
Abstract
Despite the multidisciplinary integration in the therapeutic management of glioblastoma multiforme (GBM), the prognosis of GBM patients is poor. There is growing recognition that the cells in the tumor microenvironment play a vital role in regulating the progression of glioma. Astrocytes are an important component of the blood-brain barrier (BBB) as well as the tripartite synapse neural network to promote bidirectional communication with neurons under physiological conditions. Emerging evidence shows that tumor-associated reactive astrocytes interact with glioma cells and facilitate the progression, aggression, and survival of tumors by releasing different cytokines. Communication between reactive astrocytes and glioma cells is further promoted through ion channels and ion transporters, which augment the migratory capacity and invasiveness of tumor cells by modifying H+ and Ca2+ concentrations and stimulating volume changes in the cell. This in part contributes to the loss of epithelial polarization, initiating epithelial-mesenchymal transition. Therefore, this review will summarize the recent findings on the role of reactive astrocytes in the progression of GBM and in the development of treatment-resistant glioma. In addition, the involvement of ion channels and transporters in bridging the interactions between tumor cells and astrocytes and their potential as new therapeutic anti-tumor targets will be discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is a WHO grade IV glioma and characterized as the most aggressive, primary malignant tumor [1]. GBM has an incidence of three per 100,000 adults annually, and patients with GBM survive approximately 12 months [2]. Common presenting symptoms include headache, progressive neurological disorder, increased intracranial pressure, and secondary epilepsy. These clinical symptoms not only reduce the patients’ quality of life but also shorten patients’ survival. Current treatment is surgical resection with adjuvant chemotherapy and/or radiotherapy; such multimodal treatment is necessary given the highly aggressive and progressive capacities of GBM [3]. Even so, therapy often fails to prevent tumor recurrence and patients still show significant mortality with a median survival of less than 15 months [4]. Therefore, there is an urgency to identify novel targets for more effective therapeutic development for GBM treatment.
The past effort has been centered on studying roles of genetic mutations as drivers in tumorigenesis in GBM [5]. Recently, the role of the tumor microenvironment has drawn considerable attention in GBM research [6]. Several studies suggest that the peritumoral tissue may participate in regulating the tumorigenesis, progression, invasion, angiogenesis, and treatment resistance of GBM [7, 8]. Microglia account for 10–15% of the cells in the brain and occupy about 30% of GBM tumor masses [9]. They promote the proliferation and invasion of GBM by releasing cytokines and immune adaptive factors [10, 11]. Another prominent species in the tumor microenvironment, constituting 40–50% of brain cells, are astrocytes and mature glial cells that are capable of mitosis. While they had initially been thought not to participate in tumor development, new evidence emerged to suggest that astrocytes could be activated by brain tumor cells and play a vital role in the progression, aggression, and angiogenesis of the tumor mass [12].
GBM is further propagated by the interaction between astrocytes and tumor cells via ion channels and ion transporters. They play an important role in maintaining the ionic homeostasis, establishing the membrane potential, regulating cell volume, osmotic pressure, and acid-base balance under physiological conditions. Recent studies have shown that expression and activation of Ca2+ and K+ channels regulate ion influx or efflux and are involved in proliferation, migration, and survival of glioma [13, 14]. This review will cover the role of ion channels in communication between astrocytes and glioma cells. Furthermore, the impact of ion channels and ion transporters in glioblastoma cell proliferation, migration, invasion, and apoptosis will be discussed. These new findings illustrate the potential of these ion channel and ion transporter proteins as therapeutic targets for GBM.
Astrocytes and Reactive Astrocytes in GBM
Astrocytes comprise nearly half of the total number of cells in mammalian brains [15] and play an important physiological role in the central nervous system (CNS). First, astrocytes support the neuronal network by dividing gray matter into distinct domains and augment the myelinating capacity of white matter [16, 17]. Growing evidence indicates that astrocytes also participate in the formation, development, and maintenance of neural circuits [18]. The membrane processes of astrocytes that contact other neural cells promote cell-cell communication, allowing the recycling of neurotransmitters and meeting the metabolic demands of the cells [19,20,21]. In addition, astrocyte endfeet are a component of the brain-blood barrier (BBB), which separates circulating blood from extracellular fluid by its highly selective permeability. By detecting the level of glutamatergic activity in the synapse, astrocytes can correspondingly adjust the calcium (Ca2+) concentration in their endfeet and release gliotransmitters that act on vascular smooth muscle to regulate brain blood flow [22].
In certain CNS pathologies, astrocytes are transformed into reactive astrocytes, characterized by hypertrophy, upregulation of intermediate filaments composed of nestin, vimentin, and glial fibrillary acidic protein (GFAP), as well as activation of cell proliferation [23]. Reactive astrocytes, in acute brain injuries like trauma, spinal cord injury, or stroke, form a functional barrier, termed as “glial scar,” which serves to restrict and regulate inflammation, isolate the lesion, regulate and repair the BBB, enhance synaptic plasticity, and initiate neuronal circuit reorganization [24]. Glial scar formation may interfere with axonal regeneration [23]. Furthermore, reactive astrocytes release cytokines, chemokines, interleukins, nitric oxide (NO), and other molecules, thereby exacerbating the neuroinflammatory responses [25]. Another pathology that has noted reactive astrocyte development includes the brain tumor microenvironment [6] which is further discussed below.
Glioma and Astrocyte Crosstalk
The tumor microenvironment is recognized to be crucial to the progression of tumors, with specifically the parenchymal, non-malignant cells adjacent to the mass thought to aid its development and growth [6]. The invasiveness of GBM is reflected by a significant amount of non-transformed cells adjacent to the tumor mass during examination of the surgically resected tissue, indicating close interaction between the two cell populations. Particularly, astrocytes display a reactive phenotype when they contact tumor cells, expressing a high concentration of GFAP [26]. These reactive astrocytes aid the parenchymal infiltrative capacity of glioma cells by expressing matrix metalloproteinase-2 (MMP2) and further stimulate uncontrolled proliferation of glioma cells by secreting stromal cell-derived factor-1 (SDF1) [27]. Thus, the reactive astrocytes in the tumor microenvironment augment GBM malignancy by causing aberrant cell proliferation and enhancing migration [28].
Reactive astrocyte formation in GBM can be regulated by the NF-κB signaling pathway [29]. Upon being treated with recombinant RANKL (rRANKL), a well-known activator of NF-κB signaling, astrocytes display increased GFAP, predisposing them to astrogliosis [30]. Lipopolysaccharide (LPS), another activator of NF-κB signaling, has also been shown to promote NF-κB activity by decreasing the protein levels of IκBα, a negative regulator of NF-κB [29, 30]. Inhibition of NF-κB reduced induction of GFAP expression in astrocytes by rRANKL and LPS, thus preventing their transformation into the reactive phenotype [30].
Another important player in the crosstalk between GBM and astrocytes is the tumor suppressor gene p53, found mutated in 87% of GBM cases [31]. When healthy cells suffer DNA damage, p53 is activated in the nucleus initiating cell death or growth arrest. However, the apoptosis mechanism is completely inhibited in cancerous p53−/− cells or reduced in heterozygous p53+/− cells. In addition to its autonomous function, p53 has been shown to regulate expression of proteins that are secreted to stimulate adjacent cells [32]. The extracellular matrix (ECM) of p53+/− astrocytes had a greater presence of laminin and fibronectin than the ECM of p53+/+ astrocytes. This richer tumor microenvironment formed by the neighboring p53+/− astrocytes aids the survival of GBM cells. In addition, evidence suggests that conditioned medium from GBM cells can inhibit the function of p53 in healthy p53+/+ astrocytes. Thus, there is a synergistic relation formed between GBM cells and the cells in the tumor microenvironment: GBM cells hinder p53 expression in astrocytes to predispose cells to aberrant cell proliferation, while dysfunctional p53 induces a nutritious environment in which GBM cells can thrive [28].
Reactive astrocytes can communicate with GBM cells directly or indirectly. One direct form of communication involves gap junction channel protein connexin43 (Cx43) in glioma-associated astrocytes [33]. Direct interaction is also facilitated by tunneling nanotubes (TNT), long and thin tubular structures established between GBM cells and astrocytes, participating in cell communication and influencing GBM proliferation [34]. In addition to this direct contact with glioma cells, reactive astrocytes secrete chemokines or cytokines, such as interleukin-6 (IL-6), transforming growth factor- β (TGF-β), insulin-like growth factor-1 (IGF-1), monocyte chemotactic protein-4 (MCP-4), interleukin-19 (IL-19), vascular endothelial growth factor (VEGF), and leukemia inhibitory factor (LIF), among others, to promote tumor cell invasion and migration, proliferation, and growth [35]. These mechanisms will be discussed in detail in the following sections.
Reactive Astrocytes Are Involved in Proliferation and Invasion of GBM
Tumor cells interact with one another and with astrocytes via gap junctions, integrins, and by releasing signaling molecules like growth factors and cytokines to enhance their proliferative and invasive capacity. It is thought that reactive astrocytes upregulate expression of Cx43 to confer chemotherapy resistance in addition to enhancing glioma cell proliferation and migration. In response to temozolomide (TMZ) exposure, Cx43 modulates the levels of Bcl-2 and Bax2 in LN229 human glioma cells, inhibiting the mitochondrial apoptotic response. Cx43 expression further blocks cytochrome C release from the mitochondria, which also prevents the malignant cell from undergoing apoptosis [36]. Besides resistance to TMZ treatment, Cx43 could augment the invasiveness of glioma cells. Substituting the wild-type Cx43 protein with a C-terminal truncated mutant in astrocytes reduced its communication with glioma, and the invasiveness of the glioma cells into the parenchyma was decreased [37]. Glioma cells have the capability to deliver miRNAs to astrocytes via gap junctions for intracellular communication. Thus, elimination of miRNAs in astrocytes decreases glioma invasiveness [38].
l-Glutamine (Gln) is involved in balancing the carbon and nitrogen requirements of neural tissue. In GBM, such metabolic dysfunction as Gln starvation could hinder glioma proliferation [39]. Although GBM cells can produce Gln by themselves, it is not sufficient to meet the metabolic need of the cell. To facilitate this requirement, astrocytes synthesize and secrete Gln, which was subsequently taken up by GBM cells, as seen in the astrocyte-glioma co-culture system [39]. Thus, there is an interdependence created in the tumor microenvironment between the glioma cells and the astrocytes, where the two work synergistically to promote GBM proliferation and invasiveness.
Indirectly, astrocytes secrete chemokines and cytokines to influence GBM proliferation and invasion. One such cytokine, IL-6, triggers several oncogenic factors, to create a suitable tumor microenvironment that promotes tumor proliferation, invasion, and angiogenesis [40]. Through the activation of the IL-6/p-STAT3 mechanistic pathway, IL-6 increases MMP2 expression, a transmembrane proteinase critical for the degradation of ECM to enhance the invasiveness of GBM [41]. Recent findings suggest that tumor-associated astrocytes secrete IL-6 to promote the progression of GBM in the astrocyte-glioma co-culture system, increasing its malignancy [35, 41]. IL-6 secreted by astrocytes also induces upregulation of MMP-14 and activates the Akt/p38MAPK/ERK(1/2) signaling pathway, further enhancing the invasive capacity of GBM [35]. The interaction between glioma cells and the surrounding astrocytes also activates MMP-2 via uPA-plasmin cascade [42]. In addition, reactive astrocytes secrete glial cell line-derived neurotrophic factor (GDNF), which binds to the RET/GFR1 receptor and activates the PI3K/Akt pathway to induce GBM migration. One study found that reducing GDNF concentration can decrease the size of the GBM tumor [43].
Initially identified in fetal astrocytes, astrocyte elevated gene-1 (AEG-1) presence is associated with poor survival in GBM [44] because of its involvement in the progression and migration of glioma cells [6]. Specifically, AEG-1 enhances the proliferation and the invasiveness of GBM by modulating the PI3K/Akt, NF-κB signaling pathway and upregulating MMP-2 and MMP-9 expression [6, 44,45,46]. Inhibition of AEG-1 with DYT-40, a novel synthetic 2-styryl-5-nitroimidazole derivative, was shown to reduce tumor volume and induce cell apoptosis in a rat model. Its mechanism of action involves downregulating the MAPK and PI3K/Akt signaling cascades, blocking the translocation of NF-κB, and ultimately inhibiting expression of AEG-1 [47].
Reactive Astrocytes and Resistance of Radiochemotherapy Treatment
Given the predisposition of GBM cells for aberrant proliferation and their extensive invasive capacity, the prognosis of patients is poor. Traditional clinical treatment includes surgery, radiation therapy (RT), and chemotherapy. With the advent of new medical technology, treatment became multidisciplinary with the integration of neuroimaging, surgery, neuropathology, chemotherapy, and palliative care [48]. Unfortunately, these are not sufficient to prevent tumor recurrence, and median survival of GBM remains 12–15 months. With recent evidence, suggesting the interaction between tumor cells and their microenvironment contributes to the resistance of tumor cells to therapy, therapeutic research has pivoted to find ways to combat such resistance [12].
Surgerical resection remains essential to the management of all grade gliomas, including GBM. Gross total resection is usually the frontline therapy considered because of its better clinical outcome. However, such maximal excision should only be performed if most of the patient’s neurological function can be protected [48]. Given the extent of GBM cells’ invasiveness and uncontrolled proliferation, it is difficult to remove the tumor totally [49]. Furthermore, the tumor microenvironment has a crucial role in regulating the progression, metastasis, and angiogenesis of GBM, which makes total tumor resection more difficult [12]. The tumor-associated astrocytes are a significant component of the tumor microenvironment and promote the increased proliferation of glioma cells, even after resection of tumor [50].
After surgery, patients are generally treated with combinational chemotherapy. TMZ is thought to be the most effective chemotherapeutic agent because of its high penetrance past the BBB [51]. TMZ’s mechanism of action involves increasing DNA strand breaks and triggering apoptosis in glioma cells [52]. Nevertheless, some patients exhibit resistance to TMZ treatment, in part, due to expression of O6-methylguanine-DNA methyltransference (MGMT). MGMT causes a decrease in TMZ-mediated methylation of DNA, reducing toxicity in the cell, and thus, lessens apoptotic activity [53].
Gap junction communication (GJC) between glioma cells and astrocytes has also been suggested to decrease sensitivity to TMZ chemotherapy [54]. These gap junctions allow the transfer of small molecules and microRNAs between cells that confer therapy-resistive properties [55]. Gap junction Cx43 in glioma cells can account for TMZ resistance [36]. Upon being exposed to TMZ, epidermal growth factor receptor (EGFR) is upregulated on the surface of tumor cells, which activates the JNK-ERK-AP-1 axis. This serves to increase Cx43 expression to ultimately reduce the sensitivity of glioma cells to TMZ [55]. Meanwhile, it was observed that knockdown of Cx43 in astrocytes increased TMZ-induced apoptosis of glioma cells [56]. It was noted above that upregulation of Cx43 in astrocytes promoted the proliferative and migratory capacities of glioma cells, so by experimentally downregulating this gap junction protein, TMZ was able to increase its effectiveness in combating GBM.
Another option for treatment of GBM involves radiotherapy. One model, to better understand the impact of the tumor microenvironment in treatment resistance, co-cultured human glioblastoma stem-like cells (GSCs) with astrocytes and discovered that the presence of astrocytes diminished the sensitivity of glioma cells to radiotherapy. Astrocytes were proposed to act in a paracrine mechanism to induce repair of DNA strand breaks. Chemokine profiling identified the activation of STAT3 in GSCs grown among astrocytes, which was not present in GSCs grown alone. The study proposed treating glioma cells with WP1066, a JAK/STAT3 inhibitor, and discovered that the radiosensitivity of the cancer cells increased, making them more vulnerable to therapy [57]. Therefore, studying the tumor microenvironment is crucial in understanding additional targets for therapy optimization.
Ion Channels and Ion Transporters as a Bridge Between Reactive Astrocytes and Glioma
The interaction between glioma cells and astrocytes in the tumor microenvironment is facilitated further via ion channels and ion transporters. Ion channels, considered pore-forming and multimeric proteins, are located in the plasma membrane of cells and serve to establish an electrical potential across the cell membrane. This prompts the flow of ions in response to chemical or mechanical stimuli, which regulates the cell volume. Recently, emerging new evidences suggest the role of ion channels in the progression, migration, and angiogenesis of tumor cells [58,59,60]. A multitude of ion channels are expressed in glial cells, including Na+ channels, K+ channels, Ca2+ channels, and Cl− channels [61]. This section discusses the role of ion channels and ion transporters in the communication between astrocytes and glioma cells.
Ion channels are involved in augmenting cell development, passing them through the necessary cell cycle checkpoints, and thus, playing a role in proliferation. The likely mechanism of action is by regulating the cell’s membrane potential; there is a distinct increase in K+ channel activity during the G1/S checkpoint, and similar Cl− channel activity is involved in the G2/M checkpoint [62]. As mentioned previously, GBM cells are highly invasive, and one feature that aids migration is the unregulated activity of ion channels. Water and ion movement is modulated by ion channel activity, having a great influence on the morphology of glioma cells. In addition to aiding invasiveness, ion channels contribute to the proliferative capacity of glioma cells [60].
Calcium Ion Channels in GBM
Ca2+ concentration in glioma cells helps regulate cell proliferation, migration, and apoptosis [63, 64]. GBM cells are proposed to use Ca2+ as a second messenger to aid in migration, the fluctuations in concentration regulated by ion channels. These changes in Ca2+ concentration induce Ca2+/calmodulin-dependent protein kinase II (CaMKII) to activate ClC-3 chloride channels, which begins GBM invasion [62]. The release of intercellular Ca2+ stores through inositol 1,4,5-trisphosphate receptor (IP3R) [65] and triggers Ca2+ entry into glioma cells, mediated by stromal interacting molecular 1 (STIM1) and Ca2+ channel Orai 1 [66, 67]. One study found that knockdown of STIM1 and Orai l, which decreases the intercellular Ca2+ concentration reduces GBM proliferation and invasion and induces tumor apoptosis [67, 68]. Furthermore, inhibition of IP3R reduces GBM migration and increases survival in a mouse xenograft model [65].
Cytosolic Ca2+ concentration helps modulate cell proliferation. There are several Ca2+-permeable channels observed in glioma cells, including T-type low voltage-gated calcium channels (VGCC) and transient receptor potential (TRP) channels, as well as purinergic receptors, which modulate ion flux [69]. Through managing Ca2+ influx and changing intercellular Ca2+ concentration, T-type VGCC promoted proliferation of one GSC cell line while inhibition of T-type VGCC by endostatin suppressed the tumor growth and increased tumor cell apoptosis [70]. The α1 subunit of T-type VGCC may play a key role in abnormal proliferation of GBM; siRNA-mediated knockdown of this subunit was demonstrated to decrease proliferation of GBM cells [71]. Furthermore, inhibition of T-type VGCC disrupted Akt signaling, a pathway involved in the proliferation of GBM cells [72]. In brief, T-type VGCC regulates glioma cell proliferation and apoptosis by controlling Ca2+ entry, but also via mediating Akt signaling.
Another subtype of Ca2+ channels is TRP channels, located in the plasma membrane of almost every cell type, where they act as the driving force for ion entry [73]. Subtypes of TRP channels were significantly overexpressed in GBM cells as compared to low-grade gliomas; these channels include transient receptor potential melastatin subfamily member-2 (TRPM2), TRPM3, TRPM7, TRPM8, transient receptor potential canonical channel protein-1 (TRPC1), TRPC6, transient receptor potential vanilloid subfamily member-1 (TRPV1), and TRPV2 [13, 74,75,76]. The TRPM channels are involved in the regulation of Ca2+ entry and the development of proliferation in tumor cells [73]. Studies show that TRPM7, when stimulated by hepatocyte growth factor/scatter factor (HGF/SF), induced influx of Ca2+ to increase the migration of glioma cells [77]. This mechanism involves TRPM7 modulating the PI3K/Akt and MEK/ERK signaling pathways to ultimately influence GBM cell proliferation and migration [78]. TRPM7 also works by activating the JAK/STAT3 and Notch signaling pathways to increase progression and aggression of GBM [79].
The TRPC family, activated directly by diacylglycerol (DAG) or indirectly through IP3-mediated Ca2+ release, serves to increase cytosolic Ca2+ concentration and plays a role in regulating cell growth [74]. TRPC1, a subtype of TRPC, was detected as one of the leading causes of cell migration, when stimulated by epidermal growth factor (EGF), which activates the p38MAPK/JNK signaling pathway [80]. Another subtype of TRPC, TRPC6, has been shown to be overexpressed in glioma cells [62]. It activates the Notch signaling pathway, increasing the aggression of glioma cells [81]. Inhibition of TRPC6 suppresses intracellular Ca2+ concentration, leading to inhibition of the cell cycle at the G2/M checkpoint. This response was utilized in mouse models with xenografted tumors, where blocking TRPC6 demonstrated reduced tumor volume [62].
However, not all subfamilies of TRP promote the proliferation and migration of glioma cells. Stock et al. demonstrated that neural precursor cells (NPCs) release endovanilloids that activate TRPV1 on glioma cells [76]. Activation of TRPV1 triggers glioma cell death [82], specifically via activating transcription factor-3 (ATF3), which mediates an endoplasmic reticulum stress pathway [76]. This innate defense mechanism is weakened with age, but TRPV1 agonists illustrate potential as therapeutic agents against GBM.
Potassium Ion Channels in GBM
Physiologically healthy glial cells have a very negative resting membrane potential (between − 80 and − 90 mV) and prefer K+ uptake, caused by inwardly rectifying currents. This contrasts from GBM cells, with more positive resting membrane potentials, between − 20 and − 40 mV. They tend to expel K+, meaning they express outwardly rectifying currents. Given their predisposition to expel K+, glioma cells are thought to express fewer inwardly rectifying K+ (Kir) channels than healthy glial cells [62].
Thus, glial cells experience more K+ influx and actively distribute K+ according to concentration, which involves diffusing the ions through gap junctions between cells. This K+ uptake by glial cells serves a crucial purpose: increased extracellular K+ in neural tissue would cause neurons to remain depolarized and unable to fire action potentials, needed for signaling [60]. Glioma cells have a diminished number of Kir channels and are unable to perform the function carried out by healthy glial cells to remove extracellular K+. This accrual of K+ in the extracellular space causes aberrant neuron signaling and may explain the prevalence of epileptic seizures seen in GBM patients, through an indirect mechanism of action. The presence of extracellular K+ causes the less negative resting potential seen in glioma cells (from − 40 to − 20 mV), which also weakens the Na+ gradient across the plasma membrane. Subsequently, the Na+-dependent glutamate transporters are downregulated, causing a higher glutamate presence extracellularly [62]. Excess glutamate concentration causes excitotoxicity, a condition correlated with epilepsy [83].
One type of voltage-dependent K+ channel, the human ether a-go-go-related gene potassium channel (hERG), is overxpressed in endometrial adenocarcinoma, myeloid leukemia, and colorectal cancer [84], a finding also observed in GBM cells [85]. hERG promotes VEGF secretion to initiate angiogenesis and increases the capacity for GBM proliferation [84]. When hERG was inhibited, either by siRNA knockdown or by treatment with doxazosin, apoptosis of glioma cells was observed [86]. Recent evidence indicates the involvement of microRNA in regulating hERG activity, to mediate proliferation and chemotherapy resistance in GBM cells. MiRNA-133b/miRNA-34a activates hERG, so upregulation of MiRNA-133b/miRNA-34a induces increased expression of hERG in glioma cells, leading to apoptosis resistance [87]. However, upregulation of another species of microRNA, miRNA296-3p, reduces the expression level of hERG, promoting anti-proliferation and reducing resistance to treatment [88]. Abnormal expression of other voltage-gated K+ channels, Kv1.5, Kv10.1, and Kv11.1, were also detected in GBM. Studies found that expression of these channels seemed to be associated with prognosis of GBM patients: for example, high Kv1.5 expression was correlated with a longer survival rate, while high Kv10.1 level predicted a shorter lifespan [89, 90].
Another key type of K+ channel is intermediate-conductance Ca2+-activated K+ (IK) channels. IK channels play a crucial role in maintaining the membrane potential, specifically by favoring outwardly rectifying K+ currents. Because IK channels are activated by Ca2+, increases in concentration make the potential of cells more negative [62]. Recently, overexpression of IK was detected to be correlated with the malignancy of the tumor, and thus indicative of poor patient survival [91]. Several studies indicate that chemokines like CXC-motif chemokine ligand 12 (CXCL12) can modulate GBM cell migration by activating IK channels [92, 93]. However, when cells were treated with TRAM-34, an inhibitor of IK channels, the migratory capacity of GBM cells was reduced, diminishing its invasiveness [94]. In addition, IK channels function by regulating the ERK pathway to promote tumor aggression [95]. A new study shows that ionizing radiation can activate IK channels, which subsequently enhances glioma cell proliferation and induces therapy resistance [96]. This suggests that IK channel antagonists may prove a viable therapeutic target to circumvent therapy-resistant cells and induce apoptosis.
Another subtype of Ca2+-activated K+ channels are big conductance Ca2+-activated K+ (BK) channels, which present with several isoforms because of the alternative splicing seen in their α-subunits. GBM cells express the isoform glioma BK (gBK) channels, formed as a result of a novel splice of hSlo, the gene that codes for the α-subunits [62]. gBK channels can be activated by TRPM8-mediated increases in intercellular Ca2+ concentration. Upon activation by changes in Ca2+ concentration, gBK channels stimulate changes in cell volume to promote migration and invasiveness of glioma cells [97]. Its role in migration was further demonstrated when gBK channel inhibitors like tetraethylammonium and paxilline actually impaired the migration of glioma cells [62]. gBK channels may also contribute to conferring therapy resistance to glioma cells; when cells were treated with TMZ, gBK channels were demonstrated to be activated, and the glioma cells’ apoptotic response was arrested [98]. Therefore, gBK channels are not only involved with the progression and invasion of GBM but can also enhance the glioma cells’ resistance to treatment.
Sodium Channels in GBM
Na+ ions contribute to maintaining osmotic pressure and electrolyte balance in cell. Patients with Na+ channel mutations, according to a univariate analysis, undergo a more aggressive disease course and suffer shorter survival rate than patients without Na+ channel mutations [99]. Deg/epithelial Na+ channels (ENaC) are responsible for Na+ uptake and water homeostasis under physiological conditions [100]. However, an abnormally high expression of ENaC was observed in GBM tissue [101, 102]. ENaC was shown to enhance the capacity of GBM cell migration [103]. One possible therapeutic option is the administration of psalmotoxin-1 (PcTX-1), an inhibitor of cation currents. In human GBM samples and GBM cell lines, PcTX-1 did block Na+ currents, as detected by electrophysiological properties. However, it had little effect on human astrocytes [62]. In addition, ENaC is also involved in the regulation of GBM proliferation by affecting the cell cycle. Treatment with PcTX-1 or benzamil could arrest cell growth in G0/G1 phase and reduce cell accumulation in S and G2/M phase [103]. Other antiproliferative treatments include ouabain and digoxin, which demonstrated reduced cell division when applied to GBM cell lines [62]. Additionally, acid-sensing ion channel-1 (ASIC1), a proton-gated cation channel when interacting with the ENaC family, may modulate the invasiveness of GBM [104]. ASIC1 interacts with α-ENaC and γ-ENaC, a process mediated by CaMKII [105]. Knockdown of either ASIC1, α-ENaC, or γ-ENaC reduces the glioma cell migration [102].
Chlorine Ion Channels in GBM
Cl− is essential to maintaining ion homeostasis and helps regulate cell volume and morphology [106]. Some studies discovered that upregulation of certain chloride intracellular channel proteins (CLICs) was also detected in glioma cells, such as CLIC-1, CLIC-3, and CLIC-7 [107]. CLICs are involved in the progression of GBM by promoting glioma cell proliferation and self-renewal. For instance, CLIC-1, a subtype of CLIC, takes part in regulating the cell cycle when activated during the G1/S phase, and inhibition of CLIC-1 was illustrated to cause G1 arrest [108]. In addition, CLIC-1 is involved in secreting extracellular vesicles, which play an important role in cell-cell communications, and further regulate cell growth [109]. Given their involvement in cell proliferation, high expression of CLIC1 correlate with shorter survival time for GBM patients [110].
Another subtype of CLICs, CLIC-3, is involved in regulation of glioma cell migration. Inhibition of CLIC-3 by chlorotoxin notably suppressed the invasiveness of tumor cells in vitro, although not completely. Similar results were found when CLIC-3 was blocked by siRNA-mediated knockdown [111]. CLIC-3 can also be phosphorylated by CAMKII, which serves to regulate channel activity. CAMKII inhibition in CLIC-3-expressing GBM cells reduced migration the same degree as when CLIC-3 was directly inhibited with chlorotoxin [62]. Thus, CAMKII proves to be an important player in ion transfers, which is crucial for the invasive cell migration seen in GBM.
As a part of neuron-glial network, astrocytes set up bidirectional communication with other cells by receiving and sending neurotransmitters, such as glutamate and γ-aminobutyric acid (GABA). Various mechanisms have been proposed for gliotransmission, including Ca2+-regulated vesicular exocytosis and release via plasma membrane ion channels under physiological condition [112]. In recent years, it was discovered that cell-cell communication via secreted extracellular vesicles is more common than initially thought. Exosomes are 30–100 nm in diameter and transport miRNAs and proteins that promote the progression of GBM [113]. Some ion channel proteins are circulating proteins secreted by other cell lines. In addition to CLIC-1-mediated GBM proliferation and self-renewal, CLIC-1 proteins are secreted via extracellular vesicles to promote glioma cell growth, as seen in vitro and in vivo. CLIC-1 silencing or overexpression can respectively diminish or enhance the effects of the secreted exosome [109].
Na-Dependent Ion Transporters in Glioma
Chemotherapy triggers a significant reduction in glioma cell volume resulting from loss of K+ and Cl−, which is a hallmark of apoptosis and is referred to as apoptotic volume decrease (AVD) [114]. In responding to cell volume loss triggered by apoptosis, cells will compensate by activating regulatory volume increase (RVI) pathways. RVI could be mediated by the gain of active solute such as Na+, K+, and Cl− or changes in osmotic pressure. Na-K-Cl cotransporter (NKCC1) is the main regulator of RVI, functioning by bringing Na+, K+, and Cl− into cells to maintain cell volume and counteract cell shrinkage [115]. It has been shown that bumetanide, an inhibitor of NKCC1, impairs RVI following TMZ treatment and thus enhances TMZ-mediated apoptosis in glioma cells [116].
Ion transporters also play a key role in the progression of GBM. The sodium/hydrogen exchanger (NHE) promotes migration and survival of GBM by inducing H+ extrusion, which maintains an alkaline intracellular pH and an acidic interstitial/extracellular pH [117]. Glioma cells secrete factors into the tumor microenvironment, such as IL-6, IL-10, STAT, TGF-β, bFGF, EGF, and MMP [12]. The production of proinflammatory cytokines stimulate astrocyte reactivity and disrupt ion homeostasis [118]. Cytokines secreted by glioma cells also regulate expression of ion channels in glioma-associated cells [119]. Meanwhile, reactive astrocytes release abundant chemokines into the tumor microenvironment, which further promote the progression, aggression, and tumorigenesis of GBM. Ion channels in the plasma membrane of these astrocytes are involved in regulating the release of secretion. For instance, activation of Ca2+ channels induces endothelin-1 secretion by astrocytes [120]. Blockade of Ca2+ channels prompts reduced astrocytic release of IL-6 and interleukin 1β (IL-1β) which may stimulate glioma cells [121]. Therefore, ion transporters play equally important roles in intercellular communications in glioma and glioma progression.
Conclusion
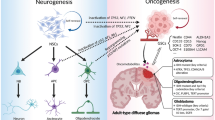
For several decades, studies of glioblastoma biology have generally focused on the tumor’s own genetic and molecular changes. However, tumor cells are not isolated, but grow in a particular environment by communicating with other cells, which influences progression, aggression, and survival of cancer [122]. Newly emerged evidence indicates that tumor-associated astrocytes can be activated by neighboring glioma cells and interact in a complex way with the tumor microenvironment to promote the proliferation, invasion, and treatment resistance of GBM. However, there are many unresolved questions and the mechanisms of interaction between tumors and astrocytes need to be explored further. In addition to gap junctions, ion channels and ion transporters play a key role in the progression, metastasis, and tumorigenesis of GBM (Fig. 1). They serve as a bridge of communication between tumor cells and the microenvironment. Blockage of ion channels and/or ion transporters have been shown to reduce the intercellular connections and decrease the proliferative and invasive nature of GBM (Fig. 1). In addition, combined treatment of TMZ with ion channel or ion transporter inhibitors enhanced apoptosis of tumor cells in vitro and/or in animal models. Collectively, further investigation of the intercellular interactive mechanisms and combined treatment of ion channel and ion transporter inhibitors may bring researchers one step closer to understanding GBM and creating a therapy that effectively combats its various defenses.

Schematic illustration of glioma and tumor associated astrocytes. Tumor-associated astrocytes interact in a complex way with glioma cells, endothelium, and the tumor microenvironment to promote the proliferation, invasion, and treatment resistance of GBM. In addition to gap junctions, ion channels and ion transporters play a key role in the progression, metastasis, and tumorigenesis of GBM. They serve as a bridge of communication between tumor cells and the microenvironment. Blockage of ion channels and/or ion transporters have been shown to reduce the intercellular connections and decrease the proliferative and invasive nature of GBM in experimental studies. Collectively, further investigation of the intercellular interactive mechanisms and combined treatment with ion channel and ion transporter inhibitors may enhance the efficacy of GBM therapies
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820. https://doi.org/10.1007/s00401-016-1545-1
Gallego O (2015) Nonsurgical treatment of recurrent glioblastoma. Curr Oncol 22(4):e273–e281. https://doi.org/10.3747/co.22.2436
Franceschi E, Minichillo S, Brandes AA (2017) Pharmacotherapy of glioblastoma: established treatments and emerging concepts. CNS Drugs 31(8):675–684. https://doi.org/10.1007/s40263-017-0454-8
Szulzewsky F, Arora S, de Witte L, Ulas T, Markovic D, Schultze JL, Holland EC, Synowitz M et al (2016) Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 64(8):1416–1436. https://doi.org/10.1002/glia.23014
Wang J, Su HK, Zhao HF, Chen ZP, To SS (2015) Progress in the application of molecular biomarkers in gliomas. Biochem Biophys Res Commun 465(1):1–4. https://doi.org/10.1016/j.bbrc.2015.07.148
Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H (2012) The brain tumor microenvironment. Glia 60(3):502–514. https://doi.org/10.1002/glia.21264
Fidoamore A, Cristiano L, Antonosante A, d'Angelo M, Di Giacomo E, Astarita C, Giordano A, Ippoliti R et al (2016) Glioblastoma stem cells microenvironment: the paracrine roles of the niche in drug and radioresistance. Stem Cells Int 2016:6809105–6809117. https://doi.org/10.1155/2016/6809105
Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, Hu J (2016) Glioma stem cells: signaling, microenvironment, and therapy. Stem Cells Int 2016:7849890. doi:https://doi.org/10.1155/2016/7849890, 1, 10
Miller IS, Didier S, Murray DW, Turner TH, Issaivanan M, Ruggieri R, Al-Abed Y, Symons M (2014) Semapimod sensitizes glioblastoma tumors to ionizing radiation by targeting microglia. PLoS One 9(5):e95885. https://doi.org/10.1371/journal.pone.0095885
Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19(1):20–27. https://doi.org/10.1038/nn.4185
Li W, Graeber MB (2012) The molecular profile of microglia under the influence of glioma. Neuro-Oncology 14(8):958–978. https://doi.org/10.1093/neuonc/nos116
Placone AL, Quinones-Hinojosa A, Searson PC (2016) The role of astrocytes in the progression of brain cancer: complicating the picture of the tumor microenvironment. Tumour Biol: J Int Soc Oncodevelopmental Biol Med 37(1):61–69. https://doi.org/10.1007/s13277-015-4242-0
Alptekin M, Eroglu S, Tutar E, Sencan S, Geyik MA, Ulasli M, Demiryurek AT, Camci C (2015) Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumour Biol: J Int Soc Oncodevelopmental Biol Med 36(12):9209–9213. https://doi.org/10.1007/s13277-015-3577-x
Bury M, Girault A, Megalizzi V, Spiegl-Kreinecker S, Mathieu V, Berger W, Evidente A, Kornienko A et al (2013) Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis 4(3):e561. https://doi.org/10.1038/cddis.2013.85
Herculano-Houzel S (2014) The glia/neuron ratio: how it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 62(9):1377–1391. https://doi.org/10.1002/glia.22683
Butt AM, Fern RF, Matute C (2014) Neurotransmitter signaling in white matter. Glia 62(11):1762–1779. https://doi.org/10.1002/glia.22674
Haroutunian V, Katsel P, Roussos P, Davis KL, Altshuler LL, Bartzokis G (2014) Myelination, oligodendrocytes, and serious mental illness. Glia 62(11):1856–1877. https://doi.org/10.1002/glia.22716
Clarke LE, Barres BA (2013) Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci 14(5):311–321. https://doi.org/10.1038/nrn3484
Pekny M, Pekna M (2014) Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev 94(4):1077–1098. https://doi.org/10.1152/physrev.00041.2013
Barres BA, Stevens B, Khakh BS, Sofroniew MV (2015) Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18(7):942–952. https://doi.org/10.1038/nn.4043
Choudhury GR, Ding S (2016) Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol Dis 85:234–244. https://doi.org/10.1016/j.nbd.2015.05.003
Bazargani N, Attwell D (2016) Astrocyte calcium signaling: the third wave. Nat Neurosci 19(2):182–189. https://doi.org/10.1038/nn.4201
Boccazzi M, Ceruti S (2016) Where do you come from and what are you going to become, reactive astrocyte? Stem Cell Investig 3:15. https://doi.org/10.21037/sci.2016.05.02
Burda JE, Bernstein AM, Sofroniew MV (2016) Astrocyte roles in traumatic brain injury. Exp Neurol 275(Pt 3):305–315. https://doi.org/10.1016/j.expneurol.2015.03.020
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14(4):388–405. https://doi.org/10.1016/s1474-4422(15)70016-5
Gagliano N, Costa F, Cossetti C, Pettinari L, Bassi R, Chiriva-Internati M, Cobos E, Gioia M et al (2009) Glioma-astrocyte interaction modifies the astrocyte phenotype in a co-culture experimental model. Oncol Rep 22(6):1349–1356
Barbero S, Bajetto A, Bonavia R, Porcile C, Piccioli P, Pirani P, Ravetti JL, Zona G et al (2002) Expression of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1 in human brain tumors and their involvement in glial proliferation in vitro. Ann N Y Acad Sci 973(1):60–69. https://doi.org/10.1111/j.1749-6632.2002.tb04607.x
Biasoli D, Sobrinho MF, da Fonseca AC, de Matos DG, Romao L, de Moraes Maciel R, Rehen SK, Moura-Neto V et al (2014) Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogene 3(10):e123. https://doi.org/10.1038/oncsis.2014.36
Lin ST, Wang Y, Xue Y, Feng DC, Xu Y, Xu LY (2008) Tetrandrine suppresses LPS-induced astrocyte activation via modulating IKKs-IkappaBalpha-NF-kappaB signaling pathway. Mol Cell Biochem 315(1–2):41–49. https://doi.org/10.1007/s11010-008-9787-4
Kim JK, Jin X, Sohn YW, Jin X, Jeon HY, Kim EJ, Ham SW, Jeon HM et al (2014) Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett 353(2):194–200. https://doi.org/10.1016/j.canlet.2014.07.034
Cancer Genome Atlas Research N (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216):1061–1068. https://doi.org/10.1038/nature07385
Rangel LP, Costa DC, Vieira TC, Silva JL (2014) The aggregation of mutant p53 produces prion-like properties in cancer. Prion 8(1):75–84. https://doi.org/10.4161/pri.27776
Theodoric N, Bechberger JF, Naus CC, Sin WC (2012) Role of gap junction protein connexin43 in astrogliosis induced by brain injury. PLoS One 7(10):e47311. https://doi.org/10.1371/journal.pone.0047311
Zhang L, Zhang Y (2015) Tunneling nanotubes between rat primary astrocytes and C6 glioma cells alter proliferation potential of glioma cells. Neurosci Bull 31(3):371–378. https://doi.org/10.1007/s12264-014-1522-4
Chen W, Xia T, Wang D, Huang B, Zhao P, Wang J, Qu X, Li X (2016) Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget 7(38):62425–62438. https://doi.org/10.18632/oncotarget.11515
Gielen PR, Aftab Q, Ma N, Chen VC, Hong X, Lozinsky S, Naus CC, Sin WC (2013) Connexin43 confers temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology 75:539–548. https://doi.org/10.1016/j.neuropharm.2013.05.002
Sin WC, Aftab Q, Bechberger JF, Leung JH, Chen H, Naus CC (2016) Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 35(12):1504–1516. https://doi.org/10.1038/onc.2015.210
Hong X, Sin WC, Harris AL, Naus CC (2015) Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 6(17):15566–15577. https://doi.org/10.18632/oncotarget.3904
Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PO et al (2015) Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol 17(12):1556–1568. https://doi.org/10.1038/ncb3272
Bharti R, Dey G, Mandal M (2016) Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: a snapshot of IL-6 mediated involvement. Cancer Lett 375(1):51–61. https://doi.org/10.1016/j.canlet.2016.02.048
Li (2010) IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol Rep 23(6). https://doi.org/10.3892/or_00000795
Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG, Rewcastle B, Yong VW (2003) Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci : Off J Soc Neurosci 23(10):4034–4043
Shabtay-Orbach A, Amit M, Binenbaum Y, Na'ara S, Gil Z (2015) Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int J Cancer 137(5):1012–1020. https://doi.org/10.1002/ijc.29380
Hu B, Emdad L, Bacolod MD, Kegelman TP, Shen XN, Alzubi MA, Das SK, Sarkar D et al (2014) Astrocyte elevated gene-1 interacts with Akt isoform 2 to control glioma growth, survival, and pathogenesis. Cancer Res 74(24):7321–7332. https://doi.org/10.1158/0008-5472.can-13-2978
Emdad L, Sarkar D, Su ZZ, Randolph A, Boukerche H, Valerie K, Fisher PB (2006) Activation of the nuclear factor kappaB pathway by astrocyte elevated gene-1: implications for tumor progression and metastasis. Cancer Res 66(3):1509–1516. https://doi.org/10.1158/0008-5472.can-05-3029
Sarkar D, Park ES, Emdad L, Lee SG, Su ZZ, Fisher PB (2008) Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res 68(5):1478–1484. https://doi.org/10.1158/0008-5472.can-07-6164
Zou M, Duan Y, Wang P, Gao R, Chen X, Ou Y, Liang M, Wang Z et al (2016) DYT-40, a novel synthetic 2-styryl-5-nitroimidazole derivative, blocks malignant glioblastoma growth and invasion by inhibiting AEG-1 and NF-kappaB signaling pathways. Sci Rep 6(1):27331. https://doi.org/10.1038/srep27331
Jiang T, Mao Y, Ma W, Mao Q, You Y, Yang X, Jiang C, Kang C et al (2016) CGCG clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett 375(2):263–273. https://doi.org/10.1016/j.canlet.2016.01.024
Hingtgen S, Figueiredo JL, Farrar C, Duebgen M, Martinez-Quintanilla J, Bhere D, Shah K (2013) Real-time multi-modality imaging of glioblastoma tumor resection and recurrence. J Neuro-Oncol 111(2):153–161. https://doi.org/10.1007/s11060-012-1008-z
Okolie O, Bago JR, Schmid RS, Irvin DM, Bash RE, Miller CR, Hingtgen SD (2016) Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro-Oncology 18(12):1622–1633. https://doi.org/10.1093/neuonc/now117
Agarwala SS, Kirkwood JM (2000) Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 5(2):144–151. https://doi.org/10.1634/theoncologist.5-2-144
Villano JL, Seery TE, Bressler LR (2009) Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother Pharmacol 64(4):647–655. https://doi.org/10.1007/s00280-009-1050-5
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG (2000) Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 343(19):1350–1354. https://doi.org/10.1056/NEJM200011093431901
Lin Q, Liu Z, Ling F, Xu G (2016) Astrocytes protect glioma cells from chemotherapy and upregulate survival genes via gap junctional communication. Mol Med Rep 13(2):1329–1335. https://doi.org/10.3892/mmr.2015.4680
Munoz JL, Rodriguez-Cruz V, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P (2014) Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis 5(3):e1145. https://doi.org/10.1038/cddis.2014.111
Chen W, Wang D, Du X, He Y, Chen S, Shao Q, Ma C, Huang B et al (2015) Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med Oncol (Northwood, London, England) 32(3):43. https://doi.org/10.1007/s12032-015-0487-0
Rath BH, Wahba A, Camphausen K, Tofilon PJ (2015) Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med 4(11):1705–1716. https://doi.org/10.1002/cam4.510
Nelson M, Yang M, Millican-Slater R, Brackenbury WJ (2015) Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget 6(32):32914–32929. https://doi.org/10.18632/oncotarget.5441
House CD, Wang BD, Ceniccola K, Williams R, Simaan M, Olender J, Patel V, Baptista-Hon DT et al (2015) Voltage-gated Na+ channel activity increases colon cancer transcriptional activity and invasion via persistent MAPK signaling. Sci Rep 5(1):11541. https://doi.org/10.1038/srep11541
Li F, Abuarab N, Sivaprasadarao A (2016) Reciprocal regulation of actin cytoskeleton remodelling and cell migration by Ca2+ and Zn2+: role of TRPM2 channels. J Cell Sci 129(10):2016–2029. https://doi.org/10.1242/jcs.179796
Olsen ML, Khakh BS, Skatchkov SN, Zhou M, Lee CJ, Rouach N (2015) New insights on astrocyte ion channels: critical for homeostasis and neuron-glia signaling. J Neurosci: Off J Soc Neurosci 35(41):13827–13835. https://doi.org/10.1523/JNEUROSCI.2603-15.2015
Molenaar RJ (2011) Ion channels in glioblastoma. ISRN Neurology 2011:590249. doi:https://doi.org/10.5402/2011/590249, 1, 7
Sforna L, Cenciarini M, Belia S, D'Adamo MC, Pessia M, Franciolini F, Catacuzzeno L (2014) The role of ion channels in the hypoxia-induced aggressiveness of glioblastoma. Front Cell Neurosci 8:467. https://doi.org/10.3389/fncel.2014.00467
Boscia F, Begum G, Pignataro G, Sirabella R, Cuomo O, Casamassa A, Sun D, Annunziato L (2016) Glial Na(+)-dependent ion transporters in pathophysiological conditions. Glia 64(10):1677–1697. https://doi.org/10.1002/glia.23030
Kang SS, Han KS, BM K, Lee YK, Hong J, Shin HY, Almonte AG, Woo DH et al (2010) Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res 70(3):1173–1183. https://doi.org/10.1158/0008-5472.can-09-2886
Potier M, Trebak M (2008) New developments in the signaling mechanisms of the store-operated calcium entry pathway. Pflugers Archiv : Eur J Physiol 457(2):405–415. https://doi.org/10.1007/s00424-008-0533-2
Motiani RK, Hyzinski-Garcia MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, Matrougui K, Mongin AA et al (2013) STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Archiv : Eur J Physiol 465(9):1249–1260. https://doi.org/10.1007/s00424-013-1254-8
Liu H, Hughes JD, Rollins S, Chen B, Perkins E (2011) Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp Mol Pathol 91(3):753–760. https://doi.org/10.1016/j.yexmp.2011.09.005
Morrone FB, Gehring MP, Nicoletti NF (2016) Calcium channels and associated receptors in malignant brain tumor therapy. Mol Pharmacol 90(3):403–409. https://doi.org/10.1124/mol.116.103770
Zhang Y, Zhang J, Jiang D, Zhang D, Qian Z, Liu C, Tao J (2012) Inhibition of T-type Ca(2)(+) channels by endostatin attenuates human glioblastoma cell proliferation and migration. Br J Pharmacol 166(4):1247–1260. https://doi.org/10.1111/j.1476-5381.2012.01852.x
Zhang Y, Wang H, Qian Z, Feng B, Zhao X, Jiang X, Tao J (2014) Low-voltage-activated T-type Ca2+ channel inhibitors as new tools in the treatment of glioblastoma: the role of endostatin. Pflugers Archiv : Eur J Physiol 466(4):811–818. https://doi.org/10.1007/s00424-013-1427-5
Valerie NC, Dziegielewska B, Hosing AS, Augustin E, Gray LS, Brautigan DL, Larner JM, Dziegielewski J (2013) Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem Pharmacol 85(7):888–897. https://doi.org/10.1016/j.bcp.2012.12.017
Nilius B, Owsianik G (2011) The transient receptor potential family of ion channels. Genome Biol 12(3):218. https://doi.org/10.1186/gb-2011-12-3-218
Bomben VC, Sontheimer H (2010) Disruption of transient receptor potential canonical channel 1 causes incomplete cytokinesis and slows the growth of human malignant gliomas. Glia 58(10):1145–1156. https://doi.org/10.1002/glia.20994
Leng TD, Li MH, Shen JF, Liu ML, Li XB, Sun HW, Branigan D, Zeng Z et al (2015) Suppression of TRPM7 inhibits proliferation, migration, and invasion of malignant human glioma cells. CNS Neurosci Ther 21(3):252–261. https://doi.org/10.1111/cns.12354
Stock K, Kumar J, Synowitz M, Petrosino S, Imperatore R, Smith ES, Wend P, Purfurst B et al (2012) Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat Med 18(8):1232–1238. https://doi.org/10.1038/nm.2827
Wondergem R, Ecay TW, Mahieu F, Owsianik G, Nilius B (2008) HGF/SF and menthol increase human glioblastoma cell calcium and migration. Biochem Biophys Res Commun 372(1):210–215. https://doi.org/10.1016/j.bbrc.2008.05.032
Chen WL, Barszczyk A, Turlova E, Deurloo M, Liu B, Yang BB, Rutka JT, Feng ZP et al (2015) Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget 6(18):16321–16340. https://doi.org/10.18632/oncotarget.3872
Liu M, Inoue K, Leng T, Guo S, Xiong ZG (2014) TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell Signal 26(12):2773–2781. https://doi.org/10.1016/j.cellsig.2014.08.020
Bomben VC, Turner KL, Barclay TT, Sontheimer H (2011) Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J Cell Physiol 226(7):1879–1888. https://doi.org/10.1002/jcp.22518
Chigurupati S, Venkataraman R, Barrera D, Naganathan A, Madan M, Paul L, Pattisapu JV, Kyriazis GA et al (2010) Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res 70(1):418–427. https://doi.org/10.1158/0008-5472.can-09-2654
Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, Giangaspero F, Santoni G (2007) Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J Neurochem 102(3):977–990. https://doi.org/10.1111/j.1471-4159.2007.04582.x
Medina-Ceja L, Pardo-Pena K, Morales-Villagran A, Ortega-Ibarra J, Lopez-Perez S (2015) Increase in the extracellular glutamate level during seizures and electrical stimulation determined using a high temporal resolution technique. BMC Neurosci 16(1):11. https://doi.org/10.1186/s12868-015-0147-5
Masi A, Becchetti A, Restano-Cassulini R, Polvani S, Hofmann G, Buccoliero AM, Paglierani M, Pollo B et al (2005) hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br J Cancer 93(7):781–792. https://doi.org/10.1038/sj.bjc.6602775
Patt S, Preussat K, Beetz C, Kraft R, Schrey M, Kalff R, Schonherr K, Heinemann SH (2004) Expression of ether a go-go potassium channels in human gliomas. Neurosci Lett 368(3):249–253. https://doi.org/10.1016/j.neulet.2004.07.001
Staudacher I, Jehle J, Staudacher K, Pledl HW, Lemke D, Schweizer PA, Becker R, Katus HA et al (2014) HERG K+ channel-dependent apoptosis and cell cycle arrest in human glioblastoma cells. PLoS One 9(2):e88164. https://doi.org/10.1371/journal.pone.0088164
Wang J, Li Y, Jiang C (2015) MiR-133b contributes to arsenic-induced apoptosis in U251 glioma cells by targeting the hERG channel. J Mol Neurosci : MN 55(4):985–994. https://doi.org/10.1007/s12031-014-0455-8
Bai Y, Liao H, Liu T, Zeng X, Xiao F, Luo L, Guo H, Guo L (2013) MiR-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-a-go-go (EAG1). Eur J Cancer (Oxford, England : 1990) 49(3):710–724. https://doi.org/10.1016/j.ejca.2012.08.020
Arvind S, Arivazhagan A, Santosh V, Chandramouli BA (2012) Differential expression of a novel voltage gated potassium channel—Kv 1.5 in astrocytomas and its impact on prognosis in glioblastoma. Br J Neurosurg 26(1):16–20. https://doi.org/10.3109/02688697.2011.583365
Martinez R, Stuhmer W, Martin S, Schell J, Reichmann A, Rohde V, Pardo L (2015) Analysis of the expression of Kv10.1 potassium channel in patients with brain metastases and glioblastoma multiforme: impact on survival. BMC Cancer 15(1):839. https://doi.org/10.1186/s12885-015-1848-y
Turner KL, Honasoge A, Robert SM, McFerrin MM, Sontheimer H (2014) A proinvasive role for the Ca(2+) -activated K(+) channel KCa3.1 in malignant glioma. Glia 62(6):971–981. https://doi.org/10.1002/glia.22655
Catacuzzeno L, Aiello F, Fioretti B, Sforna L, Castigli E, Ruggieri P, Tata AM, Calogero A et al (2011) Serum-activated K and Cl currents underlay U87-MG glioblastoma cell migration. J Cell Physiol 226(7):1926–1933. https://doi.org/10.1002/jcp.22523
Sciaccaluga M, Fioretti B, Catacuzzeno L, Pagani F, Bertollini C, Rosito M, Catalano M, D'Alessandro G et al (2010) CXCL12-induced glioblastoma cell migration requires intermediate conductance Ca2+-activated K+ channel activity. Am J Physiol Cell Physiol 299(1):C175–C184. https://doi.org/10.1152/ajpcell.00344.2009
So EC, Huang YM, Hsing CH, Liao YK, Wu SN (2015) Arecoline inhibits intermediate-conductance calcium-activated potassium channels in human glioblastoma cell lines. Eur J Pharmacol 758:177–187. https://doi.org/10.1016/j.ejphar.2015.03.065
Fioretti B, Castigli E, Micheli MR, Bova R, Sciaccaluga M, Harper A, Franciolini F, Catacuzzeno L (2006) Expression and modulation of the intermediate- conductance Ca2+-activated K+ channel in glioblastoma GL-15 cells. Cell Physiol Biochem : Int J Exp Cell Physiol, Biochem Pharmacol 18(1–3):47–56. https://doi.org/10.1159/000095135
Stegen B, Butz L, Klumpp L, Zips D, Dittmann K, Ruth P, Huber SM (2015) Ca2+-activated IK K+ channel blockade radiosensitizes glioblastoma cells. Mol Cancer Res : MCR 13(9):1283–1295. https://doi.org/10.1158/1541-7786.mcr-15-0075
Wondergem R, Bartley JW (2009) Menthol increases human glioblastoma intracellular Ca2+, BK channel activity and cell migration. J Biomed Sci 16(1):90. https://doi.org/10.1186/1423-0127-16-90
Hoa NT, Ge L, Martini F, Chau V, Ahluwalia A, Kruse CA, Jadus MR (2016) Temozolomide induces the expression of the glioma big potassium (gBK) ion channel, while inhibiting fascin-1 expression: possible targets for glioma therapy. Expert Opin Ther Targets 20(10):1155–1167. https://doi.org/10.1080/14728222.2016.1208172
Joshi AD, Parsons DW, Velculescu VE, Riggins GJ (2011) Sodium ion channel mutations in glioblastoma patients correlate with shorter survival. Mol Cancer 10(1):17. https://doi.org/10.1186/1476-4598-10-17
Hanukoglu I, Hanukoglu A (2016) Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579(2):95–132. https://doi.org/10.1016/j.gene.2015.12.061
Vila-Carriles WH, Kovacs GG, Jovov B, Zhou ZH, Pahwa AK, Colby G, Esimai O, Gillespie GY et al (2006) Surface expression of ASIC2 inhibits the amiloride-sensitive current and migration of glioma cells. J Biol Chem 281(28):19220–19232. https://doi.org/10.1074/jbc.M603100200
Kapoor N, Bartoszewski R, Qadri YJ, Bebok Z, Bubien JK, Fuller CM, Benos DJ (2009) Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J Biol Chem 284(36):24526–24541. https://doi.org/10.1074/jbc.M109.037390
Rooj AK, McNicholas CM, Bartoszewski R, Bebok Z, Benos DJ, Fuller CM (2012) Glioma-specific cation conductance regulates migration and cell cycle progression. J Biol Chem 287(6):4053–4065. https://doi.org/10.1074/jbc.M111.311688
Kapoor N, Lee W, Clark E, Bartoszewski R, McNicholas CM, Latham CB, Bebok Z, Parpura V et al (2011) Interaction of ASIC1 and ENaC subunits in human glioma cells and rat astrocytes. Am J Physiol-Cell Physiol 300(6):C1246–C1259. https://doi.org/10.1152/ajpcell.00199.2010
Sun X, Zhao D, Li YL, Sun Y, Lei XH, Zhang JN, MM W, Li RY et al (2013) Regulation of ASIC1 by Ca2+/calmodulin-dependent protein kinase II in human glioblastoma multiforme. Oncol Rep 30(6):2852–2858. https://doi.org/10.3892/or.2013.2777
Peretti M, Angelini M, Savalli N, Florio T, Yuspa SH, Mazzanti M (2015) Chloride channels in cancer: Focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochimica et biophysica acta 1848 (10 Pt B):2523-2531. doi:https://doi.org/10.1016/j.bbamem.2014.12.012
Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H (2003) Expression of voltage-gated chloride channels in human glioma cells. J Neurosci : Off J Soc Neurosci 23(13):5572–5582
Gritti M, Wurth R, Angelini M, Barbieri F, Peretti M, Pizzi E, Pattarozzi A, Carra E et al (2014) Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 5(22):11252–11268. https://doi.org/10.18632/oncotarget.2617
Setti M, Osti D, