenesis and synapse formation, enhancing mitochondrial function and suppressing oxidative stress and apoptosis [178]. HD is an autosomal dominant neurodegenerative disease, resulting from polyglutamine expansion in the Huntingtin gene [179]. In HD patients, neuropathology leads to progressive motor dysfunction, cognitive impairment, psychiatric symptom, and peripheral organ disturbances [180]. Exercise training can exert a beneficial role in motor behavior by reducing deficits in mitochondrial function in a HD rodent model [181]. Moreover, a series of studies with physical activity have displayed an improvement in motor function and specific tasks, suggesting that exercise is safe and feasible treatment for HD patients [182]. To sum up, exercise is an effective treatment for various neurodegenerative diseases. More research is required to solve the detailed problems, including the exercise type, appropriate intensity, duration, and so on.
Conclusion
As the elderly population gradually increases and no disease-modifying treatments are available, AD has been one of the central global medical issues in the twenty-first century. AD proceeding is closely related to extracellular accumulation of Aβ in the brain. Exercise provides a cost-effective and non-invasive way to influence many of the mechanisms that have been displayed to reduce Aβ levels and alter AD progression, which can serve as the basis for non-pharmacological means to combat neurodegeneration in AD.
In Table 1, we have summarized some details in the abovementioned literatures on the mechanisms of exercise on Aβ, including models, AD ages/stages (no matter animals or humans), and exercise time and strength in the referenced studies. In addition, most longitudinal prospective studies have confirmed that higher levels of physical exercise are protective against AD dementia and, conversely, lower levels of physical activity are relevant to higher risks of AD development [183,184,185]. Of note, at very high intensity, exercise may become a stressor that has negative effects on human because of the diminished protective response to oxidative stress in brain [187]. Therefore, a medium amount of physical exercise seems to be beneficial for AD patients. Although exercise has multiple positive effects on regulating Aβ and AD, a few studies reported no changes of Aβ after exercise. Such discrepancies are probably explained by the age of mice, the phase of AD progression and different exercise protocols, and environment. Moreover, vigorous exercise might do more harm than good for the elderly. Exercise strategies should be recommended by experts to avoid excessive intensity.
In summary, this review summarized the related mechanisms that are involved in the effect of exercise on AD. However, it remains unclear whether the intensity or duration of the exercise affects Aβ clearance, degradation, and APP processing; it is also unknown how exercise initiates the changes to remove Aβ. Moreover, it needs to be stated that the aggregation of all these mechanisms might be important and acting on a single target might not be sufficient, and exercise might be of interest in that it may act on multiple targets concurrently. The application of exercise to alleviate AD abnormalities still needs further and more detailed research in the future.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I (2010) Role of presenilins in neuronal calcium homeostasis. J Neurosci 30(25):8566–8580. https://doi.org/10.1523/jneurosci.1554-10.2010
Klomparens K, Ding Y (2020) Updates on the association of brain injury and Alzheimer’s disease. Brain Circ 6(2):65–69. https://doi.org/10.4103/bc.bc_18_20
Ferreira D, Perestelo-Pérez L, Westman E, Wahlund LO, Sarría A, Serrano-Aguilar P (2014) Meta-review of CSF core biomarkers in Alzheimer’s disease: the state-of-the-art after the new revised diagnostic criteria. Front Aging Neurosci 6:47. https://doi.org/10.3389/fnagi.2014.00047
Silva MVF, Loures CMG, Alves LCV, de Souza LC, Borges KBG, Carvalho MDG (2019) Alzheimer’s disease: risk factors and potentially protective measures. J Biomed Sci 26(1):33. https://doi.org/10.1186/s12929-019-0524-y
McGurran H, Glenn J, Madero E, Bott N (2019) Prevention and treatment of Alzheimer’s disease: biological mechanisms of exercise. Journal of Alzheimer’s disease : JAD 69(2):311–338. https://doi.org/10.3233/jad-180958
Mao Z-Q, Wang X, Xu X, Cui Z-Q, Pan L-S, Ning X-J, Xu B-X, Ma L, Ling Z-P, Jia J-J, Yu X-G (2018) Partial improvement in performance of patients with severe Alzheimer’s disease at an early stage of fornix deep brain stimulation. Neural Regen Res 13(12):2164–2172. https://doi.org/10.4103/1673-5374.241468
Al-Kuraishy HM, Abdulhadi MH, Hussien NR, Al-Niemi MS, Rasheed HA, Al-Gareeb AI (2020) Involvement of orexinergic system in psychiatric and neurodegenerative disorders: a scoping review. Brain Circ 6(2):70–80. https://doi.org/10.4103/bc.bc_42_19
Klimova B (2017) Mobile phone apps in the management and assessment of mild cognitive impairment and/or mild-to-moderate dementia: an opinion article on recent findings. Front Hum Neurosci 11:461. https://doi.org/10.3389/fnhum.2017.00461
Kalra J, Khan A (2015) Reducing Aβ load and tau phosphorylation: emerging perspective for treating Alzheimer’s disease. Eur J Pharmacol 764:571–581. https://doi.org/10.1016/j.ejphar.2015.07.043
Herrmann N, Chau S, Kircanski I, Lanctôt K (2011) Current and emerging drug treatment options for Alzheimer’s disease: a systematic review. Drugs 71(15):2031–2065. https://doi.org/10.2165/11595870-000000000-00000
Shinohara M, Sato N, Shimamura M, Kurinami H, Hamasaki T, Chatterjee A, Rakugi H, Morishita R (2014) Possible modification of Alzheimer’s disease by statins in midlife: interactions with genetic and non-genetic risk factors. Frontiers in aging neuroscience 6:71. https://doi.org/10.3389/fnagi.2014.00071
