
Abstract
Alzheimer’s disease (AD) is characterized by the deposition of aggregated amyloid-beta (Aβ), which triggers a cellular stress response called the unfolded protein response (UPR). The UPR signaling pathway is a cellular defense system for dealing with the accumulation of misfolded proteins but switches to apoptosis when endoplasmic reticulum (ER) stress is prolonged. ER stress is involved in neurodegenerative diseases including AD, but the molecular mechanisms of neuronal apoptosis and inflammation by Aβ-induced ER stress to exercise training are not fully understood. Here, we demonstrated that treadmill exercise (TE) prevented PS2 mutation-induced memory impairment and reduced Aβ-42 deposition through the inhibition of β-secretase (BACE-1) and its product, C-99 in cortex and/or hippocampus of aged PS2 mutant mice. We also found that TE down-regulated the expression of GRP78/Bip and PDI proteins and inhibited activation of PERK, eIF2α, ATF6α, sXBP1 and JNK-p38 MAPK as well as activation of CHOP, caspase-12 and caspase-3. Moreover, TE up-regulated the expression of Bcl-2 and down-regulated the expressions of Bax in the hippocampus of aged PS2 mutant mice. Finally, the generation of TNFα and IL-1α and the number of TUNEL-positive cells in the hippocampus of aged PS2 mutant mice was also prevented or decreased by TE. These results showed that TE suppressed the activation of UPR signaling pathways as well as inhibited the apoptotic pathways of the UPR and inflammatory response following Aβ-induced ER stress. Thus, therapeutic strategies that modulate Aβ-induced ER stress through TE could represent a promising approach for the prevention or treatment of AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer disease (AD) is a serious form of dementia, and has become a growing health concern. One of the hallmarks of AD is the aggregation of amyloid-beta (Aβ) or plaques in the brain. This process is highly toxic to neurons, and causes inflammation that contributes to the death of brain cells. The endoplasmic reticulum (ER) is the primary intracellular organelle where misfolded or abnormal proteins accumulate, and these are recognized by several ER chaperone proteins for maintenance of cellular homeostasis. The accumulation of unfolded or misfolded proteins in the ER activates a cellular stress response known as the unfolded protein response (UPR) and initiates the removal of toxic misfolded proteins as a way to protect the cell. However, an excessive buildup of misfolded protein prolongs ER stress and induces oxidative stress, aberrant ER Ca2+ regulation, impaired proteasomal and lysosomal activity, and inflammation. This phenomenon extends the UPR and eventually triggers neuronal cell death [1, 2]. Not surprisingly, therefore, ER stress-induced neuronal cell death plays a crucial role in the pathogenesis of neurodegenerative diseases like AD [3–5].
The ER contains three stress sensors: inositol-requiring kinase 1 (IRE1), protein kinase-like ER kinase (PERK), and activating transcription factor 6 alpha (ATF6α) transducers; these are normally maintained in an inactive state via binding with an ER chaperone, glucose regulated protein 78 (GRP78) [also called binding immunoglobulin protein (BiP), thus written as GRP78/Bip]. However, ER stress triggers the release of GRP78/BiP from the complexes, and the stress sensors recognize the misfolded proteins in the ER and activate a complex signaling network of UPR [6, 7]. Another ER chaperone, protein disulfide isomerase (PDI), is also induced during ER stress, and plays a critical role in either cell survival or death, depending upon the degree of ER stress [8].
UPR signaling is a multifaceted cascade designed to maintain ER homeostasis—either by reducing overall protein synthesis to mitigate the ER overload caused by accumulation of misfolded proteins or by selectively increasing the production of ER chaperones to promote refolding of misfolded/unfolded proteins to attenuate ER stress [7, 9]. In contrast, under severe and prolonged ER stress, UPR continuously activates ER stress sensors. For example, extended phosphorylation of PERK subsequently results in phosphorylation and inactivation of its effector eukaryotic initiation factor 2α (eIF2α), causing a repression of overall gene translation and leading to cell death [10, 11]. Interaction also occurs between two other ER stress sensors: ATF6α expresses the messenger RNA (mRNA) of transcription factor X-box binding protein 1 (XBP1), and IRE1 splices its gene product to generate spliced XBP1 (sXBP1), which modulates apoptotic signaling [12]. In addition, IRE1 alone activates c-Jun NH(2)-terminal protein Kinases (JNK) that reduce activities of anti-apoptotic proteins [13]. Prolonged ER stress then promotes the upregulation of a transcription factor C/EBP homologous protein (CHOP), which down-regulates the expression of anti-apoptotic protein Bcl-2, further contributing to apoptosis [14]. Accordingly, several studies have demonstrated that neuronal tissues from AD patients or animals showed increased phosphorylation of PERK and eIF2α [11], disrupted ER Ca2+ regulation [15], and upregulation of CHOP [16], thereby providing evidence for UPR activation in AD.
Aβ is generated through a proteolytic process of amyloid precursor protein (APP). β-secretase, also known as beta-site APP cleaving enzyme 1 (BACE-1), and γ-secretase are involved in cleaving APP and releasing Aβ into extracellular compartments. Depending upon the cleavage site of APP acted upon by γ-secretase, either the short length Aβ-40 form (which is non-toxic) or the full length Aβ-42 form (which is toxic and aggregated) can be generated [17]. Genetic studies with mouse models showed that gene mutations encoding APP or presenilin (one of three subcomponents comprising γ-secretase) cause overproduction of Aβ-42 and early onset of AD [18].
The accumulation of extracellular aggregates of Aβ-42 in the senile plaques and the presence of intracellular aggregates of hyperphosphorylated tau in the neurofibrillary tangles (NTFs) are typical observations in the brains of AD patients. However, several studies show that Aβ-42 is localized not only in the extracellular compartment, but also in intracellular compartments such as the ER at the onset of AD, which suggests that Aβ-42 can directly induce ER stress and play a crucial role in the pathogenesis of AD [3, 19–24]. Importantly, recent studies have reported that aggregations of Aβ and tau protein cause neuronal cell death in AD [25–28]. Aggregations of Aβ-42 are also linked to the activation of inflammation through several intertwined cellular pathways; particularly reactive oxygen species (ROS) and proinflammatory cytokines [29]. However, clear-cut roles of inflammation in pathogenesis of AD are still elusive [22]. Nevertheless, the use of anti-inflammatory approaches is suggested for consideration as a treatment option for AD patients.
Physical activity has been recommended as a both preventive and a therapeutic regimen in the management of patients with AD. Evidence is accumulating that physical activity may be sufficient to delay cognitive impairment and repress neuronal apoptosis in both animal models and patients of AD, indicating that physical activity has profound benefits for maintaining normal function of the brain [30–34]. Despite this common perception, the molecular mechanisms by which physical activity protects the brain against the onset of AD remain to be fully elucidated. Therefore, we investigated the potential mechanistic connections between the exercise training and Aβ-induced ER stress using a well-controlled mouse model of AD.
