
Abstract
Although Alzheimer’s disease (AD) has been shown to be associated with a true primary optic neuropathy, the underlying pathophysiology of this disease and in particular the optic nerve disorder is still poorly understood. The receptor for advanced glycation end products (RAGE) has been implicated in the pathogenesis of AD by mediating the transport of plasma amyloid-β into the brain. Once ligated, RAGE can play a role in signal transduction, leading to amplification and perpetuation of inflammatory processes. As a key player in the reaction to CNS injury, astrocytes have been shown to associate with RAGE in a number of diseases, including AD. To investigate the role of RAGE and astrocytes in the pathogenesis of AD optic neuropathy, we conducted immunohistochemical studies to examine the presence of RAGE in donor eyes from patients with AD (n = 10) and controls (n = 3). Both qualitative observation and quantitative analyses using imaging software were used to document the extent of RAGE in the neural tissues. The intensity and extent of RAGE expression was more prominent in AD nerves compared to controls (P < 0.05). The RAGE immunoreactivity was observed in the microvasculature and in close proximity to astrocytic processes. While RAGE immunoreactivity increased with age, the increase was more precipitous in the AD group compared to the controls. The up-regulation of RAGE in the AD optic nerves indicates that RAGE may play a role in the pathophysiology of AD optic neuropathy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly, afflicting 40% of individuals aged 85 or older [13]. AD is a neurodegenerative disorder characterized by impairment of cognitive and behavioral functions. Histopathological examination of the brain often reveals neurofibrillary tangles and extracellular senile plaques. Even though the optic nerve is part of the central nervous system and the effect of AD on the cerebral function has been well established, the fact that AD is associated with a true primary optic neuropathy has been largely overlooked [28]. Furthermore, the primary AD associated optic neuropathy produces visual effects that largely spare visual acuity [28, 30]. As a result, visual complaints are often disregarded and simply attributed to impaired cognitive function. In spite of normal visual acuity, patients with mild AD have demonstrated delayed initiation of saccadic eye movements and deficits in both visual evoked responses and contrast sensitivity [16]. Severe AD is often associated with impairments of vision, involving visual acuity, visual fields, and color vision [29]. Our previous histopathological studies have demonstrated a widespread axonal degeneration in AD optic nerves with damage primarily affecting the largest class of retinal ganglion cells, the M-cells [14, 30]. This selective involvement of M-cells leads to the clinically measurable neuro-ophthalmic impairments of AD optic neuropathy.
Although amyloid was traditionally thought to cause dysfunction in AD by disturbing the integrity of cell membranes, the amyloid deposits and fragments are now believed to be biologically active via their interaction with specific cell surface receptors [33]. Previous research has introduced the receptor for advanced glycation end products (RAGE) as one such receptor involved in the mediation of AD pathogenesis [6, 19, 23, 36]. Upregulation of RAGE in the cerebral microvasculature has been demonstrated in both AD brain and transgenic AD animal models, supporting its role in mediating the transport of amyloid-β from the circulation into the brain [1, 6, 7, 36, 37].
RAGE is a multiligand transmembrane receptor of the immunoglobulin superfamily, which binds not only advanced glycation end products, but also amyloid beta protein (Aβ), amphoterin, S100b/calgranulins, and other pro-inflammatory ligands [5, 32]. RAGE is expressed at low levels in cells such as neurons and astrocytes in its normal physiologic state [4]. Once ligated, RAGE mediates a signaling cascade which leads to its own up-regulation and a chronic inflammatory state amplified and perpetuated by the RAGE cascade [33]. Indeed, RAGE upregulation has been implicated in a diverse group of diseases, such as diabetes, inflammatory disorders, tumorigenesis, Huntington’s disease, AD, and other neurodegenerative diseases [5, 18, 27, 33, 34].