Mucke L (2009) Neuroscience: Alzheimer’s disease. Nature 461(7266):895–897. https://doi.org/10.1038/461895a
Vorobyeva AG, Saunders AJ (2018) Amyloid-β interrupts canonical Sonic hedgehog signaling by distorting primary cilia structure. Cilia 7:5. https://doi.org/10.1186/s13630-018-0059-y
Gupta A, Goyal R (2016) Amyloid beta plaque: a culprit for neurodegeneration. Acta Neurol Belg 116(4):445–450. https://doi.org/10.1007/s13760-016-0639-9
Martinelli AHS, Lopes FC, John EBO, Carlini CR, Ligabue-Braun R (2019) Modulation of disordered proteins with a focus on neurodegenerative diseases and other pathologies. Int J Mol Sci 20(6):1322. https://doi.org/10.3390/ijms20061322
Ebrahimi K, Majdi A, Baghaiee B, Hosseini SH, Sadigh-Eteghad S (2017) Physical activity and beta-amyloid pathology in Alzheimer’s disease: a sound mind in a sound body. EXCLI J 16:959–972. https://doi.org/10.17179/excli2017-475
Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2(7):a006338. https://doi.org/10.1101/cshperspect.a006338
Lazarov O, Robinson J, Tang Y, Hairston I, Korade-Mirnics Z, Lee V, Hersh L, Sapolsky R, Mirnics K, Sisodia S (2005) Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120(5):701–713. https://doi.org/10.1016/j.cell.2005.01.015
Sobol NA, Dall CH, Høgh P, Hoffmann K, Frederiksen KS, Vogel A, Siersma V, Waldemar G, Hasselbalch SG, Beyer N (2018) Change in fitness and the relation to change in cognition and neuropsychiatric symptoms after aerobic exercise in patients with mild Alzheimer’s disease. J Alzheimer’s Diss: JAD 65(1):137–145. https://doi.org/10.3233/jad-180253
Kemoun G, Thibaud M, Roumagne N, Carette P, Albinet C, Toussaint L, Paccalin M, Dugué B (2010) Effects of a physical training programme on cognitive function and walking efficiency in elderly persons with dementia. Dement Geriatr Cogn Disord 29(2):109–114. https://doi.org/10.1159/000272435
Holthoff VA, Marschner K, Scharf M, Steding J, Meyer S, Koch R, Donix M (2015) Effects of physical activity training in patients with Alzheimer’s dementia: results of a pilot RCT study. PLoS ONE 10(4):e0121478. https://doi.org/10.1371/journal.pone.0121478
Hoffmann K, Sobol NA, Frederiksen KS, Beyer N, Vogel A, Vestergaard K, Brændgaard H, Gottrup H, Lolk A, Wermuth L, Jacobsen S, Laugesen LP, Gergelyffy RG, Høgh P, Bjerregaard E, Andersen BB, Siersma V, Johannsen P, Cotman CW, Waldemar G, Hasselbalch SG (2016) Moderate-to-high intensity physical exercise in patients with Alzheimer’s disease: a randomized controlled trial. J Alzheimer’s Dis : JAD 50(2):443–453. https://doi.org/10.3233/jad-150817
Toots A, Littbrand H, Boström G, Hörnsten C, Holmberg H, Lundin-Olsson L, Lindelöf N, Nordström P, Gustafson Y, Rosendahl E (2017) Effects of exercise on cognitive function in older people with dementia: a randomized controlled trial. J Alzheimer’s Dis: JAD 60(1):323–332. https://doi.org/10.3233/jad-170014
Sobol NA, Hoffmann K, Frederiksen KS, Vogel A, Vestergaard K, Brændgaard H, Gottrup H, Lolk A, Wermuth L, Jakobsen S, Laugesen L, Gergelyffy R, Høgh P, Bjerregaard E, Siersma V, Andersen BB, Johannsen P, Waldemar G, Hasselbalch SG, Beyer N (2016) Effect of aerobic exercise on physical performance in patients with Alzheimer’s disease. Alzheimers Dement 12(12):1207–1215. https://doi.org/10.1016/j.jalz.2016.05.004
Panza GA, Taylor BA, MacDonald HV, Johnson BT, Zaleski AL, Livingston J, Thompson PD, Pescatello LS (2018) Can exercise improve cognitive symptoms of Alzheimer’s disease? J Am Geriatr Soc 66(3):487–495. https://doi.org/10.1111/jgs.15241
Colcombe S, Kramer AF (2003) Fitness effects on the cognitive function of older adults: a meta-analytic study. Psychol Sci 14(2):125–130. https://doi.org/10.1111/1467-9280.t01-1-01430
Lancioni GE, Singh NN, O’Reilly MF, Sigafoos J, D’Amico F, Addante LM, Pinto K (2017) Persons with advanced Alzheimer’s disease engage in mild leg exercise supported by technology-aided stimulation and prompts. Behav Modif 41(1):3–20. https://doi.org/10.1177/0145445516649581
Lancioni GE, Singh NN, O’Reilly MF, Sigafoos J, D’Amico F, Turnone B, Laporta D, Scordamaglia A, Pinto K (2019) Smartphone-based interventions to foster simple activity and personal satisfaction in people with advanced Alzheimer’s disease. Am J Alzheimers Dis Other Demen 34(7–8):478–485. https://doi.org/10.1177/1533317519844144
Öhman H, Savikko N, Strandberg T, Kautiainen H, Raivio M, Laakkonen ML, Tilvis R, Pitkälä KH (2016) Effects of exercise on functional performance and fall rate in subjects with mild or advanced Alzheimer’s disease: secondary analyses of a randomized controlled study. Dement Geriatr Cogn Disord 41(3–4):233–241. https://doi.org/10.1159/000445712
Lu Y, Dong Y, Tucker D, Wang R, Ahmed ME, Brann D, Zhang Q (2017) Treadmill exercise exerts neuroprotection and regulates microglial polarization and oxidative stress in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. J Alzheimer’s Dis : JAD 56(4):1469–1484. https://doi.org/10.3233/JAD-160869
McGurran H, Glenn JM, Madero EN, Bott NT (2019) Prevention and treatment of Alzheimer’s disease: biological mechanisms of exercise. J Alzheimer’s Dis: JAD 69(2):311–338. https://doi.org/10.3233/JAD-180958
Rosa J, Camargo A, Wolin I, Kaster M, Rodrigues A (2020) Physical exercise prevents amyloid β-induced disturbances in NLRP3 inflammasome pathway in the hippocampus of mice. Metab Brain Dis. https://doi.org/10.1007/s11011-020-00646-8
Alkadhi KA, Dao AT (2018) Exercise decreases BACE and APP levels in the hippocampus of a rat model of Alzheimer’s disease. Mol Cell Neurosci 86:25–29. https://doi.org/10.1016/j.mcn.2017.11.008
Cho J, Shin MK, Kim D, Lee I, Kim S, Kang H (2015) Treadmill running reverses cognitive declines due to Alzheimer Disease. Med Sci Sports Exerc 47(9):1814–1824. https://doi.org/10.1249/MSS.0000000000000612
Zhang J, Guo Y, Wang Y, Song L, Zhang R, Du Y (2018) Long-term treadmill exercise attenuates Abeta burdens and astrocyte activation in APP/PS1 mouse model of Alzheimer’s disease. Neurosci Lett 666:70–77. https://doi.org/10.1016/j.neulet.2017.12.025
Rao SK, Ross JM, Harrison FE, Bernardo A, Reiserer RS, Reiserer RS, Mobley JA, McDonald MP (2015) Differential proteomic and behavioral effects of long-term voluntary exercise in wild-type and APP-overexpressing transgenics. Neurobiol Dis 78:45–55. https://doi.org/10.1016/j.nbd.2015.03.018
Yu F, Xu B, Song C, Ji L, Zhang X (2013) Treadmill exercise slows cognitive deficits in aging rats by antioxidation and inhibition of amyloid production. NeuroReport 24(6):342–347. https://doi.org/10.1097/WNR.0b013e3283606c5e
Zhao G, Liu HL, Zhang H, Tong XJ (2015) Treadmill exercise enhances synaptic plasticity, but does not alter beta-amyloid deposition in hippocampi of aged APP/PS1 transgenic mice. Neurosci 298:357–366. https://doi.org/10.1016/j.neuroscience.2015.04.038
Souza LC, Filho CB, Goes ATR, Fabbro LD, de Gomes MG, Savegnago L, Oliveira MS, Jesse CR (2013) Neuroprotective effect of physical exercise in a mouse model of Alzheimer’s disease induced by β-amyloid1–40 peptide. Neurotox Res 24(2):148–163. https://doi.org/10.1007/s12640-012-9373-0
Maliszewska-Cyna E, Xhima K, Aubert I (2016) A comparative study evaluating the impact of physical exercise on disease progression in a mouse model of Alzheimer’s disease. J Alzheimer’s Dis: JAD 53(1):243–257. https://doi.org/10.3233/JAD-150660
Wang X, Zhou X, Li G, Zhang Y, Wu Y, Song W (2017) Modifications and trafficking of APP in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci 10:294. https://doi.org/10.3389/fnmol.2017.00294
Acevedo KM, Hung YH, Dalziel AH, Li QX, Laughton K, Wikhe K, Rembach A, Roberts B, Masters CL, Bush AI, Camakaris J (2011) Copper promotes the trafficking of the amyloid precursor protein. J Biol Chem 286(10):8252–8262. https://doi.org/10.1074/jbc.M110.128512
Liu X, Wang Z, Wu Y, Wang J, Song W (2013) BACE2 degradation mediated by the macroautophagy-lysosome pathway. Eur J Neurosci 37(12):1970–1977. https://doi.org/10.1111/ejn.12204
Gandy S (2005) The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Investig 115(5):1121–1129. https://doi.org/10.1172/jci25100
Hannaoui S, Shim SY, Cheng YC, Corda E, Gilch S (2014) Cholesterol balance in prion diseases and Alzheimer’s disease. Viruses 6(11):4505–4535. https://doi.org/10.3390/v6114505
Martorana A, Di Lorenzo F, Belli L, Sancesario G, Toniolo S, Sallustio F, Sancesario GM, Koch G (2015) Cerebrospinal fluid Aβ42 levels: when physiological become pathological state. CNS Neurosci Ther 21(12):921–925. https://doi.org/10.1111/cns.12476
De-Paula VJ, Radanovic M, Diniz BS, Forlenza OV (2012) Alzheimer’s disease. Subcell Biochem 65:329–352. https://doi.org/10.1007/978-94-007-5416-4_14
Hung S-Y, Fu W-M (2017) Drug candidates in clinical trials for Alzheimer’s disease. J Biomed Sci 24(1):47. https://doi.org/10.1186/s12929-017-0355-7
Chen H, Dang Y, Liu X, Ren J, Wang H (2019) Exogenous brain-derived neurotrophic factor attenuates neuronal apoptosis and neurological deficits after subarachnoid hemorrhage in rats. Exp Ther Med 18(5):3837–3844. https://doi.org/10.3892/etm.2019.8029
He W, Tian X, Yuan B, Chu B, Gao F, Wang H (2019) Rosuvastatin improves neurite extension in cortical neurons through the Notch 1/BDNF pathway. Neurol Res 41(7):658–664. https://doi.org/10.1080/01616412.2019.1610226
O’Brien R, Wong P (2011) Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34:185–204. https://doi.org/10.1146/annurev-neuro-061010-113613
Weinstein G, Beiser A, Choi S, Preis S, Chen T, Vorgas D, Au R, Pikula A, Wolf P, DeStefano A, Vasan R, Seshadri S (2014) Serum brain-derived neurotrophic factor and the risk for dementia: the Framingham Heart Study. JAMA Neurol 71(1):55–61. https://doi.org/10.1001/jamaneurol.2013.4781
Nigam SM, Xu S, Kritikou JS, Marosi K, Brodin L, Mattson MP (2017) Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J Neurochem 142(2):286–296. https://doi.org/10.1111/jnc.14034
Zagaar M, Alhaider I, Dao A, Levine A, Alkarawi A, Alzubaidy M, Alkadhi K (2012) The beneficial effects of regular exercise on cognition in REM sleep deprivation: behavioral, electrophysiological and molecular evidence. Neurobiol Dis 45(3):1153–1162. https://doi.org/10.1016/j.nbd.2011.12.039
Dao AT, Zagaar MA, Levine AT, Salim S, Eriksen JL, Alkadhi KA (2013) Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr Alzheimer Res 10(5):507–515. https://doi.org/10.2174/1567205011310050006
Lin TW, Shih YH, Chen SJ, Lien CH, Chang CY, Huang TY, Chen SH, Jen CJ, Kuo YM (2015) Running exercise delays neurodegeneration in amygdala and hippocampus of Alzheimer’s disease (APP/PS1) transgenic mice. Neurobiol Learn Mem 118:189–197. https://doi.org/10.1016/j.nlm.2014.12.005
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450(7170):712–716. https://doi.org/10.1038/nature06261
Koo JH, Kang EB, Oh YS, Yang DS, Cho JY (2017) Treadmill exercise decreases amyloid-beta burden possibly via activation of SIRT-1 signaling in a mouse model of Alzheimer’s disease. Exp Neurol 288:142–152. https://doi.org/10.1016/j.expneurol.2016.11.014
Koronowski KB, Perez-Pinzon MA (2015) Sirt1 in cerebral ischemia. Brain Circ 1(1):69–78. https://doi.org/10.4103/2394-8108.162532
Russo E, Nguyen H, Lippert T, Tuazon J, Borlongan CV, Napoli E (2018) Mitochondrial targeting as a novel therapy for stroke. Brain Circ 4(3):84–94. https://doi.org/10.4103/bc.bc_14_18
Donmez G (2012) The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol Sci 33(9):494–501. https://doi.org/10.1016/j.tips.2012.05.007
Zhang Z, Shen Q, Wu X, Zhang D, Xing D (2020) Activation of PKA/SIRT1 signaling pathway by photobiomodulation therapy reduces Abeta levels in Alzheimer’s disease models. Aging Cell 19(1):e13054. https://doi.org/10.1111/acel.13054
Shah S, Yoon G, Chung S, Abid M, Kim T, Lee H, Kim M (2017) Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol Psychiatry 22(3):407–416. https://doi.org/10.1038/mp.2016.23
Porquet D, Casadesús G, Bayod S, Vicente A, Canudas A, Vilaplana J, Pelegrí C, Sanfeliu C, Camins A, Pallàs M, del Valle J (2013) Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age (Dordr) 35(5):1851–1865. https://doi.org/10.1007/s11357-012-9489-4
Xia D, Huang X, Bi C, Mao L, Peng L, Qian H (2017) PGC-1α or FNDC5 is involved in modulating the effects of Aβ oligomers on suppressing the expression of BDNF, a beneficial factor for inhibiting neuronal apoptosis, Aβ deposition and cognitive decline of APP/PS1 Tg mice. Front aging Neurosci 9:65. https://doi.org/10.3389/fnagi.2017.00065
Albrecht E, Norheim F, Thiede B, Holen T, Ohashi T, Schering L, Lee S, Brenmoehl J, Thomas S, Drevon C, Erickson H, Maak S (2015) Irisin—a myth rather than an exercise-inducible myokine. Sci Rep 5:8889. https://doi.org/10.1038/srep08889
Lourenco M, Frozza R, de Freitas G, Zhang H, Kincheski G, Ribeiro F, Gonçalves R, Clarke J, Beckman D, Staniszewski A, Berman H, Guerra L, Forny-Germano L, Meier S, Wilcock D, de Souza J, Alves-Leon S, Prado V, Prado M, Abisambra J, Tovar-Moll F, Mattos P, Arancio O, Ferreira S, De Felice F (2019) Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat Med 25(1):165–175. https://doi.org/10.1038/s41591-018-0275-4
Noda Y, Kuzuya A, Tanigawa K, Araki M, Kawai R, Ma B, Sasakura Y, Maesako M, Tashiro Y, Miyamoto M, Uemura K, Okuno Y, Kinoshita A (2018) Fibronectin type III domain-containing protein 5 interacts with APP and decreases amyloid beta production in Alzheimer’s disease. Mol Brain 11(1):61. https://doi.org/10.1186/s13041-018-0401-8
Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, Kim E, Rompala A, Oram MK, Asselin C, Aronson J, Zhang C, Miller SJ, Lesinski A, Chen JW, Kim DY, van Praag H, Spiegelman BM, Gage FH, Tanzi RE (2018) Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 361(6406):eaan8821. https://doi.org/10.1126/science.aan8821
Cui L, Jeong H, Borovecki F, Parkhurst C, Tanese N, Krainc D (2006) Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127(1):59–69. https://doi.org/10.1016/j.cell.2006.09.015
Liu Y, Chu J, Yan T, Zhang Y, Chen Y, Chang R, Wong G (2020) Short-term resistance exercise inhibits neuroinflammation and attenuates neuropathological changes in 3xTg Alzheimer’s disease mice. J Neuroinflammation 17(1):4. https://doi.org/10.1186/s12974-019-1653-7
Jodeiri Farshbaf M, Ghaedi K, Megraw T, Curtiss J, Shirani Faradonbeh M, Vaziri P, Nasr-Esfahani M (2016) Does PGC1α/FNDC5/BDNF elicit the beneficial effects of exercise on neurodegenerative disorders? NeuroMol Med 18(1):1–15. https://doi.org/10.1007/s12017-015-8370-x
Wang R, Li J, Diao S, Kwak Y, Liu L, Zhi L, Büeler H, Bhat N, Williams R, Park E, Liao F (2013) Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab 17(5):685–694. https://doi.org/10.1016/j.cmet.2013.03.016
Wrann C, White J, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, Lin J, Greenberg M, Spiegelman B (2013) Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab 18(5):649–659. https://doi.org/10.1016/j.cmet.2013.09.008
Azimi M, Gharakhanlou R, Naghdi N, Khodadadi D, Heysieattalab S (2018) Moderate treadmill exercise ameliorates amyloid-β-induced learning and memory impairment, possibly via increasing AMPK activity and up-regulation of the PGC-1α/FNDC5/BDNF pathway. Peptides 102:78–88. https://doi.org/10.1016/j.peptides.2017.12.027
Y J, DM S, J C, H K, SR L, Y H (2018) Molecular and functional interaction of the myokine irisin with physical exercise and Alzheimer’s disease. Molecules (Basel, Switzerland) 23(12):3229. https://doi.org/10.3390/molecules23123229
Lee H, Khummuang S, Youn H, Park J, Choi J, Shin T, Cho S, Kim B, Seo J, Kim M, Park T, Cho B (2019) The effect of heat stress on frame switch splicing of X-box binding protein 1 gene in horse. Asian Australas J Anim Sci 32(8):1095–1103. https://doi.org/10.5713/ajas.18.0757
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13(2):89–102. https://doi.org/10.1038/nrm3270
Ma T, Klann E (2014) PERK: a novel therapeutic target for neurodegenerative diseases? Alzheimer’s research & therapy 6(3):30. https://doi.org/10.1186/alzrt260
Lee D, Lee K, Lee H, Kim D, Noh Y, Yu K, Jung H, Lee S, Lee J, Youn Y, Jeong Y, Kim D, Lee W, Kim S (2010) Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS ONE 5(5):e10489. https://doi.org/10.1371/journal.pone.0010489
Kang E, Kwon I, Koo J, Kim E, Kim C, Lee J, Yang C, Lee Y, Cho I, Cho J (2013) Treadmill exercise represses neuronal cell death and inflammation during Aβ-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis : an international journal on programmed cell death 18(11):1332–1347. https://doi.org/10.1007/s10495-013-0884-9
Devi L, Ohno M (2014) PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol Aging 35(10):2272–2281. https://doi.org/10.1016/j.neurobiolaging.2014.04.031
Ohta K, Mizuno A, Li S, Itoh M, Ueda M, Ohta E, Hida Y, Wang M, Furoi M, Tsuzuki Y, Sobajima M, Bohmoto Y, Fukushima T, Kobori M, Inuzuka T, Nakagawa T (2011) Endoplasmic reticulum stress enhances γ-secretase activity. Biochem Biophys Res Commun 416:362–366. https://doi.org/10.1016/j.bbrc.2011.11.042
Xia J, Li B, Yin L, Zhao N, Yan Q, Xu B (2019) Treadmill exercise decreases β-amyloid burden in APP/PS1 transgenic mice involving regulation of the unfolded protein response. Neurosci Lett 703:125–131. https://doi.org/10.1016/j.neulet.2019.03.035
Ready D, Yagiz K, Amin P, Yildiz Y, Funari V, Bozdag S, Cinar B (2017) Mapping the STK4/Hippo signaling network in prostate cancer cell. PLoS ONE 12(9):e0184590. https://doi.org/10.1371/journal.pone.0184590
Motoki K, Kume H, Oda A, Tamaoka A, Hosaka A, Kametani F, Araki W (2012) Neuronal β-amyloid generation is independent of lipid raft association of β-secretase BACE1: analysis with a palmitoylation-deficient mutant. Brain and behavior 2(3):270–282. https://doi.org/10.1002/brb3.52
Ehehalt R, Keller P, Haass C, Thiele C, Simons K (2003) Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160(1):113–123. https://doi.org/10.1083/jcb.200207113
Zhang X-L, Zhao N, Xu B, Chen X-H, Li T-J (2019) Treadmill exercise inhibits amyloid-β generation in the hippocampus of APP/PS1 transgenic mice by reducing cholesterol-mediated lipid raft formation. NeuroReport 30(7):498–503. https://doi.org/10.1097/wnr.0000000000001230
Yang L, Wang H, Shen Q, Feng L, Jin H (2017) Long non-coding RNAs involved in autophagy regulation. Cell Death Dis 8(10):e3073. https://doi.org/10.1038/cddis.2017.464
Dong W, Wang R (2016) [Effects of resveratrol-induced cellular autophagy in control of neurodegenerative diseases]. Yao xue xue bao = Acta pharmaceutica Sinica 51(1):18–22
Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, Ginsberg SD, Nixon RA (2016) Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12(12):2467–2483. https://doi.org/10.1080/15548627.2016.1239003
Kou X, Chen D, Chen N (2019) Physical activity alleviates cognitive dysfunction of Alzheimer’s disease through regulating the mTOR signaling pathway. Int J Mol Sci 20(7):1591. https://doi.org/10.3390/ijms20071591
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Investig 118(6):2190–2199. https://doi.org/10.1172/jci33585
Li Y, Sun D, Zheng Y, Cheng Y (2020) Swimming exercise activates aortic autophagy and limits atherosclerosis in ApoE mice. Obes Res Clin Pract. https://doi.org/10.1016/j.orcp.2020.04.008
Zhao N, Zhang X, Song C, Yang Y, He B, Xu B (2018) The effects of treadmill exercise on autophagy in hippocampus of APP/PS1 transgenic mice. NeuroReport 29(10):819–825. https://doi.org/10.1097/wnr.0000000000001038
Kang E-B, Cho J-Y (2015) Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. J Exerc Nutr Biochem 19(3):199–209. https://doi.org/10.5717/jenb.2015.15090806
Xing Y, Yang S-D, Wang M-M, Feng Y-S, Dong F, Zhang F (2019) The beneficial roles of exercise training via autophagy in neurological diseases and possible mechanisms. Life Sci 221:130–134. https://doi.org/10.1016/j.lfs.2019.02.026
Xia M, Yang L, Sun G, Qi S, Li B (2017) Mechanism of depression as a risk factor in the development of Alzheimer’s disease: the function of AQP4 and the glymphatic system. Psychopharmacol 234(3):365–379. https://doi.org/10.1007/s00213-016-4473-9
Plog BA, Nedergaard M (2018) The glymphatic system in central nervous system health and disease: past, present, and future. Annu Rev Pathol 13:379–394. https://doi.org/10.1146/annurev-pathol-051217-111018
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes including amyloid. Sci Transl Med 4(147):147ra111-147ra111. https://doi.org/10.1126/scitranslmed.3003748
Iliff JJ, Nedergaard M (2013) Is there a cerebral lymphatic system? Stroke 44(6 Suppl 1):S93-95. https://doi.org/10.1161/strokeaha.112.678698
Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, Regan S, Kasper T, Peng S, Ding F, Benveniste H, Nedergaard M, Deane R (2016) Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol Dis 93:215–225. https://doi.org/10.1016/j.nbd.2016.05.015
Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M (2014) Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 76(6):845–861. https://doi.org/10.1002/ana.24271
Mee-Inta O, Zhao ZW, Kuo YM (2019) Physical exercise inhibits inflammation and microglial activation(2019) Cells 8(7):691. https://doi.org/10.3390/cells8070691
Jessen NA, Munk AS, Lundgaard I, Nedergaard M (2015) The glymphatic system: a beginner’s guide. Neurochem Res 40(12):2583–2599. https://doi.org/10.1007/s11064-015-1581-6
Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nat Rev Neurosci 14(4):265–277. https://doi.org/10.1038/nrn3468
Ea N, Ottersen OP (2013) Physiological roles of aquaporin-4 in brain. Physiol Rev 93(4):1543–1562. https://doi.org/10.1152/physrev.00011.2013
Burfeind KG, Murchison CF, Westaway SK, Simon MJ, Erten-Lyons D, Kaye JA, Quinn JF, Iliff JJ (2017) The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 3(3):348–359. https://doi.org/10.1016/j.trci.2017.05.001
Xu Z, Xiao N, Chen Y, Huang H, Marshall C, Gao J, Cai Z, Wu T, Hu G, Xiao M (2015) Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol Neurodegener 10:58. https://doi.org/10.1186/s13024-015-0056-1
von Holstein-Rathlou S, Petersen NC, Nedergaard M (2018) Voluntary running enhances glymphatic influx in awake behaving, young mice. Neurosci Lett 662:253–258. https://doi.org/10.1016/j.neulet.2017.10.035
He XF, Liu DX, Zhang Q, Liang FY, Dai GY, Zeng JS, Pei Z, Xu GQ, Lan Y (2017) Voluntary exercise promotes glymphatic clearance of amyloid beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci 10:144. https://doi.org/10.3389/fnmol.2017.00144
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M (2013) Sleep drives metabolite clearance from the adult brain. Science 342(6156):373–377. https://doi.org/10.1126/science.1241224
Rainey-Smith SR, Mazzucchelli GN, Villemagne VL, Brown BM, Porter T, Weinborn M, Bucks RS, Milicic L, Sohrabi HR, Taddei K, Ames D, Maruff P, Masters CL, Rowe CC, Salvado O, Martins RN, Laws SM, Group AR (2018) Genetic variation in Aquaporin-4 moderates the relationship between sleep and brain Abeta-amyloid burden. Transl Psychiatry 8(1):47. https://doi.org/10.1038/s41398-018-0094-x
Burke SL, Hu T, Spadola CE, Burgess A, Li T, Cadet T (2019) Treatment of sleep disturbance may reduce the risk of future probable Alzheimer’s disease. J Aging Health 31(2):322–342. https://doi.org/10.1177/0898264318795567
Brown BM, Rainey-Smith SR, Villemagne VL, Weinborn M, Bucks RS, Sohrabi HR, Laws SM, Taddei K, Macaulay SL, Ames D, Fowler C, Maruff P, Masters CL, Rowe CC, Martins RN (2016) The relationship between sleep quality and brain amyloid burden. Sleep 39(5):1063–1068. https://doi.org/10.5665/sleep.5756
Memon AA, Coleman JJ, Amara AW (2020) Effects of exercise on sleep in neurodegenerative disease. Neurobiol Dis 140:104859. https://doi.org/10.1016/j.nbd.2020.104859
Nascimento C, Ayan C, Cancela J, Gobbi L, Gobbi S, Stella F (2014) Effect of a multimodal exercise program on sleep disturbances and instrumental activities of daily living performance on Parkinson’s and Alzheimer’s disease patients. Geriatr Gerontol Int 14(2):259–266. https://doi.org/10.1111/ggi.12082
Jia R-X, Liang J-H, Xu Y, Wang Y-Q (2019) Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: a meta-analysis. BMC Geriatr 19(1):181. https://doi.org/10.1186/s12877-019-1175-2
Fragoulis A, Siegl S, Fendt M, Jansen S, Soppa U, Brandenburg L, Pufe T, Weis J, Wruck C (2017) Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol 12:843–853. https://doi.org/10.1016/j.redox.2017.04.024
Schimmel SJ, Acosta S, Lozano D (2017) Neuroinflammation in traumatic brain injury: a chronic response to an acute injury. Brain Circ 3(3):135–142. https://doi.org/10.4103/bc.bc_18_17
Kelly AM (2018) Exercise-induced modulation of neuroinflammation in models of Alzheimer’s disease. Brain plasticity 4(1):81–94. https://doi.org/10.3233/BPL-180074
Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J (2004) Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging 25(5):663–74. https://doi.org/10.1016/j.neurobiolaging.2004.01.007
Lian H, Litvinchuk A, Chiang A, Aithmitti N, Jankowsky J, Zheng H (2016) Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci 36(2):577–589. https://doi.org/10.1523/jneurosci.2117-15.2016
Li W, Yang S (2016) Targeting oxidative stress for the treatment of ischemic stroke: upstream and downstream therapeutic strategies. Brain Circ 2(4):153–163. https://doi.org/10.4103/2394-8108.195279
Tang Y, Le W (2016) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53(2):1181–1194. https://doi.org/10.1007/s12035-014-9070-5
Choy F, Klarić T, Koblar S, Lewis M (2015) The role of the neuroprotective factor Npas4 in cerebral ischemia. Int J Mol Sci 16(12):29011–29028. https://doi.org/10.3390/ijms161226144
Borges-Machado F, Ribeiro O, Sampaio A, Marques-Aleixo I, Meireles J, Carvalho J (2019) Feasibility and impact of a multicomponent exercise intervention in patients with Alzheimer’s disease: a pilot study. Am J Alzheimers Dis Other Demen 34(2):95–103. https://doi.org/10.1177/1533317518813555
Roura LC, Arulkumaran SS (2015) Facing the noncommunicable disease (NCD) global epidemic—the battle of prevention starts in utero—the FIGO challenge. Best Pract Res Clin Obstet Gynaecol 29(1):5–14. https://doi.org/10.1016/j.bpobgyn.2014.04.018
Zhang X, He Q, Huang T, Zhao N, Liang F, Xu B, Chen X, Li T, Bi J (2019) Treadmill exercise decreases Abeta deposition and counteracts cognitive decline in APP/PS1 mice, possibly via hippocampal microglia modifications. Front aging Neurosci 11:78. https://doi.org/10.3389/fnagi.2019.00078
Abd El-Kader SM, Al-Jiffri OH (2016) Aerobic exercise improves quality of life, psychological well-being and systemic inflammation in subjects with Alzheimer’s disease. Afr Health Sci 16(4):1045–1055. https://doi.org/10.4314/ahs.v16i4.22
Morris JK, Vidoni ED, Johnson DK, Van Sciver A, Mahnken JD, Honea RA, Wilkins HM, Brooks WM, Billinger SA, Swerdlow RH, Burns JM (2017) Aerobic exercise for Alzheimer’s disease: A randomized controlled pilot trial. PLoS One 12(2):e0170547. https://doi.org/10.1371/journal.pone.0170547
Jensen CS, Bahl JM, Ostergaard LB, Hogh P, Wermuth L, Heslegrave A, Zetterberg H, Heegaard NHH, Hasselbalch SG, Simonsen AH (2019) Exercise as a potential modulator of inflammation in patients with Alzheimer’s disease measured in cerebrospinal fluid and plasma. Exp Gerontol 121:91–98. https://doi.org/10.1016/j.exger.2019.04.003
Najjar S, Pahlajani S, De Sanctis V, Stern JNH, Najjar A, Chong D (2017) Neurovascular unit dysfunction and blood-brain barrier hyperpermeability contribute to schizophrenia neurobiology: a theoretical integration of clinical and experimental evidence. Front Psych 8:83. https://doi.org/10.3389/fpsyt.2017.00083
Vandenhaute E, Dehouck L, Boucau M-C, Sevin E, Uzbekov R, Tardivel M, Gosselet F, Fenart L, Cecchelli R, Dehouck M-P (2011) Modelling the neurovascular unit and the blood-brain barrier with the unique function of pericytes. Curr Neurovasc Res 8(4):258–269. https://doi.org/10.2174/156720211798121016
Cai Z, Qiao P-F, Wan C-Q, Cai M, Zhou N-K, Li Q (2018) Role of blood-brain barrier in Alzheimer’s disease. J Alzheimers Dis 63(4):1223–1234. https://doi.org/10.3233/jad-180098
Trigiani LJ, Hamel E (2017) An endothelial link between the benefits of physical exercise in dementia. J Cereb Blood Flow Metab 37(8):2649–2664. https://doi.org/10.1177/0271678X17714655
Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, Martins RN (2008) Clearance mechanisms of Alzheimer’s amyloid-β peptide: implications for therapeutic design and diagnostic tests. Mol Psychiatry 14(5):469–486. https://doi.org/10.1038/mp.2008.96
Choi BR, Cho WH, Kim J, Lee HJ, Chung C, Jeon WK, Han JS (2014) Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp Mol Med 46(2):e75. https://doi.org/10.1038/emm.2013.147
Wang MY, Ross-Cisneros FN, Aggarwal D, Liang CY, Sadun AA (2009) Receptor for advanced glycation end products is upregulated in optic neuropathy of Alzheimer’s disease. Acta Neuropathol 118(3):381–389. https://doi.org/10.1007/s00401-009-0513-4
Estrada LD, Ahumada P, Cabrera D, Arab JP (2019) Liver dysfunction as a novel player in Alzheimer’s progression: looking outside the brain. Front aging Neurosci 11:174. https://doi.org/10.3389/fnagi.2019.00174
Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA (2010) Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer’s amyloid β-peptide elimination from the brain. J Neurochem 115(5):1077–1089. https://doi.org/10.1111/j.1471-4159.2010.07002.x
Patel P, Shah J (2017) Role of vitamin D in amyloid clearance via LRP-1 upregulation in Alzheimer’s disease: a potential therapeutic target? J Chem Neuroanat 85:36–42. https://doi.org/10.1016/j.jchemneu.2017.06.007
Herring A, Yasin H, Ambree O, Sachser N, Paulus W, Keyvani K (2008) Environmental enrichment counteracts Alzheimer’s neurovascular dysfunction in TgCRND8 mice. Brain Pathol 18(1):32–39. https://doi.org/10.1111/j.1750-3639.2007.00094.x
Khodadadi D, Gharakhanlou R, Naghdi N, Salimi M, Azimi M, Shahed A, Heysieattalab S (2018) Treadmill exercise ameliorates spatial learning and memory deficits through improving the clearance of peripheral and central amyloid-beta levels. Neurochem Res 43(8):1561–1574. https://doi.org/10.1007/s11064-018-2571-2
Zeng B, Zhao G, Liu HL (2020) The differential effect of treadmill exercise intensity on hippocampal soluble Aβ and lipid metabolism in APP/PS1 mice. Neuroscience 430:73–81. https://doi.org/10.1016/j.neuroscience.2020.01.005
Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, Fossati S (2018) Traumatic brain injury and Alzheimer’s disease: the cerebrovascular link. EBioMedicine 28:21–30. https://doi.org/10.1016/j.ebiom.2018.01.021
Sagare AP, Deane R, Zlokovic BV (2012) Low-density lipoprotein receptor-related protein 1: a physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacol Ther 136(1):94–105. https://doi.org/10.1016/j.pharmthera.2012.07.008
Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV (2007) Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med 13(9):1029–1031. https://doi.org/10.1038/nm1635
Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM (1997) Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA 94(10):5296–5301. https://doi.org/10.1073/pnas.94.10.5296
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97(7):889–901. https://doi.org/10.1016/s0092-8674(00)80801-6
Prasad K (2016) Is elevated levels of serum soluble receptor for advanced glycation end products harmful in cigarette smokers? Int J Angiol 25(3):199–202. https://doi.org/10.1055/s-0036-1579691
Gao L, Jiang Y, Wei S, Shang S, Li P, Chen C, Dang L, Wang J, Huo K, Deng M, Wang J, Zhang R, Qu Q (2018) The Level of plasma amyloid-β40 is correlated with peripheral transport proteins in cognitively normal adults: a population-based cross-sectional study. J Alzheimers Dis 65(3):951–961. https://doi.org/10.3233/jad-180399
Sponder M, Campean IA, Emich M, Fritzer-Szekeres M, Litschauer B, Graf S, Dalos D, Strametz-Juranek J (2018) Long-term physical activity leads to a significant increase in serum sRAGE levels: a sign of decreased AGE-mediated inflammation due to physical activity? Heart Vessels 33(8):893–900. https://doi.org/10.1007/s00380-018-1125-5
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12(12):723–738. https://doi.org/10.1038/nrn3114
Pang RQ, Wang XF, Pei FF, Zhang W, Shen J, Gao X, Chang C (2019) Regular exercise enhances cognitive function and intracephalic GLUT expression in Alzheimer’s disease model mice. J Alzheimer’s Dis : JAD. https://doi.org/10.3233/JAD-190328
Bonfili L, Cecarini V, Gogoi O, Berardi S, Scarpona S, Angeletti M, Rossi G, Eleuteri AM (2020) Gut microbiota manipulation through probiotics oral administration restores glucose homeostasis in a mouse model of Alzheimer’s disease. Neurobiol Aging 87:35–43. https://doi.org/10.1016/j.neurobiolaging.2019.11.004
Bloom GS (2014) Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71(4):505–508. https://doi.org/10.1001/jamaneurol.2013.5847
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P, Nelson AR, Sullivan JS, Zhao Z, Meiselman HJ, Wendy RB, Soto J, Abel ED, Makshanoff J, Zuniga E, De Vivo DC, Zlokovic BV (2015) GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18(4):521–530. https://doi.org/10.1038/nn.3966
Nalivaeva NN, Belyaev ND, Kerridge C, Turner AJ (2014) Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer’s disease. Frontiers in aging neuroscience 6:235. https://doi.org/10.3389/fnagi.2014.00235
Cheng S, LeBlanc KJ, Li L (2014) Triptolide preserves cognitive function and reduces neuropathology in a mouse model of Alzheimer’s disease. PLoS ONE 9(9):e108845. https://doi.org/10.1371/journal.pone.0108845
Adlard PA (2005) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci 25(17):4217–4221. https://doi.org/10.1523/jneurosci.0496-05.2005
Liu Y, Chu JMT, Yan T, Zhang Y, Chen Y, Chang RCC, Wong GTC (2020) Short-term resistance exercise inhibits neuroinflammation and attenuates neuropathological changes in 3xTg. Alzheimer’s disease mice 17(1):4. https://doi.org/10.1186/s12974-019-1653-7
Tapia-Rojas C, Aranguiz F, Varela-Nallar L, Inestrosa NC (2016) Voluntary running attenuates memory loss, decreases neuropathological changes and induces neurogenesis in a mouse model of Alzheimer’s disease. Brain Pathol 26(1):62–74. https://doi.org/10.1111/bpa.12255
Luca A, Calandra C, Luca M (2018) Molecular bases of Alzheimer’s disease and neurodegeneration: the role of neuroglia. Aging and disease 9(6):1134–1152. https://doi.org/10.14336/ad.2018.0201
Llorens-Martín M (2018) Exercising new neurons to vanquish Alzheimer disease. Brain plasticity 4(1):111–126. https://doi.org/10.3233/bpl-180065
Liu HL, Zhao G, Zhang H, Shi LD (2013) Long-term treadmill exercise inhibits the progression of Alzheimer’s disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav Brain Res 256:261–272. https://doi.org/10.1016/j.bbr.2013.08.008
Kang EB, Cho JY (2015) Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. J Exerc Nutr Biochem 19(3):199–209. https://doi.org/10.5717/jenb.2015.15090806
Koldamova R, Lefterov I (2007) Role of LXR and ABCA1 in the pathogenesis of Alzheimer’s disease—implications for a new therapeutic approach. Curr Alzheimer Res 4(2):171–178. https://doi.org/10.2174/156720507780362227
Thiele DK, Anderson CM (2016) Developmental origins of health and disease: a challenge for nurses. J Pediatr Nurs 31(1):42–46. https://doi.org/10.1016/j.pedn.2015.10.020
Barnes MD, Heaton TL, Goates MC, Packer JM (2016) Intersystem implications of the developmental origins of health and disease: advancing health promotion in the 21st century. Healthcare 4(3):45. https://doi.org/10.3390/healthcare4030045
Weissgerber TL, Wolfe LA, Davies GA, Mottola MF (2006) Exercise in the prevention and treatment of maternal-fetal disease: a review of the literature. Appl Physiol Nutr Metab 31(6):661–674. https://doi.org/10.1139/h06-060
Clapp JF 3rd (1996) Morphometric and neurodevelopmental outcome at age five years of the offspring of women who continued to exercise regularly throughout pregnancy. J Pediatr 129(6):856–863. https://doi.org/10.1016/s0022-3476(96)70029-x
Herring A, Donath A, Yarmolenko M, Uslar E, Conzen C, Kanakis D, Bosma C, Worm K, Paulus W, Keyvani K (2012) Exercise during pregnancy mitigates Alzheimer-like pathology in mouse offspring. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 26(1):117–128. https://doi.org/10.1096/fj.11-193193
Greenamyre JT, Hastings TG (2004) Biomedicine. Parkinson’s—divergent causes, convergent mechanisms. Sci 304(5674):1120–1122. https://doi.org/10.1126/science.1098966
Li L, Guo P, Ding D, Lian T, Zuo L, Du F, Zhang W (2019) Parkinson’s disease with orthostatic hypotension: analyses of clinical characteristics and influencing factors. Neurol Res 41(8):734–741. https://doi.org/10.1080/01616412.2019.1610224
Feng YS, Yang SD, Tan ZX, Wang MM, Xing Y, Dong F, Zhang F (2020) The benefits and mechanisms of exercise training for Parkinson’s disease. Life Sci 245:117345. https://doi.org/10.1016/j.lfs.2020.117345
Zigmond M, Cameron J, Leak R, Mirnics K, Russell V, Smeyne R, Smith A (2009) Triggering endogenous neuroprotective processes through exercise in models of dopamine deficiency. Park Relat Disord Suppl 3:S42–45. https://doi.org/10.1016/s1353-8020(09)70778-3
McColgan P, Tabrizi SJ (2018) Huntington’s disease: a clinical review. Eur J Neurol 25(1):24–34. https://doi.org/10.1111/ene.13413
Steventon JJ, Collett J, Furby H, Hamana K, Foster C, O’Callaghan P, Dennis A, Armstrong R, Nemeth AH, Rosser AE, Murphy K, Quinn L, Busse M, Dawes H (2018) Alterations in the metabolic and cardiorespiratory response to exercise in Huntington’s disease. Parkinsonism Relat Disord 54:56–61. https://doi.org/10.1016/j.parkreldis.2018.04.014
Caldwell CC, Petzinger GM, Jakowec MW, Cadenas E (2020) Treadmill exercise rescues mitochondrial function and motor behavior in the CAG140 knock-in mouse model of Huntington’s disease. Chem Biol Interact 315:108907. https://doi.org/10.1016/j.cbi.2019.108907
Mueller SM, Petersen JA, Jung HH (2019) Exercise in Huntington’s disease: current state and clinical significance. Tremor and other hyperkinetic movements (New York, NY) 9:601. https://doi.org/10.7916/tm9j-f874
de Bruijn RF, Schrijvers EM, de Groot KA, Witteman JC, Hofman A, Franco OH, Koudstaal PJ, Ikram MA (2013) The association between physical activity and dementia in an elderly population: the Rotterdam Study. Eur J Epidemiol 28(3):277–283. https://doi.org/10.1007/s10654-013-9773-3
Kishimoto H, Ohara T, Hata J, Ninomiya T, Yoshida D, Mukai N, Nagata M, Ikeda F, Fukuhara M, Kumagai S, Kanba S, Kitazono T, Kiyohara Y (2016) The long-term association between physical activity and risk of dementia in the community: the Hisayama Study. Eur J Epidemiol 31(3):267–274. https://doi.org/10.1007/s10654-016-0125-y
Tan ZS, Spartano NL, Beiser AS, DeCarli C, Auerbach SH, Vasan RS, Seshadri S (2017) Physical activity, brain volume, and dementia risk: the Framingham Study. J Gerontol A Biol Sci Med Sci 72(6):789–795. https://doi.org/10.1093/gerona/glw130
Russell VA, Zigmond MJ, Dimatelis JJ, Daniels WMU, Mabandla MV (2014) The interaction between stress and exercise, and its impact on brain function. Metab Brain Dis 29(2):255–260. https://doi.org/10.1007/s11011-013-9479-y
Funding
The present study was supported by the National Natural Science Foundation of China (no. 82072531; the recipient is Feng Zhang).
Author information
Authors and Affiliations
Contributions
ZXT, FD, and FZ had the idea for the article. YSF, ZXT, LYW, and FD performed the literature search and data analysis. ZXT and FZ drafted and critically revised the work.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Zi-Xuan Tan and Fang Dong contributed equally to this Article.
Rights and permissions
About this article

Cite this article
Tan, ZX., Dong, F., Wu, LY. et al. The Beneficial Role of Exercise on Treating Alzheimer’s Disease by Inhibiting β-Amyloid Peptide. Mol Neurobiol 58, 5890–5906 (2021). https://doi.org/10.1007/s12035-021-02514-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02514-7