Mutations in PSEN2, a gene encoding presenilin 2 (PS2), which is one of three components of γ-secretase, lead to the incomplete function of γ-secretase and results in a loss of cleavage of APP; this generates Aβ-42, and causes AD [35]. We used transgenic (Tg) mice under the control of the neuron-specific enolase promoter, which overexpresses human mutant PS2 (N141I), to investigate the effect of 12 weeks of treadmill exercise (TE) on ER stress-induced neuronal apoptosis and inflammation in the brain of 24-month-old mice. This PS2 mutant transgenic (Tg) mouse model of AD has been used in several studies [36, 37], and has provided highly convincing results that are comparable to those derived using other mouse models of AD, including APP mutant and PS1 mutant Tg mice. We found that TE suppresses the production of Aβ-42 as well as expression of the ER stress chaperones, GRP78/BiP and PDI. In addition, TE inhibits UPR-mediated apoptotic signaling pathways involving CHOP, caspase-12, and caspase-3, thereby preventing apoptotic neuronal cell death. TE also significantly reduces the levels of proinflammatory cytokines, tumor necrosis factor-α (TNFα), and interleukin-1α (IL-1α) in aged PS2 mutant mice. Collectively, these findings suggest that TE-mediated blockade of ER stress and inflammation via suppression of Aβ-42 is a potential mechanism responsible for the observation of exercise-induced neuronal protection against AD. Consequently, this study provides crucial insight for the development of practical and effective non-pharmaceutical therapeutic strategies for retardation and prevention of the progression of AD.
Materials and methods
Transgenic mice
All animal experimental procedures used in this study were approved by the Institutional Animal Care and Use Committee at Korea National Sport University and by the Korea FDA. Transgenic mice, PS2 mutant mice expressing the human PS2 mutant under the control of neuron-specific enolase (NSE) were maintained in the genetic background of C57BL/6 × DBA/2 mice, as previously described [38]. The mice were maintained at a 12:12 h dark–light cycle, housed at 22 ± 2 °C with 50 % relative humidity, and had free access to standard chow diet (Purina Mills, Seoul, Korea) ad libitum. Mice were handled in an accredited Korea FDA animal facility in accordance with the AAALAC International Animal Care Policies (Accredited Unit-Korea Food and Drug Administration: Unit Number-000996).
Experimental design and treadmill exercise
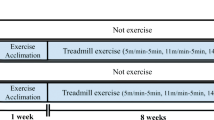
PS2 mutant transgenic (Tg) mice and their control non-transgenic (non-Tg) mice at 24 months of age, were divided into one of the following groups: sedentary non-Tg mice (SED/non-Tg, n = 8), sedentary PS2 mutant mice (SED/Tg, n = 8) or treadmill-exercised PS2 mutant mice (TE/Tg, n = 8). Pre-exercise was performed at 5 m/min, 10 min/day for 5 days for the familiarization of the treadmill-exercise environment, as previously conducted [34]. After this period, TE was performed at 12 m/min, 60 min/day, for 5 days/week on a 0 % gradient for a total of 12 weeks. However, a sedentary group remained in their home cage throughout the course of the experiment.
Water maze test
The water maze test is a widely accepted method for testing memory. Thus, we performed this test to assess memory impairment as described elsewhere [33]. The maze test was performed using the SMART-CS (Panlab, Barcelona, Spain) program and equipment. A circular plastic pool (height 35 cm, diameter 100 cm) was filled with water (containing dark ink) kept at 22–25 °C. An escape platform (height 14.5 cm, diameter 4.5 cm) was positioned and submerged 0.5–1 cm below the surface of the water. The test was performed three times per day for 5 days. Each trial lasted for 60 s or ended as soon as the mouse reached the submerged platform and was allowed to remain on the platform for 10 s. Escape latency, escape distance and swimming pattern of each mouse were monitored by a camera above the center of the pool connected to a SMART-LD program (Panlab, Barcelona, Spain). A quiet environment, consistent lighting, constant water temperature, and a fixed spatial frame were maintained throughout the period of the experiment.
Brain tissue collection and preservation
After the behavioral test, the five mice of each group were sacrificed, after which the brains were rapidly removed and the hemispheres were separated on ice. The cortex and hippocampus from hemispheres, selected at random were snap-frozen on dry ice and then stored at −80 °C for Western blot analysis. The other three mice of each group were perfused transcardially with 50 mM phosphate buffered saline (PBS) followed by 4 % paraformaldehyde in 0.1 M sodium phosphate buffer at pH 7.4 for immunohistochemistry, immunofluorescence staining and TUNEL assay.
Western blotting
Western blot analyses were conducted as previously described [33]. Briefly, proteins (30 μg) were separated by electrophoresis on a 12 % polyacrylamide gel for 90 min, after which they were transferred to a polyvinylidene fluoride membrane (Immuno-Blot, PVDF membrane, Bio-Rad, CA, USA) for 1 h at a constant voltage of 60 V. Each membrane was then separately incubated overnight at 4 °C with specific antibodies: Aβ-42 (SIG-39320) and C-99 (SIG-39152) antibodies (Covance, dilution: 1:1,000); Caspase-3 (#9662), CHOP (#2895), eIF2α (#9722), phospho-eIF2α (#9721), JNK (#9252), phospho-JNK, (#9251), p38MAPK (#9212), phospho-p38MAPK (#9211) and PDI (#3501) antibodies (Cell signaling, dilution: 1:1,000); PERK (sc-13073), phospho-PERK (sc-32577), ATF6α (sc-22799), TNFα (sc-8301), sXBP1 (sc7160) and GAPDH (sc-20357) antibodies (Santa Cruz, dilution: 1:1,000); IL-1α (Sigma, I3392, dilution: 1:1,000); GRP78/BiP (ab21685) and caspase-12 (ab18766) antibodies (Abcam, dilution: 1:1,000); Bcl-2 (B9804) and Bax (B8429) antibodies (Sigma, dilution: 1:1,000); BACE-1 (Millipore, MAB5308, dilution: 1:1,000) antibody. The membranes were then washed with a washing buffer, after which they were incubated with secondary antibodies. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit (invitrogen, 656120) for GRP78/BiP, PDI, PERK, phospho-PERK, eIF2α, phospho-eIF2α, ATF6α, Caspase-12, Caspase-3, sXBP1, JNK, phospho-JNK, p38MAPK, phospho-p38MAPK and TNFα (dilution: 1:5,000). Horseradish peroxidase (HRP)-conjugated rabbit anti-goat (invitrogen, 611620) for IL-1α and GAPDH (dilution: 1:5,000). Horseradish peroxidase-conjugated goat anti-mouse (Santa Cruz, sc-2005) was used for Aβ-42, C-99, BACE-1, CHOP, Bcl-2, and Bax (dilution: 1:5,000). Immunoreactive proteins were detected with the ECL Western blotting detection system (Santa Cruz Biotechnology, CA, USA). The density of the developed bands was determined using a ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA).
Immunohistochemistry
Samples for immunohistochemical analyses were embedded in paraffin and sections at 6 μm thickness and performed as previously described [30, 39]. The β-amyloid plaques in the sections were pre-treated with DAB (Invitrogen, CA, USA) for 40 min at room temperature with a blocking buffer containing 10 % goat serum in phosphate buffer solution for 1 h, followed by incubation with a primary unconjugated mouse monoclonal anti-β-amyloid 42 (Covance, SIG-39320, USA) at 1:300 in a blocking buffer at room temperature overnight. The β-amyloid plaques in the sections were washed in a washing buffer and then incubated with a secondary antibody, HRP-conjugated goat anti-mouse IgG Santa Cruz, sc-2005) at 1:200 for 2 h at room temperature. The peroxidase activity was visualized with diaminobenzidine (DAB) substrate kit (Vector Laboratories, Burlingame, CA, USA). The slides were observed under the light microscope (Olympus U-LH 100HG, Tokyo, Japan).
Immunofluorescence staining
Immunofluorescence staining was performed as previously described [39]. Tissue sections were deparaffinized, cleared, and hydrated to PBS using a descending series of ethanol. The sections were blocked for 40 min at 37 °C with 3 % goat serum in PBS followed by quenching endogenous peroxidase activity by exposing slides to 0.3 % H2O2 and 10 % methanol for 5 min. Primary antibodies for GRP78/BiP (1:100; ab21685) were added and incubated overnight at 4 °C. The following day, the slides were washed in PBS three times for 5 min and pre-incubated in 2 % normal donkey serum (NDS) for 15 min, and then transferred to a mixture of the secondary antibodies (GRP78/BiP, Alexa 488 conjugated donkey anti-rabbit; 1:200 dilution, Jackson Immunochemicals, West Grove, PA, USA) for 3 h at room temperature. After several rinses, the sections were mounted on cover slides slipped with Vectashield (Vector Laboratories, Burlingame, CA, USA), and examined under an immunofluorescence microscope (Leica Microsystems, Wetzlar, Germany). For negative controls, the primary antibodies were omitted.
Detection of apoptosis by TUNEL
In situ detection of apoptotic cells was performed with commercially available ApoTag Peroxidase In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA) by TUNEL assay in paraffin sections according to the company’s manual. Briefly, the slides were incubated with 20 μg/ml proteinase K (Chemicon, Temecula, CA, USA) in PBS for 15 min followed by washing in distilled water. After quenching in 3 % H2O2 and applying the equilibrium buffer for 10 min, the sections were incubated in TdT enzyme for 1 hr at 37 °C and put in stop/wash buffer for 10 min. The slides were rinsed in PBS three times and then anti-digoxigenin peroxidase conjugate was applied on the tissues for 30 min at room temperature. After washing in PBS, the peroxidase activity was visualized with DAB substrate kit (Vector Laboratories, Burlingame, CA, USA). The nuclei were counter-stained with 0.5 % methyl green. The slides were observed under the light microscope (Olympus U-LH 100HG, Tokyo, Japan).
Histological scoring of the apoptosis
TUNEL positive cells in the hippocampus had nuclei with dark brown and pointed apoptosis. The number of apoptotic cells per 0.025 mm2 was calculated by dividing the number of positive apoptotic cells within a 0.025 mm2 at 200× magnifications using an image analyzer (Analysis Pro 3.2, Sis Co., Munster, Germany). The results were counted as the number per mm2 in the hippocampus. Results were counted as mean ± SEM.
Statistical analysis
Data were analyzed using SPSS version 18.0 (SPSS, Inc., Chicago, IL, USA). All values are expressed as mean ± SEM. Statistical significance was determined using a one-way ANOVA when comparing the groups. A Bonferroni post hoc test was followed for all pairwise multiple comparisons if a statistically significant group main effect was found. Differences were considered statistically significant at α = 0.05.
Results
The effects of treadmill exercise on cognitive impairment in aged PS2 mutant mice
Previous studies demonstrated that over-expressing the PS2 mutant under control of the NSE promoter in transgenic mice is associated with several AD-like pathogenic phenotypes such as impairment of cognitive performance and Aβ-42 deposition in the brain at 12 months of age [38]. Our previous studies showed that TE improved cognitive performance in APPsw transgenic mice or the PS2 mutant mouse model [33, 34]. Thus, in these studies, TE was performed for 3 months to 12-month-old or 24-month-old APPsw and PS2 mutant mice. The cognitive functions of the transgenic mice were then tested for 5 consecutive days (3 times/day) in the location of and escape onto a platform, and their learning scores were recorded. Statistical analysis (ANOVA) of the data on day 5 showed the significance of the cognitive performance improving effect of TE (escape distance, F(2, 23) = 24.40, p < 0.001; escape latency F(2, 23) = 28.47, p < 0.001, Fig. 1a, b). The non-transgenic control mice and treadmill exercised PS2 mutant mice exhibited shorter escape distance and shorter escape latency time than the sedentary PS2 mutant mice during the water maze test. Taken together, these results demonstrate that 3 months of TE significantly ameliorated cognitive impairment in aged PS2 mutant mice.

Effect of treadmill exercise on memory dysfunction in aged PS2 mutant mice. All mice were tested for 5 consecutive days to locate and escape onto the platform, and their escape latency and escape distance were recorded. After 3 months of exercise on the treadmill in PS2 mutant mice, escape distance (a), escape latency (b) and patterns of swimming (c) until arriving at the platform were recorded. Treadmill-exercised PS2 mutant mice were ameliorated in their cognitive abilities to learn the task and showed consistently shorter escape distances and lower escape latencies throughout testing relative to sedentary PS2 mutant mice. Fisher’s LSD post hoc test after ANOVA. Values are presented as mean ± SEM from eight mice/groups
The effects of treadmill exercise on Aβ-42 levels in the cortex and hippocampus of aged PS2 mutant mice
We determined the effect of TE on Aβ-42 expression in the cortex and hippocampus of aged PS2 mutant mice because deposition of Aβ-42 is implicated in cognitive dysfunction, and our results were similar to previous data [30, 33]. One-way ANOVA of Aβ-42 data indicated significant effects for the group [cortex, F(2, 14) = 15.09, p < 0.001; hippocampus F(2, 14) = 22.88, p < 0.001]. As shown in Fig. 2a–d, treadmill exercised PS2 mutant mice had a lower level of Aβ-42 than PS2 mutant mice in the sedentary condition. In addition, we analyzed Aβ immunoreactivity in the cortex and hippocampus with higher magnification. As shown in Fig. 2e, immunostaining with an Aβ-42 specific antibody revealed that the Aβ-42 plaques in the cortex and hippocampus region of PS2 mutant mice were notably increased whereas the enhanced Aβ-42 plaques were reduced after TE. These results suggest that TE leads to a reduction in the levels of Aβ-42 in the cortex and hippocampus, possibly by rectifying the functional processes of the amyloid precursor protein (APP) through the reduction of β-secretase (BACE-1).

Expression and immunostaining analysis of Aβ-42 level in the cortex and hippocampus of aged PS2 mutant mice after 3 months of treadmill exercise. Western blot (n = 5) and immuno staining analysis (n = 3) of Aβ-42 deposition. a–d Nitrocellulose filters transferring 30 μg of protein from the cortex and hippocampus of each group, incubated with anti-human Aβ-42 antibody. The bands were quantified by densitometry to obtain relative levels of Aβ-42. GAPDH was probed as an internal control. e Immunostaining of Aβ-42. 6 μm-thick sections of brains from each group were incubated with anti-human Aβ-42 primary antibody and HRP-conjugated goat anti-rabbit IgG. The resulting tissues were viewed with a microscope. The narrow distribution and low intensity of Aβ-42 deposition were shown in the cortex and hippocampus tissue of treadmill-exercised PS2 mutant mice. Five mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate on Western blot analysis and three mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate by immunohistochemistry assay. Fisher’s LSD post hoc test after ANOVA. Values are presented as mean ± SEM
The effects of treadmill exercise on BACE-1 and C-99 expression in the hippocampus of aged PS2 mutant mice
We determined the effect of TE on the levels of β-secretase (BACE-1) and its product C-99 in the hippocampus of 24 month-old PS2 mutant mice. The one-way ANOVA on BACE-1 and C-99 data indicated significant effects for the group [BACE-1, F(2, 14) = 158.90, p < 0.001; C-99, F(2, 14) = 19.74, p < 0.001]. Western blot analysis showed that TE leads to a reduction in the levels of BACE-1 and its product C-99 in the hippocampus, suggesting that TE reduced Aβ-42 level, thereby reducing β-secretase and its product C-99 (Fig. 3a–c).

Effect of treadmill exercise on expression of BACE-1 and C-99 in the hippocampus of aged PS2 mutant mice. Inhibitory effect of TE on expression of BACE-1 and C-99 in the hippocampus of PS2 mutant mice brains. a–c Hippocampal BACE-1 and C-99 proteins were up-regulated in PS2 mutant mice compared with non-transgenic mice, and the levels of those proteins in PS2 mutant mice were down-regulated after TE. The data shown in the Western blot were means from five mice brains. GAPDH was probed as an internal control. Fisher’s LSD post hoc test after ANOVA. Values are presented as mean ± SEM
Treadmill exercise down-regulates GRP78/BiP and PDI in the hippocampus of aged PS2 mutant mice
We have examined whether TE reduced expression or intensity of GRP78/BiP and PDI in the hippocampus of 24 month-old PS2 mutant mice. The one-way ANOVA on GRP78/Bip data indicated significant effects for the group [F(2, 14) = 72.93, p < 0.001]. The one-way ANOVA on PDI data indicated significant effects for the group [F(2, 14) = 91.23, p < 0.001]. The level of GRP78/Bip, an ER chaperone, and PDI, a family of enzymes that catalyze disulfide bond formation, reduction, or isomerization of newly synthesized proteins in the lumen of the ER, were markedly increased in the hippocampus of aged PS2 mutant mice. However, those proteins were down-regulated after TE (Fig. 4a–c). Fluorescence photomicrograph revealed that intensity of GRP78/Bip in the hippocampus (CA-2 and CA-3) of PS2 mutant mice was notably enhanced, whereas the enhanced expression was suppressed after TE (Fig. 4d). These results indicate that TE leads to the down-regulation of Aβ-induced UPR activation in the hippocampus of aged PS2 mutant mice.

Treadmill exercise inhibited GRP78/BiP and PDI expression in the hippocampus of aged PS2 mutant mice. Representative Western blot showing hippocampal GRP78/Bip and PDI levels for all groups of mice were presented. a–c Hippocampal GRP78/Bip and PDI proteins were up-regulated in PS2 mutant mice compared with non-transgenic mice, and the levels of those proteins in PS2 mutant mice were down-regulated after TE. GAPDH was probed as an internal control. Fluorescence photomicrographs of cortical and hippocampal sections from all groups of mice immunolabeled with anti-GRP78/BiP antibody (left panel, green). d Note that GRP78/Bip expression was decreased in CA-2 and CA-3 regions of hippocampus after TE in PS2 mutant mice, 6 μm. Five mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate on Western blot analysis and three mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate by immunofluorescence assay. Fisher’s LSD post hoc test after ANOVA. Values are mean ± SEM
Treadmill exercise down-regulates PERK-eIF2α pathway in the hippocampus of aged PS2 mutant mice
We have examined whether TE reduced the expression of PERK-eIF2α and ATF6α in the hippocampus of aged PS2 mutant mice. The one-way ANOVA on phospho-ERK/t-PERK ratio and phospho-eIF2α/t-eIF2α ratio data indicated significant effects for the group [phospho-PERK/t-PERK ratio, F(2, 14) = 56.07, p < 0.001; phospho-eIF2α/t-eIF2α ratio, F(2, 14) = 14.96, p < 0.001]. As shown in Fig. 5a–c, PS2 mutant mice that were maintained under sedentary conditions had a higher phospho-PERK/t-PERK ratio and phospho-eIF2α/t-eIF2α ratio than those of non-transgenic mice. However, phospho-PERK and phospho-eIF2α were down-regulated after TE. Thus, these results suggest that down-regulation of the GRP78/BiP-dependent PERK-eIF2α pathway by TE seem to play a neuroprotective role against Aβ-induce neurotoxicity, suggesting that TE possibly reduces susceptibility to Aβ-induced ER stress by altering the UPR signaling pathway.

Treadmill exercise inhibited GRP78/BiP-dependent PERK-eIF2α activation in the hippocampus of aged PS2 mutant mice. Representative Western blot showing hippocampal PERK-eIF2α levels of all groups of mice were presented. a–c PERK-eIF2α phosphorylation was expressed as p-PERK/t-PERK and p-eIF2α/t-eIF2α, respectively. Hippocampal PERK-eIF2α phosphorylation showed a significant difference between non-transgenic mice and PS2 mutant mice. In addition, treadmill-exercised PS2 mutant mice revealed significantly lower hippocampal PERK-eIF2α phosphorylation relative to sedentary PS2 mutant mice. GAPDH was probed as an internal control. Five mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate on Western blot analysis. Fisher’s LSD post hoc test after ANOVA. Values are mean ± SEM
Treadmill exercise represses ATF6α, sXBP1 and JNK-p38 MAPK in the hippocampus in aged PS2 mutant mice
ER stress can induce apoptotic proteases such as CHOP, caspase-12 and caspase-3, and activate JNK-p38 MAPK via the IRE1-XBP1 pathway, which induces neuronal apoptosis. In addition, ATF6α also collaborates with IRE1 to induce sXBP1 expression. Therefore, it was of interest to characterize the potential signaling events following TE in the hippocampus of aged PS2 mutant mice. The one-way ANOVA on ATF6α data indicated significant effects for the group [F(2, 14) = 33.85, p < 0.001]. The one-way ANOVA on sXBP1 data indicated significant effects for the group [sXBP1, F(2, 14) = 116.27, p < 0.001]. In addition, the one-way ANOVA on JNK and p38 MAPK data indicated significant effects for the group [JNK 54, F(2, 14) = 612.22, p < 0.001; JNK 46, F(2, 14) = 103.76, p < 0.001; p38, F(2, 14) = 213.05, p < 0.001]. As shown in Fig. 6a–f, treadmill-exercised PS2 mutant mice had lower levels of ATF6α and sXBP1 than those of sedentary PS2 mutant mice. In addition, PS2 mutant mice that were subjected to TE had lower phosphorylation levels of JNK 54, JNK 46 and p38MAPK than those of PS2 mutant mice in sedentary conditions. These results suggest that TE leads to the down-regulation of Aβ-induced ATF6α and sXBP1, which in turn reduces JNK and p38 activation in the hippocampus of aged PS2 mutant mice.

Treadmill exercise reduced activation of ATF6α, sXBP1 and JNK-p38MAPK in the hippocampus of aged PS2 mutant mice. Representative Western blot showing hippocampal ATF6α, sXBP1 and JNK-p38MAPK of all groups of mice were presented. a, b ATF6α levels were evaluated in protein lysates derived from the hippocampus of non-transgenic mice and PS2 mutant mice. In addition, treadmill-exercised PS2 mutant mice revealed significantly lower hippocampal ATF6α proteins relative to sedentary PS2 mutant mice. a, c Hippocampal sXBP1 protein shows the significant difference between non-transgenic mice and PS2 mutant mice. In addition, TE reduced the activation level of sXBP1 in the hippocampus of PS2 mutant mice. a, d, e JNK activation. The top demonstrates 30 μg of protein per sample, with an antibody against phospho-JNK. The bottom demonstrates the total JNK protein, which was assessed using antibodies recognizing these proteins regardless of their phosphorylated state. a, f p38MAPK activation. The top demonstrates 30 μg of protein incubated with an antibody for phospho-p38. The bottom demonstrates the total p38 protein at relatively constant levels. TE reduced the activation level of phospho-JNK (p54/p46) and phospho-p38MAPK in the hippocampus of PS2 mutant mice. GAPDH was probed as an internal control. Five mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate on Western blot analysis. Fisher’s LSD post hoc test after ANOVA. Values are mean ± SEM
Reduction of CHOP, caspase-12 and caspase-3 and TNFα and IL-1α expression in response to treadmill exercise in the hippocampus of aged PS2 mutant mice
We have examined whether TE reduced the expression of CHOP, caspase-12 and caspase-3 in the hippocampus of aged PS2 mutant mice. The one-way ANOVA on CHOP and caspase-12 data indicated significant effects for the group [CHOP, F(2, 14) = 25.96, p < 0.001; caspase-12, F(2, 14) = 63.16, p < 0.001]. The one-way ANOVA on caspase-3 data indicated significant effects for the group [hippocampus, F(2, 14) = 261.15, p < 0.001]. In addition, the one-way ANOVA on TNFα and IL-1α data indicated significant effects for group [TNFα, F(2, 14) = 80.17, p < 0.001; IL-1α, F(2, 14) = 201.73, p < 0.001]. As shown in Fig. 7a–e, these results indicate that TE significantly reduced the expression of CHOP and cleaved forms of caspase-12 and caspase-3, suggesting that the protective effect of TE against Aβ-induced ER stress can be significantly attributed to the inactivation of ER stress-specific neuronal cell death proteases. In addition, TE represses local inflammatory responses through the reduction of chronic overexpression of TNFα and IL-1α.

Treadmill exercise inhibited Aβ-induced apoptotic pathway and inflammatory response of the UPR in the hippocampus of aged PS2 mutant mice. Levels of CHOP, caspase-12, caspase-3, TNFα and IL-1α proteins in the hippocampus were analyzed by Western blot analysis. a–f CHOP, caspase-12, caspase-3, TNFα and IL-1α proteins showed a significant difference between non-transgenic mice and PS2 mutant mice. In addition, CHOP, caspase-12, caspase-3, TNFα and IL-1α proteins in the hippocampus were significantly reduced in PS2 mutant mice after TE. GAPDH was probed as an internal control. Five mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate on Western blot analysis. Fisher’s LSD post hoc test after ANOVA. Values are mean ± SEM
Down-regulation of hippocampal Bax protein and up-regulation of the Bcl-2 protein in response to treadmill exercise in aged PS2 mutant mice
We have examined whether TE up-regulated the level of the anti-apoptotic factor, Bcl-2 and down-regulated the level of the pro-apoptotic factor, Bax in the hippocampus of aged PS2 mutant mice, with special attention to the possible relationship among Bax and Bcl-2 proteins and TE. The one-way ANOVA on Bax and Bcl-2 data indicated significant effects for the group [Bax, F(2, 14) = 317.284, p < 0.001; Bcl-2, F(2, 14) = 61.99, p < 0.001]. As shown in Fig. 8a–c, the hippocampus of treadmill-exercised PS2 mutant mice had lower levels of Bax than that of sedentary PS2 mutant mice whereas the hippocampus of treadmill-exercised PS2 mutant mice had higher levels of Bcl-2 than that of sedentary PS2 mutant mice. Taken together, these results demonstrate that up-regulation of Bcl-2 and down-regulation of Bax in hippocampus regions containing Aβ deposits following TE was associated with neuroprotection.

The effects of treadmill exercise on Bax and Bcl-2 proteins in the hippocampus of aged PS2 mutant mice. Levels of Bax and Bcl-2 proteins in the hippocampus were analyzed by Western blot analysis. a–c Bax and Bcl-2 show a significant difference between non-transgenic mice and PS2 mutant mice. In addition, Bax protein in the hippocampus was significantly reduced in PS2 mutant mice after TE, whereas Bcl-2 protein in the hippocampus was significantly increased in PS2 mutant mice after TE. GAPDH was probed as an internal control. Five mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate on Western blot analysis. Fisher’s LSD post hoc test after ANOVA. Values are mean ± SEM
The effects of treadmill exercise on the number of TUNEL-positive cells in the hippocampus of aged PS2 mutant mice
To examine the effect of TE on neuronal apoptotic cell death in PS2 mutant mice hippocampus, apoptotic cells of the hippocampus were stained using TUNEL methods. The TUNEL-positive cells were stained brown color in hippocampus (dentate gyrus) and immune-labeling scores were shown in Fig. 9a, b. One-way ANOVA for immune-labeling scores of the hippocampus indicated significant effects for the group (hippocampus, F(2, 8) = 272.39, p < 0.001). The treadmill-exercised PS2 mutant mice showed a significant decrease in apoptotic neuronal cell death compared to the sedentary PS2 mutant mice (hippocampus gyrus region), suggesting that TE repressed neuronal apoptosis by down-regulation of UPR signaling pathways and ER stress-specific apoptotic protease, CHOP, caspase-12 and caspase-3 in the hippocampus.

Treadmill exercise repressed neuronal apoptosis in the hippocampus of aged PS2 mutant mice. a Photomicrograph of neuronal apoptotic cells in the hippocampus (see arrows). b The mean number of TUNEL positive cells. Bar size 200 μm, magnification ×400. Three mice per non-Tg and PS2 mutant mice subgroups were assayed in triplicate by TUNEL assay. The mean number of TUNEL-positive cells in the hippocampus, respectively was significantly reduced in PS2 mutant mice after TE. Three mice per non-transgenic mice and PS2 mutant mice subgroups were assayed in triplicate on TUNEL assay. Fisher’s LSD post hoc test after ANOVA. Values are mean ± SEM
Discussion
Accumulation of Aβ or aggregation of Aβ are hallmarks of AD and are associated with the progression of AD symptoms. Recent studies have reported the striking finding that exercise training prevents the progression of AD in both human and animal models of AD. This has prompted the use of exercise training as a non-pharmacological and practical therapeutic countermeasure against AD. Currently, however, the molecular mechanisms responsible for exercise-induced prevention of AD are poorly understood. In this report, we have used a mouse model to investigate the effects of TE on the repression of neuronal cell death caused by AD-induced ER stress. This mouse model has a mutation in the PS2 gene and therefore shows early onset AD.
Accumulation of Aβ, memory impairment, inflammation, and neuronal cell death are commonly observed in AD mice and humans [30, 33, 38, 40]. The pathological activation of UPR due to Aβ-induced ER stress also becomes prominent cause of neuronal cell death in the brain of AD mice and humans [22, 41–48]. We also found that aged PS2 mutant mice showed abundant Aβ protein and inflammation, impaired cognitive function, and a greater extent of neuronal apoptosis. In particular, we found that accumulation of Aβ caused chronic ER stress, thereby contributing to AD-mediated neuronal apoptosis by activation of the UPR signaling pathways.
Moderate ER stress promotes the protein expression of the ER resident chaperones such as GRP78/BiP and PDI by UPR signaling. This enhances protein refolding as a cellular defense response. In contrast, chronic ER stress induced by accumulation of Aβ switches UPR signaling from a pro-survival to a pro-apoptotic pathway. In this regard, mitigation of ER stress, possibly via the reduction of Aβ deposition, appears to be a promising strategy to help prevent the progression of AD. We found that 12 weeks of TE reduced Aβ-42 deposition in both cortex and hippocampus and prevented cognitive impairment. Since Aβ and C-99 (a cell membrane-bound protein fragment) are produced as cleavage products of amyloid precursor protein (APP) via β-secretase (BACE-1) activity, we examined the level of BACE-1 and C-99. A finding of suppression of these protein levels in response to TE increases the probability that reduction in Aβ-42 deposition is due to the suppression of BACE-1. Surprisingly, TE repressed the expression of neuronal BACE-1 in conjunction with concomitant reduction in C-99 production. These results suggest that the reduction in Aβ-42 deposition in the brain tissue following TE is due to the down-regulation of BACE-1 expression. Similar findings have been reported for voluntary wheel running exercise, which also prevents accumulation of Aβ and improves cognitive function [49, 50].
The basic mechanisms by which Aβ is regulated appear distinctive. For example, our PS2 mutant Tg mouse model showed that the reduction in Aβ following TE is induced by repression of β-secretase, whereas Adlard et al. [49] (APP double mutant Tg mouse model; voluntary wheel running) did not observe any decrease in the level of β-secretase, nor changes in Aβ-degrading enzymes such as neprilysin and insulin dependent enzyme (IDE). These findings suggested that some other mechanism, such as increased proteosomal activity via increased neuronal activity, may enhance the removal of Aβ. Similarly, Maesako et al. [50] (APP mutant Tg mouse model; voluntary wheel running) observed an exercise-mediated improvement in cognitive function, but found no exercise-mediated repression of β-secretase. However, in contrast to the results of Adlard et al., they found that a voluntary wheel running exercise increased the activity of a metalloprotease enzyme, neprilysin, and the increase was correlated with elimination of aggregated Aβ [49]. A recent study using the triple mutation (APP, PS1, and Tau) transgenic mouse model of AD showed that, aside from inducing changes in Aβ, voluntary wheel running exercise was neuroprotective against the progression of AD, by improvements in antioxidative capacity [51]. Collectively, although the mechanism of exercise-induced neuronal protection seems to differ depending on the strains of animals and modality of exercise (i.e., treadmill vs. wheel running), all studies agree that endurance exercise ameliorates the progression of AD, regardless of the stage of AD. To the best of our knowledge, our study is the first to demonstrate that treadmill running prevents the progression of AD by reducing Aβ accumulation through repression of BACE-1.
Accumulation of Aβ can trigger ER stress and result in prolonged activation of UPR in AD; this leads to neuronal apoptosis by activation of three classes of ER sensors including UPR kinases (PERK, IRE1, ATF6) [10, 11, 52–55]. Dissociation of the ER stress chaperone protein GRP78/BiP from the ER membrane initiates UPR kinase activity under ER stress. This activation in turn increases expression of GRP78/BiP and PDI as a means of counteracting ER stress [56]. For example, activation of IRE1 induces splicing of transcription factor XBP1 to generate sXBP1, which increases expression of GRP78/BiP [57]. Elevation of GRP78/BiP and PDI, and phosphorylation of UPR kinases, are therefore considered to be ER stress hallmarks. Our examination of the effects of TE on activation (phosphorylation) of UPR kinases and their downstream targets showed that, in the hippocampus of aged PS2 mutant mice, TE reduced the phosphorylation of PERK-eIF2α, which are two critical components of UPR, and it suppressed the expression of ATF6α and sXBP1. Consequently, TE prevented and attenuated the upregulation of GRP78/BiP and PDI, respectively, in aged PS2 mutant mice, suggesting that reduction of Aβ-42 levels following TE is involved in ameliorating Aβ-induced ER stress.
Chronic ER stress caused by accumulation of Aβ promotes prolonged UPR, which activates a pro-apoptotic transcriptional factor, CHOP (also called GADD153) and GADD34 (a cofactor of eIF2α phosphatase) mainly via the PERK-eIF2α pathway [58]. In particular, strong activation of PERK-eIF2α, IRE1-XBP1 and JNK-p38MAPK is apparently necessary to induce CHOP and neuronal apoptosis [53, 59, 60]. CHOP acts as a pro-apoptotic protein by suppressing transcription of the anti-apoptotic protein Bcl-2 [60, 61]. Pro-apoptotic kinases such as apoptosis signal-regulating kinase 1 (ASK-1) and c-Jun NH2 terminal kinase (JNK)-p38MAPK are also activated through the IRE1-XBP1 pathway [62–66]. In this regard, inhibition of the activation of PERK-eIF2α, ATF6α, sXBP1, JNK-p38MAPK, and CHOP would prevent the Aβ-induced neuronal apoptosis seen in AD. Surprisingly, when compared to sedentary PS2 mutant mice, treadmill exercised PS2 mutant mice showed suppression of a series of pro-apoptotic signaling pathways, reduction in phosphorylation levels of JNK-p38MAPK, and reduced expression of sXBP1 and CHOP. TE caused attenuation of the AD-induced suppression of Bcl-2 in parallel with the repression of CHOP. These results complement the findings of McCullough et al. [61], who showed that CHOP reduces the expression of Bcl-2, and that a deficiency in CHOP prevents ER stress-induced apoptosis [67]. These findings indicated that exercise-mediated inhibition of PERK-eIF2α pathways that activate CHOP is a potential mechanism for the prevention of AD-induced apoptosis.
A potent pro-apoptotic protein, Bax, in addition to Bcl-2, further contributes to neuronal apoptosis via opening of the mitochondrial permeability transition pore [68]. A recent study has demonstrated that Bax localizes to the ER and this causes ER stress-mediated apoptosis [69]. We did not examine the localization of Bax (i.e. ER or mitochondria), but Bax was markedly overexpressed in the hippocampus (whole lysate) of sedentary aged PS2 mutant mice, while TE significantly prevented this overexpression. Collectively, our data indicate that TE inhibits AD-induced apoptosis by improving antiapoptotic reprogramming; i.e., exercise induces an alteration from a proapoptotic to an antiapoptotic response.
Cysteine-aspartic protease (caspase)-12, a homologous protein to human caspase-4, is localized in the ER and activated (cleaved) by ER stress, which subsequently initiates ER-mediated apoptosis including Aβ-induced neuronal apoptosis [52–54, 70, 71]. Once activated, caspase-12 transmits its apoptotic signal by activating (cleaving) a downstream effector, caspase-9, which then activates (cleaves) a cell death executioner caspase-3 [64, 72, 73]. We found that both caspase-12 (p36) and caspase-3 (p17) were activated in the hippocampus of aged PS2 mutant mice, in agreement with the findings of other studies that showed marked activation of caspase-12 and caspase-3 in the neurons of AD transgenic mice and in the brain sections of AD patients [33, 59, 64, 72–75]. We determined whether TE inhibits ER stress-mediated neuronal apoptosis against AD by measuring the expression of cleaved forms of caspase-12 and caspase-3 and examining apoptotic cell death. TE clearly resulted in a reduction in the cleaved forms of caspase-12 and caspase-3, which consequently prevented neuronal apoptosis (as evidenced by TUNEL assays) in the hippocampus of aged PS2 mutant mice. This result is consistent with our previous study [33]. We believe that anti-apoptotic reprogramming induced by TE is initially associated with a reduced production of Aβ through suppression of BACE-1 expression and repression of Bax expression, with the overall effect of reducing ER stress and neuronal cell death.
Inflammation contributes to this ER-mediated activation of a caspase cascade, and it also contributes to neuronal cell death [25, 28]. For example, activation of ATF6 by ER stress leads to activation of nuclear factor kappa B (NF-kB), a key transcription factor involved in triggering the inflammatory response [76] and TNFα receptor-mediated apoptotic cell death [29]. Li and coauthors [77] have reported that induction of UPR causes an elevation of proinflammatory cytokines such as TNFα and IL-6. A recent study demonstrated that a pharmacological TNF-inhibitor, 3,6-dithiothalidomide, prevented memory deficit and Aβ plaque formation in a 3× Tg mutant mouse model of AD [78]. In the present study, we showed that TNFα, and IL-1α were greatly elevated in the hippocampus of aged PS2 mutant mice, while TE significantly attenuated the production of these proinflammatory cytokines. We suggest that the mitigation of Aβ-induced ER stress via TE prevents neuroinflammation, thereby possibly exerting a protective effect against inflammation-directed apoptotic cell death in the late stages of AD.
Taken together, the present study shows that dual activation of cell death pathway (by UPR and inflammation) through pathological ER stress triggers a vicious cell death cycle in the brain of AD mice. For example, the activation of UPR pathways (i.e., PERK-eIF2, ATF6α, sXBP1 and JNK-p38MAPK) in PS2 mutant mice due to accumulation of Aβ is associated with induction of CHOP, caspase-12, and caspase-3 activation, and also with activation of inflammation (TNFα and IL-1α); both of these responses are clearly engaged in activation of caspase-3. TE interferes with this dual activation by pathological UPR and inflammation.
We suggest that neuroprotection against AD due to TE is caused by down-regulation of BACE-1 expression, which results in reduction of Aβ-42, ER stress, and inflammation as well as down-regulation of Bax and up-regulation of Bcl-2. This finding is of special importance because treadmill running not only inhibits new production of Aβ but it also promotes the removal of Aβ previously accumulated during the progression of AD in the brains of aged PS2 mutant mice. We did not investigate how previously accumulated Aβ during the progression of AD is removed by exercise training; however, this interesting and critical question warrants future investigation. Currently, several possible mechanisms have been proposed to explain Aβ degradation in response to treadmill running, including, but not limited to, neprilysin degradation, ubiquitin–proteasome effects, and autophagy. A recent well-designed study has demonstrated that 30 min of TE is sufficient to promote autophagy in the cerebral cortex [79]. Nevertheless, the importance of TE-induced autophagy in mediating Aβ degradation has not been elucidated. Therefore, further studies in this area will provide crucial insight into the development of a practical and powerful non-pharmacological therapeutic strategy that can be applied at various stages of AD, to supplement current pharmaceutical clinical trials that prove unsuccessful.
References
Lai E, Teodoro T, Volchuk A (2007) Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda) 22:193–201. doi:10.1152/physiol.00050.2006
Malhotra JD, Kaufman RJ (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18(6):716–731. doi:10.1016/j.semcdb.2007.09.003
LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8(7):499–509. doi:10.1038/nrn2168
Lindholm D, Wootz H, Korhonen L (2006) ER stress and neurodegenerative diseases. Cell Death Differ 13(3):385–392. doi:10.1038/sj.cdd.4401778
Scheper W, Hoozemans JJ (2009) Endoplasmic reticulum protein quality control in neurodegenerative disease: the good, the bad and the therapy. Curr Med Chem 16(5):615–626
Bernales S, Papa FR, Walter P (2006) Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22:487–508. doi:10.1146/annurev.cellbio.21.122303.120200
Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569(1–2):29–63. doi:10.1016/j.mrfmmm.2004.06.056
Wang SB, Shi Q, Xu Y, Xie WL, Zhang J, Tian C, Guo Y, Wang K, Zhang BY, Chen C, Gao C, Dong XP (2012) Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS One 7(6):e38221. doi:10.1371/journal.pone.0038221
Rutkowski DT, Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol 14(1):20–28
Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W (2005) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110(2):165–172. doi:10.1007/s00401-005-1038-0
Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J pathol 174(4):1241–1251. doi:10.2353/ajpath.2009.080814
Yamaguchi Y, Larkin D, Lara-Lemus R, Ramos-Castaneda J, Liu M, Arvan P (2008) Endoplasmic reticulum (ER) chaperone regulation and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J Biol Chem 283(25):17020–17029. doi:10.1074/jbc.M802466200
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287(5453):664–666
Zhang ZY, Liu XH, Ye YJ, Sun S, Rong F, Guo XS, Hu WC (2009) C/EBP homologous protein-mediated endoplasmic reticulum stress-related apoptosis pathway is involved in abdominal aortic constriction-induced myocardium hypertrophy in rats. Sheng li xue bao 61(2):161–168
Verkhratsky A (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85(1):201–279. doi:10.1152/physrev.00004.2004
Sato N, Urano F, Yoon Leem J, Kim SH, Li M, Donoviel D, Bernstein A, Lee AS, Ron D, Veselits ML, Sisodia SS, Thinakaran G (2000) Upregulation of BiP and CHOP by the unfolded-protein response is independent of presenilin expression. Nat Cell Biol 2(12):863–870. doi:10.1038/35046500
Wu G, Sankaranarayanan S, Wong J, Tugusheva K, Michener MS, Shi X, Cook JJ, Simon AJ, Savage MJ (2012) Characterization of plasma beta-secretase (BACE1) activity and soluble amyloid precursor proteins as potential biomarkers for Alzheimer’s disease. J Neurosci Res 90(12):2247–2258. doi:10.1002/jnr.23122
Emilien G, Maloteaux JM, Beyreuther K, Masters CL (2000) Alzheimer disease: mouse models pave the way for therapeutic opportunities. Arch Neurol 57(2):176–181
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21(3):383–421
Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M (2004) Induction of neuronal death by ER stress in Alzheimer’s disease. J Chem Neuroanat 28(1–2):67–78. doi:10.1016/j.jchemneu.2003.12.004
Mattson MP, Chan SL (2003) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 34(4–5):385–397
Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J (2009) ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation 6:41. doi:10.1186/1742-2094-6-41
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120(4):545–555. doi:10.1016/j.cell.2005.02.008
Abbas N, Bednar I, Mix E, Marie S, Paterson D, Ljungberg A, Morris C, Winblad B, Nordberg A, Zhu J (2002) Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J Neuroimmunol 126(1–2):50–57
Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C (2008) The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr Mol Med 8(3):157–172
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2009) Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87(3):181–194
Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, Carter DB, Chin JE (2001) Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res Bull 56(6):581–588
Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140(6):900–917. doi:10.1016/j.cell.2010.02.034
Cho JY, Hwang DY, Kang TS, Shin DH, Hwang JH, Lim CH, Lee SH, Lim HJ, Min SH, Seo SJ, Song YS, Nam KT, Lee KS, Cho JS, Kim YK (2003) Use of NSE/PS2m-transgenic mice in the study of the protective effect of exercise on Alzheimer’s disease. J Sports Sci 21(11):943–951. doi:10.1080/0264041031000140365
Cotman CW, Berchtold NC (2002) Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci 25(6):295–301
Fratiglioni L, Paillard-Borg S, Winblad B (2004) An active and socially integrated lifestyle in late life might protect against dementia. Lancet Neurol 3(6):343–353. doi:10.1016/s1474-4422(04)00767-7
Um HS, Kang EB, Koo JH, Kim HT, Jin L, Kim EJ, Yang CH, An GY, Cho IH, Cho JY (2011) Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s disease. Neurosci Res 69(2):161–173. doi:10.1016/j.neures.2010.10.004
Um HS, Kang EB, Leem YH, Cho IH, Yang CH, Chae KR, Hwang DY, Cho JY (2008) Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer’s disease in an NSE/APPsw-transgenic model. Int J Mol Med 22(4):529–539
Placanica L, Tarassishin L, Yang G, Peethumnongsin E, Kim SH, Zheng H, Sisodia SS, Li YM (2009) Pen2 and presenilin-1 modulate the dynamic equilibrium of presenilin-1 and presenilin-2 gamma-secretase complexes. J Biol Chem 284(5):2967–2977. doi:10.1074/jbc.M807269200
Nguyen HN, Hwang DY, Kim YK, Yoon DY, Kim JH, Lee MS, Lee MK, Yun YP, Oh KW, Hong JT (2005) Mutant presenilin 2 increases acetylcholinesterase activity in neuronal cells. Arch Pharm Res 28(9):1073–1078
Nguyen HN, Son DJ, Lee JW, Hwang DY, Kim YK, Cho JS, Lee US, Yoo HS, Moon DC, Oh KW, Hong JT (2006) Mutant presenilin 2 causes abnormality in the brain lipid profile in the development of Alzheimer’s disease. Arch Pharm Res 29(10):884–889
Hwang DY, Chae KR, Kang TS, Hwang JH, Lim CH, Kang HK, Goo JS, Lee MR, Lim HJ, Min SH, Cho JY, Hong JT, Song CW, Paik SG, Cho JS, Kim YK (2002) Alterations in behavior, amyloid beta-42, caspase-3, and Cox-2 in mutant PS2 transgenic mouse model of Alzheimer’s disease. FASEB J 16(8):805–813. doi:10.1096/fj.01-0732
Lee J, Cho HS, Park S, Kim WK (2009) Regular exercise produced cardioprotective effects on rat’s heart with hypertension induced by L-NAME administration. Clin Exp Hypertens 31(4):364–375
Nguyen HN, Lee MS, Hwang DY, Kim YK, Yoon do Y, Lee JW, Yun YP, Lee MK, Oh KW, Hong JT (2007) Mutant presenilin 2 increased oxidative stress and p53 expression in neuronal cells. Biochem Biophys Res Commun 357(1):174–180. doi:10.1016/j.bbrc.2007.03.119
Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292(5521):1552–1555. doi:10.1126/science.292.5521.1552
Benn SC, Woolf CJ (2004) Adult neuron survival strategies: slamming on the brakes. Nat Rev Neurosci 5(9):686–700. doi:10.1038/nrn1477
Forloni G, Terreni L, Bertani I, Fogliarino S, Invernizzi R, Assini A, Ribizzi G, Negro A, Calabrese E, Volonte MA, Mariani C, Franceschi M, Tabaton M, Bertoli A (2002) Protein misfolding in Alzheimer’s and Parkinson’s disease: genetics and molecular mechanisms. Neurobiol Aging 23(5):957–976
Kikuchi H, Almer G, Yamashita S, Guegan C, Nagai M, Xu Z, Sosunov AA, McKhann GM II, Przedborski S (2006) Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci USA 103(15):6025–6030. doi:10.1073/pnas.0509227103
Paschen W, Mengesdorf T (2005) Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium 38(3–4):409–415. doi:10.1016/j.ceca.2005.06.019
Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4(1):49–60. doi:10.1038/nrn1007
Viana RJ, Nunes AF, Rodrigues CM (2012) Endoplasmic reticulum enrollment in Alzheimer’s disease. Mol Neurobiol 46(2):522–534. doi:10.1007/s12035-012-8301-x
Yoshida H (2007) Unconventional splicing of XBP-1 mRNA in the unfolded protein response. Antioxid Redox Signal 9(12):2323–2333
Adlard PA, Perreau VM, Pop V, Cotman CW (2005) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci 25(17):4217–4221. doi:10.1523/jneurosci.0496-05.2005
Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Hayashida N, Asada-Utsugi M, Watanabe K, Uemura M, Kihara T, Takahashi R, Shimohama S, Kinoshita A (2012) Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J Biol Chem 287(27):23024–23033. doi:10.1074/jbc.M112.367011
Garcia-Mesa Y, Gimenez-Llort L, Lopez LC, Venegas C, Cristofol R, Escames G, Acuna-Castroviejo D, Sanfeliu C (2012) Melatonin plus physical exercise are highly neuroprotective in the 3 × Tg-AD mouse. Neurobiol Aging 33(6):1124
Yu Z, Luo H, Fu W, Mattson MP (1999) The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol 155(2):302–314. doi:10.1006/exnr.1998.7002
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403(6765):98–103. doi:10.1038/47513
Ferreiro E, Resende R, Costa R, Oliveira CR, Pereira CM (2006) An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol Dis 23(3):669–678. doi:10.1016/j.nbd.2006.05.011
Chafekar SM, Hoozemans JJ, Zwart R, Baas F, Scheper W (2007) Abeta 1-42 induces mild endoplasmic reticulum stress in an aggregation state-dependent manner. Antioxid Redox Signal 9(12):2245–2254
Hosoi T, Ozawa K (2010) Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci 118(1):19–29. doi:10.1042/cs20080680
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7):881–891
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7(9):880–885. doi:10.1038/sj.embor.7400779
Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M (2004) Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol 165(3):347–356. doi:10.1083/jcb.200310015
Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D (1996) Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol 16(8):4273–4280
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21(4):1249–1259. doi:10.1128/mcb.21.4.1249-1259.2001
Hussain SG, Ramaiah KV (2007) Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun 355(2):365–370. doi:10.1016/j.bbrc.2007.01.156
Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332(6163):462–464. doi:10.1038/332462a0
Lee JH, Won SM, Suh J, Son SJ, Moon GJ, Park UJ, Gwag BJ (2010) Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp Mol Med 42(5):386–394
Resende R, Ferreiro E, Pereira C, Oliveira CR (2008) ER stress is involved in Abeta-induced GSK-3beta activation and tau phosphorylation. J Neurosci Res 86(9):2091–2099. doi:10.1002/jnr.21648
Zhang K, Kaufman RJ (2006) Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb Exp Pharmacol 172:69–91
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12(7):982–995
Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G (2008) Viral control of mitochondrial apoptosis. PLoS Pathog 4(5):e1000018. doi:10.1371/journal.ppat.1000018
Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162(1):59–69. doi:10.1083/jcb.200302084
Seyb KI, Ansar S, Bean J, Michaelis ML (2006) β-Amyloid and endoplasmic reticulum stress responses in primary neurons: effects of drugs that interact with the cytoskeleton. J Mol Neurosci 28(2):111–123
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276(17):13935–13940. doi:10.1074/jbc.M010677200
Kang YK, Park MK (2005) Endoplasmic reticulum Ca2+ store: regulation of Ca2+ release and reuptake by intracellular and extracellular Ca2+ in pancreatic acinar cells. Mol Cells 19(2):268–278
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y (2002) An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277(37):34287–34294. doi:10.1074/jbc.M204973200
Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EM Jr, Roth KA (1999) In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol 58(9):1020–1026
Yukioka F, Matsuzaki S, Kawamoto K, Koyama Y, Hitomi J, Katayama T, Tohyama M (2008) Presenilin-1 mutation activates the signaling pathway of caspase-4 in endoplasmic reticulum stress-induced apoptosis. Neurochem Int 52(4–5):683–687. doi:10.1016/j.neuint.2007.08.017
Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW, Paton JC, Kitamura M (2009) Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol 183(2):1480–1487. doi:10.4049/jimmunol.0900017
Chen L, Jarujaron S, Wu X, Sun L, Zha W, Liang G, Gurley EC, Studer EJ, Hylemon PB, Pandak WM, Zhang L, Wang G, Li X, Dent P, Zhou H (2009) HIV protease inhibitor lopinavir-induced TNF-a and IL-6 expression is coupled to the unfolded protein response and ERK signaling pathways in macrophages. Biochem Pharmacol 78(1):70–77. doi:10.1016/j.bcp.2009.03.022
Tweedie D, Ferguson RA, Fishman K, Frankola KA, Van Praag H, Holloway HW, Luo W, Li Y, Caracciolo L, Russo I, Barlati S, Ray B, Lahiri DK, Bosetti F, Greig NH, Rosi S (2012) Tumor necrosis factor-alpha synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J Neuroinflammation 9:106. doi:10.1186/1742-2094-9-106
He C, Sumpter R Jr, Levine B (2012) Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 8(10):1548–1551. doi:10.4161/auto.21327
Acknowledgments
We thank the animal technicians Yo-W Choi, Dong-H Choi, and Seok-M, Hong for directing the animal facility at Korea National Sport University. This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2011-013-G00016).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kang, EB., Kwon, IS., Koo, JH. et al. Treadmill exercise represses neuronal cell death and inflammation during Aβ-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis 18, 1332–1347 (2013). https://doi.org/10.1007/s10495-013-0884-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-013-0884-9