Regardless of its etiology, gliosis is a hallmark of CNS injury. Astrocytes play a primary role in a wide range of normal functions, including acting as metabolic buffers, detoxifiers, nutrient suppliers, and electrical insulators, as well as participating in repair and scar formation in the brain [11]. In response to injury, astrocytes undergo both hypertrophy and hyperplasia with prominent fibrous ramifying processes, enhanced immunoreactivity for glial fibrillary acidic protein (GFAP), and increased release of bioactive molecules. The active role of astrocytes in AD brain has been well documented in the literature [8, 9, 12, 20, 26]. While RAGE is expressed by astrocytes in the normal human brain, this expression is increased in AD [3, 17, 31]. Hence, astrogliosis may play a role in the neuronal and extracellular changes seen in the optic nerve of patients with AD.
In this immunohistochemical study, we characterized the presence and extent of RAGE and its association with microvasculature and astrocytes in human AD optic nerves. This study attempts to investigate the role of RAGE in the pathophysiology of AD optic neuropathy.
Materials and methods
Human autopsy specimens
Optic nerve specimens were obtained from ten patients with AD (79.2 ± 10.9 years, Braak’s stage III–VI) and three control subjects (68.7 ± 9.2 years) at autopsy (postmortem interval 3–13 h). All tissues were supplied by the Alzheimer’s Disease Research Center, Lions Eye Bank of Oregon, and Doheny Eye Bank following the guidelines of the local ethics committee. The diagnosis of AD was confirmed clinicopathologically in accordance with modified Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) or National Institute on Aging (NIA)-Reagan criteria and Braak–Braak Alzheimer classification [3, 15, 22]. Control optic nerves were obtained from subjects with no history of AD or other neurodegenerative diseases. Neither the patients with AD nor the patients from whom the control tissues were obtained had any history of diabetes mellitus, metastatic cancers, or sepsis. Positive controls were prepared from the hippocampus of patients with AD and control patients. A description of the patient data is summarized in Table 1.
Tissue processing
All tissues were immersion-fixed in 10% neutral buffered formalin immediately following enucleation of eyes with optic nerves attached. Nerves were oriented for superior and temporal landmarks with tissue ink and dissected away from the eyes. Dissection of the retrobulbar optic nerves into cross-sectional profiles approximately 3 mm thick immediately posterior to the globe was performed. Small nicks were placed at these superior (single) and temporal (double) landmarks with a razor blade for histological orientation. Tissues were then processed for paraffin embedding, cut at 5 μm on a microtome, and placed on coated (electrostatic attractant) “plus” glass microscope slides for greater adherence of paraffin sections for immunohistochemistry (IHC).
IHC: immunoperoxidase labeling
Using an indirect method for immunostaining for the human RAGE antigen, tissue sections on glass slides were placed in a 60°C oven for 1 h, then deparaffinized in xylene and rehydrated through graded alcohol, distilled water, and tris-buffered saline (TBS). Antigen retrieval was performed on these tissues by placing these tissue sections in a 1× citrate buffer, pH 6.2 (BioGenex, San Ramon, CA) within a steamer bath. The bath was then microwaved at 1,200 W for 15 min followed by a decrease in power at 480 W for another 15 min. After the sections were rinsed with TBS, endogenous peroxidase activity was blocked by 0.3% hydrogen peroxide. Tissue sections were incubated with primary antibody (mouse monoclonal anti-human RAGE antibody; US Biological, Cleveland, OH) at a dilution of 1:250 at room temperature in a humidity chamber for 1 h and rinsed in TBS. Sections were then exposed to a horseradish peroxidase-labeled secondary antibody (goat anti-mouse conjugated to HRP; Dako) at room temperature for 30 min and rinsed with TBS. The substrate 3,3′-diaminobenzidine (Dako) was used to visualize the reaction product (brown chromagen) and the tissue was counterstained with Mayer’s hematoxylin Lillie’s Modification (Dako), rinsed with distilled water, and incubated in TBS. Tissues were dehydrated through graded alcohols and cleared in xylene. Slides were cover-slipped with a permanent mounting medium and observed on a Zeiss Axioskop light microscope (Zeiss, Thornwood, NY). Images were captured by a Spot II digital camera (Diagnostic Instruments, Sterling Heights, MI) coupled to a computer. Negative control sections were incubated in antibody diluent (Dako) or non-immune serum in the absence of primary antibody.
IHC: double-immunofluorescence labeling
Using an indirect method for double-labeled immunostaining, histological sections were deparaffinized, rehydrated, and subjected to antigen retrieval as described previously. Sections were washed with phosphate-buffered saline solution then incubated with 2% BSA with 0.2% Triton X-100 in phosphate-buffered saline for 15 min. Tissues were then serially incubated with the first primary antibody (mouse monoclonal anti-human RAGE antibody, US Biological, Cleveland, OH) at a dilution of 1:250 at 37°C for 1 h in a humidity chamber, followed by the secondary antibody (goat anti-mouse conjugated to FITC, Dako) at a dilution of 1:20 at room temperature for 45 min. To determine the association of RAGE with astrocytes, tissue sections were incubated with a second primary antibody (rabbit anti-GFAP antibody, Dako) at a dilution of 1:8,000 at 37°C for 1 h followed by a secondary antibody (swine anti-rabbit conjugated to TRITC, Dako) at a dilution of 1:60 at room temperature for 45 min, then washed with phosphate-buffered saline. Tissue sections were mounted and cover-slipped with Vectashield mounting medium with DAPI for nuclear staining (Vector Laboratories, Burlingame, CA). To label myelin, a primary antibody (rabbit anti-myelin basic protein antibody, Dako) at a dilution of 1:2,500 was used. To determine the association of RAGE with endothelial cells, the same protocol was followed by using a primary anti-RAGE antibody (goat polyclonal anti-human RAGE antibody, Fitzgerald Industries International, Concord, MA) at a dilution of 1:250 and a primary anti-CD31 antibody (mouse monoclonal anti-human CD31 antibody, DAKO) at a dilution of 1:30. The secondary antibodies applied were rabbit anti-goat conjugated to FITC (Chemicon International, Temecula, CA) at a dilution of 1:100 and horse anti-mouse conjugated to Texas red (Vector Laboratories, Burlingame, CA) at a dilution of 1:50, respectively. Slides were viewed under a confocal microscope (Zeiss LSM 510, Zeiss), and cell types were identified based on the fluorescent label and cellular morphology.
Qualitative and quantitative analyses
Immunohistochemically stained slides were viewed under a light microscope (Zeiss Axioskop, Zeiss) at 25×, 200×, and 1,000× magnifications. The intensity of immunoreactivity was initially graded qualitatively as negative (−), minimal (+), moderate (++), and strong (+++). Negative staining was defined as no staining, minimal staining as sparse staining of the axons or glial cells, moderate staining as obvious staining limited to part of the axons or cells, and strong staining as prominent staining of axons and glia with most of the structures stained. Immunolabeling was quantitatively graded by scanning slides with a light microscope (Olympus Vanox-T, Olympus, Center Valley, PA) at 400× magnification. For every slide, ten microscopic images were captured and submitted for the quantitative analysis by analySIS image analysis software (Soft Imaging System Corp, Lakewood, CO). The average immunopositive area for every slide and the average ratio of the specific immunolabeled area to the total area analyzed were recorded. Statistical analysis was performed using the Wilcoxon Rank Sum test, Fisher’s exact test, Pearson test, and Spearman test. Two-tailed P values of 0.05 or less were considered statistically significant.
Results
Immunoperoxidase localization of RAGE in AD and control optic nerves
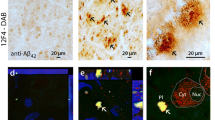
Immunoperoxidase staining was performed to examine the distribution and intensity of RAGE within the optic nerves of AD and controls which appeared in the form of punctuate granules. At low magnification, RAGE expression was mildly detectable in controls compared to the moderate-to-strong staining in AD (Fig. 1a, b). At high magnification (1,000×), very faint RAGE expression was seen in control nerves (Fig. 1c). In contrast, robust RAGE expression was evident in AD tissue (Fig. 1d). Although all nerves showed at least some expression of RAGE, control nerves never demonstrated a strong immunoreactivity for RAGE.

Immunoperoxidase labeling of the optic nerves for RAGE counterstained with hematoxylin in AD and control groups. a, c Represent control optic nerve sections from a 74-year-old male while (b) represents AD optic nerve sections from a 86-year-old male and (d) represents AD optic nerve sections from a 71-year-old male. At low magnification, RAGE expression was minimal in control (a) compared to moderate-strong staining in AD (b). At high magnification, very faint RAGE immunoreactivity (arrow) was observed in the control (c). In contrast, strong RAGE expression was evident in AD (arrow) (d). S Superior, T temporal, N nasal, I inferior
Based on our qualitative observations, the immunoreactivity for RAGE was much more prominent and widespread in the AD optic nerves compared to the control samples. The immunolabeling of RAGE in AD optic nerves was graded as moderate-to-strong as opposed to the minimal to moderate staining in the control nerves (Tables 2, 3).
Our quantitative analysis confirmed that the expression of RAGE was significantly greater in the AD group compared to the controls (P < 0.01; Table 4). The extent of RAGE immunolabeling (mean ± SD) was 6.78 ± 2.99% in the AD optic nerves and 0.57 ± 0.30% in the control optic nerves. Since our control tissues were slightly younger than AD tissues, we conducted another quantitative analysis on all tissues younger than 80 years to exclude the confounding factor of aging. The age-adjusted RAGE immunolabeling was 3.69 ± 1.00% in the AD group compared to the 0.57 ± 0.30% in the control (P < 0.05).
The correlation of RAGE expression with age
Our qualitative and quantitative analyses showed, as is evident from Fig. 2, that the intensity of RAGE grading increased with age in both AD and control groups. However, the increase in RAGE was significantly greater in the AD group compared to the controls (Fig. 2). The impact of age on the increased RAGE expression in AD optic nerves appeared in a linear fashion with a positive correlation of 0.89 (P < 0.001). The intensity of RAGE expression was stronger in the all age AD group compared to the <80-year-old AD group. However, the intensity of RAGE expression in either AD group was stronger than in the control group (Fig. 3).

The intensity of RAGE immunolabeling increased with age in both AD and control groups. However, this increase in RAGE was significantly more precipitous in the AD group compared to the control (P < 0.01). The increase in RAGE expression in the AD optic nerves increased with age in a linear fashion with a positive correlation of 0.89 (P < 0.001)

The mean values of RAGE immunolabeling from three different groups are shown. AD optic nerves from all age group demonstrated the strongest RAGE expression followed by AD optic nerves from less than 80-year-old group. Control optic nerves had the least RAGE immunoreactivity
In the control group, our analyses did not demonstrate a significant correlation between age and RAGE. In the AD group, we found no statistically significant correlation between RAGE and gender, duration of AD, pathologic stage, clinical dementia rating (CDR) score, or mini mental state examination (MMSE) score. However, our analyses were based on the limited information available on our patient’s past history data bases.
Double-immunofluorescence labeling for RAGE and astrocytes
Double-immunofluorescence labeling was performed to co-localize RAGE and GFAP-positive astrocytes. Our results demonstrated that staining for RAGE appeared in close proximity to GFAP in both AD and control groups (Fig. 4a, b). The extent of RAGE immunoreactivity was observed to be both qualitatively and quantitatively greater in the AD group compared to the control group. Double-immunofluorescence labeling of the optic nerves for RAGE and myelin basic protein (MBP) demonstrated a weak association between RAGE and myelin rings in both AD and control groups (Fig. 4c, d). The extent of RAGE expression was again stronger in the AD group. Double-immunofluorescence labeling for RAGE and CD31 demonstrated RAGE expression in the microvasculature of AD optic nerves (Fig. 5).

Double-immunofluorescence labeling of the optic nerves for RAGE coupled to FITC (green) and GFAP coupled to TRITC (red) counterstained with DAPI. a Represents control optic nerve section from a 74-year-old male and (b) represents AD optic nerve section from a 80-year-old male. RAGE expression was seen in close proximity to the GFAP-positive astrocytes in both groups (arrows). However, the intensity of RAGE immunoreactivity was much stronger in the AD optic nerves compared to the controls. Double-immunofluorescence labeling of the optic nerves for RAGE coupled to FITC (green) and MBP coupled to TRITC (red) counterstained with DAPI. c Represents control optic nerve section from a 74-year-old male and (d) represents AD optic nerve section from a 80-year-old male. The association of RAGE with myelin rings was weak in both groups (arrowheads). The intensity of RAGE expression was much stronger in the AD optic nerves compared to the controls. CT Connective tissues, FITC fluorescein isothiocyanate, GFAP glial fibrillary acidic protein, MBP myelin basic protein, TRITC tetramethyl rhodamine isothiocyanate, DAPI 4′,6-diamidino-2-phenylindole

Double immunofluorescence labeling of the AD optic nerves for RAGE coupled to FITC (green) and CD31 coupled to Texas red (red). Microvasculature in this tissue obtained from a 71-year-old male was stained with both RAGE and CD31
Discussion
We demonstrated that there is an increased expression of RAGE in AD optic nerves compared to controls. Indeed, RAGE increased with age but much more so with AD. This result is supported by similar studies on RAGE in brains of AD patients [31, 36]. RAGE normally plays a role in inflammatory and immune systems. In response to injury and inflammation, RAGE is up-regulated to mediate local tissue repair and restore homeostasis [27]. The RAGE-mediated signaling cascade induces the secretion of pro-inflammatory cytokines such as tumor necrosis factor-α and interleukin-1 and 6 by the RAGE expressing cells [31]. In addition to promoting inflammation, it also leads to the synthesis of growth factors, extracellular matrix proteins, and vascular adhesion molecules, as well as to an increase in procoagulant activities [32, 33]. Perhaps once past a threshold, these downstream events then up-regulate RAGE and activate more RAGE expressing cells in a positive feedback loop, eventually resulting in a pathophysiological cascade [33]. At the nuclear level, RAGE can activate transcriptional factors such as Nuclear Factor-κB, which not only can contribute to further pro-inflammatory cytokine release, but also can up-regulate RAGE [24]. Together, these events can lead to sustained cellular activation and proliferation, chemotaxis, angiogenesis, reactive oxygen species generation, and apoptosis. More interestingly, RAGE is upregulated in the presence of its own ligands thereby further sustains cellular inflammation [2]. In addition, since RAGE is a multiligand receptor that recognizes three-dimensional molecular structures instead of specific amino acid sequences [5, 33], it can bind to a wide variety of cells and ligands. This widespread effect and relative non-specificity and redundancy could further aggravate the sustained inflammation. Therefore, this increase in RAGE could be partially attributable to a compensatory phenomenon and a failed cellular adaptation. In this context, RAGE may be a causative agent in the pathophysiology of AD optic neuropathy. Alternatively, since RAGE is up-regulated in the presence of elevated ligands [32], the increase in RAGE might merely illustrate an end-stage consequence. Since RAGE normally plays a role in the inflammatory and immune systems, it could theoretically be beneficial when the inflammatory ligand is transient. Perhaps the problem occurs only when the inflammation is prolonged, and at a certain point RAGE serves as a trigger that converts transient inflammation into a chronically activated cellular dysfunction. Animals in the RAGE knockout models showed normal development and phenotype [27], suggesting a redundant system wherein other receptors or molecules could substitute for RAGE in its absence. The beneficial effects of RAGE could then be masked by this redundant system. The question of whether RAGE is pro-survival or detrimental still remains to be elucidated.
The increase in RAGE demonstrated by our study also appears to be age-dependent in both AD and control groups. RAGE is normally up-regulated in response to injury and oxidant stress [25]. With aging, many cellular structures deteriorate and reactive oxygen species accumulate in the cells, thereby increasing both physical and oxidant stresses. RAGE might be up-regulated to counteract such stress. Furthermore, RAGE is up-regulated during development and downregulated in adult life [4]. It is elevated again during aging, suggesting that it may have unknown cytoprotective properties against aging. However, not all individuals with the presence of RAGE develop pathology. Some links between the presence of RAGE and the final trigger to disease appear to be missing. Despite an apparent trend of RAGE to increase with aging, our younger AD optic nerves demonstrated much more RAGE labeling than the older control optic nerves. Even when the aging factor was taken into consideration, RAGE was still significantly more prevalent in the AD group, again suggesting that RAGE may play a role in the neurodegenerative process by accelerating pathology. As a result, the stronger immunoreactivity of RAGE in AD optic nerves may suggest an overlap of both aging and pathological processes.
Our results showed strong RAGE staining in close proximity to the astrocytes of AD, which parallels the results of another human AD studies of RAGE in other regions of the central nervous system [31]. Since RAGE colocalized with Aβ, Sasaki et al. suggests that RAGE could mediate degradation of Aβ in astrocytes. Increasing evidence indicates that Aβ accumulation in AD might result from both increased production and impaired elimination. Previous studies have shown that astrocytes could play an active role in the clearance of Aβ by both direct ingestion and degradation [35]. In this context, RAGE could be beneficial to prevent disease progression at its earliest stage. Perhaps RAGE initially carries a cellular protective capacity as it is expressed in the major phagocytic cells. When the system is overwhelmed by the increasing accumulation of Aβ, RAGE switches its role from neuroprotective to neurodestructive. Once the sustained cell mediated signaling pathway is activated, RAGE axis perpetuates the disease progression.
Astrocytes are the brain cells of the mononuclear phagocytic system. They function to protect neurons, supply nutrients, provide electrical insulation, and repair in gliosis [11]. Astrocytes are normally neuroprotective; but when chronically activated, they can become destructive by releasing neuro-toxic substances or activating deleterious pathways [26]. Hence, in the pathologic state, RAGE expression on astrocytes could not only mitigate their protective functions but also promote directly to neuronal injury and death. In addition, since the inflammatory response is believed to contribute significantly to neuronal dysfunction, and ultimately to neuronal death, chronic inflammation sustained by RAGE axis could aggravate the situation further.
Amyloid-β can accumulate in AD brain from increased rate of production and impaired clearance. Growing evidence suggests that amyloid-β accumulation through increased receptor-mediated transport can be another mechanism [38]. RAGE has been demonstrated to be such receptor able to actively transport amyloid-β across the blood–brain barrier into the central nervous system in animal models [1, 6]. In support of this observation, RAGE up-regulation has also been shown in human AD cerebral microvasculature associated with amyloid-β accumulation in the brain [7, 36, 37]. Another study examining the correlation between microvascular RAGE expressions with AD pathology has found an increased RAGE signal in advanced AD endothelium compared to early AD and control endothelium [21]. Our double-immunofluorescence labeling results also demonstrated co-localization of RAGE and CD31 in the AD optic nerve tissues, which also supported the receptor-mediated transport mechanism. These findings suggest that RAGE expression in the microvasculature could be a major pathogenic event in AD optic neuropathy by increasing the inward flux of amyloid-β into the optic nerves.
A weak association of RAGE with myelin rings was demonstrated in this study. Since oligodendrocytes mainly function to myelinate whereas astrocytes play a major role in tissue repair in response to injury and inflammation, astrocytes are expected to be more active in AD optic neuropathy.
The present study demonstrated only a poor correlation between RAGE and the pathoclinical stage of AD. This lack of tight correlation could be due to either the lack of documented clinical information available or the small sample size that did not carry enough statistical power to show the correlation. In the future, we will examine retinal staining to determine the distribution of RAGE in the retinal ganglion cells where the axons of the optic nerve originate. Because of its close interaction with RAGE and its critical role in AD pathophysiology, amyloid-β distribution and association with RAGE would be another area of interest. It would be interesting to analyze ocular conditions showing gliosis of other etiologies to confirm the association between RAGE and astrocytes. Future studies examining RAGE immunolabeling with microglial markers and neurofilament may further elucidate the role of RAGE axis in the pathogenesis of optic neuropathy in AD. To determine whether RAGE is the cause or consequence of AD optic neuropathy, studies using anti-RAGE may be conducted in AD optic nerve murine model. Monoclonal anti-RAGE and soluble RAGE (sRAGE) have already been shown to suppress AD-like pathology and reduce the transport of Aβ across the blood–brain barrier in AD animal models [1, 6, 10]. If RAGE indeed plays a role in the etiology of AD optic neuropathy, anti-RAGE holds therapeutic promise.
In conclusion, our findings demonstrate a significantly greater expression of RAGE in AD optic nerves than our normal controls. This result along with the RAGE expression in the AD microvasculature supports the hypothesis that RAGE may play a role in the underlying pathophysiology of AD optic neuropathy most likely by transporting amyloid-β into the optic nerve from the circulating bloodstream. It would also be of interest to determine if antagonism of this receptor, pharmacologically, could mitigate both the ongoing optic neuropathy and other neurodegenerative changes often found in patients with AD.
References
Arancio O, Zhang HP, Chen X et al (2004) RAGE potentiates abeta-induced perturbation of neuronal function in transgenic mice. EMBO J 23:4096–4105. doi:10.1038/sj.emboj.7600415
Bierhaus A, Humpert PM, Morcos M et al (2005) Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 83:876–886. doi:10.1007/s00109-005-0688-7
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. doi:10.1007/s00401-006-0127-z
Brett J, Schmidt AM, Yan SD et al (1993) Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 143:1699–1712
Bucciarelli LG, Wendt T, Rong L et al (2002) RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci 59:1117–1128. doi:10.1007/s00018-002-8491-x
Deane R, Du Yan S, Submamaryan RK et al (2003) RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med 9:907–913. doi:10.1038/nm890
Donahue JE, Flaherty SL, Johanson CE et al (2006) RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol 112:405–415. doi:10.1007/s00401-006-0115-3
Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH (1988) Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol 132:86–101
Duffy PE, Rapport M, Graf L (1980) Glial fibrillary acidic protein and Alzheimer-type senile dementia. Neurology 30:778–782
Emanuele E, D’Angelo A, Tomaino C et al (2005) Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol 62:1734–1736. doi:10.1001/archneur.62.11.1734
Frosch MP, Anthony DC, de Girolami U (2005) The central nervous system. In: Kumar V, Abbas AK, Fausto N (eds) Robbins and Cotran pathologic basis of disease, 7th edn. Elsevier Saunders, Philadelphia, pp 1347–1420
Griffin WS, Stanley LC, Ling C et al (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86:7611–7615. doi:10.1073/pnas.86.19.7611
Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60:1119–1122. doi:10.1001/archneur.60.8.1119
Hinton DR, Sadun AA, Blanks JC, Miller CA (1986) Optic-nerve degeneration in Alzheimer’s disease. N Engl J Med 315:485–487
Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095–1097. doi:10.1097/00005072-199710000-00002
Katz B, Rimmer S (1989) Ophthalmologic manifestations of Alzheimer’s disease. Surv Ophthalmol 34:31–43. doi:10.1016/0039-6257(89)90127-6
Li JJ, Dickson D, Hof PR, Vlassara H (1998) Receptors for advanced glycosylation endproducts in human brain: role in brain homeostasis. Mol Med 4:46–60
Ma L, Nicholson LF (2004) Expression of the receptor for advanced glycation end products in Huntington’s disease caudate nucleus. Brain Res 1018:10–17. doi:10.1016/j.brainres.2004.05.052
Mackic JB, Stins M, McComb JG et al (1998) Human blood-brain barrier receptors for Alzheimer’s amyloid-beta 1–40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest 102:734–743. doi:10.1172/JCI2029
Mandybur TI, Chuirazzi CC (1990) Astrocytes and the plaques of Alzheimer’s disease. Neurology 40:635–639
Miller MC, Tavares R, Johanson CE et al (2008) Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res 1230:273–280. doi:10.1016/j.brainres.2008.06.124
Mirra SS, Heyman A, McKeel D et al (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Mruthinti S, Buccafusco JJ, Hill WD et al (2004) Autoimmunity in Alzheimer’s disease: increased levels of circulating IgGs binding Abeta and RAGE peptides. Neurobiol Aging 25:1023–1032. doi:10.1016/j.neurobiolaging.2003.11.001
Onyango IG, Tuttle JB, Bennett JP Jr (2005) Altered intracellular signaling and reduced viability of Alzheimer’s disease neuronal cybrids is reproduced by beta-amyloid peptide acting through receptor for advanced glycation end products (RAGE). Mol Cell Neurosci 29:333–343. doi:10.1016/j.mcn.2005.02.012
Pichiule P, Chavez JC, Schmidt AM, Vannucci SJ (2007) Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J Biol Chem 282:36330–36340. doi:10.1074/jbc.M706407200
Pike CJ, Cummings BJ, Monzavi R, Cotman CW (1994) Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience 63:517–531. doi:10.1016/0306-4522(94)90547-9
Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15:16R–28R. doi:10.1093/glycob/cwi053
Sadun AA (1989) The optic neuropathy of Alzheimer’s disease. Metab Pediatr Syst Ophthalmol 12:64–68
Sadun AA, Borchert M, DeVita E, Hinton DR, Bassi CJ (1987) Assessment of visual impairment in patients with Alzheimer’s disease. Am J Ophthalmol 104:113–120
Sadun AA, Bassi CJ (1990) Optic nerve damage in Alzheimer’s disease. Ophthalmology 97:9–17
Sasaki N, Toki S, Chowei H et al (2001) Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res 888:256–262. doi:10.1016/S0006-8993(00)03075-4
Schmidt AM, Yan SD, Yan SF, Stern DM (2000) The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta 1498:99–111. doi:10.1016/S0167-4889(00)00087-2
Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108:949–955
Stern D, Yan SD, Yan SF, Schmidt AM (2002) Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Deliv Rev 54:1615–1625. doi:10.1016/S0169-409X(02)00160-6
Wyss-Coray T, Loike JD, Brionne TC et al (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457. doi:10.1038/nm838
Yan SD, Chen X, Fu J et al (1996) RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691. doi:10.1038/382685a0
Zlokovic BV (2004) Clearing amyloid through the blood–brain barrier. J Neurochem 89:807–811. doi:10.1111/j.1471-4159.2004.02385.x
Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28:202–208. doi:10.1016/j.tins.2005.02.001
Acknowledgments
The authors would like to express their gratitude to the Alzheimer’s Disease Research Center, Lions Eye Bank of Oregon, and Doheny Eye Bank for supplying the tissues used in the experiments. This work was supported by Research to Prevent Blindness, Oakley Alzheimer Foundation, and NIH Grant EY03040.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, M.Y., Ross-Cisneros, F.N., Aggarwal, D. et al. Receptor for advanced glycation end products is upregulated in optic neuropathy of Alzheimer’s disease. Acta Neuropathol 118, 381–389 (2009). https://doi.org/10.1007/s00401-009-0513-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0513-4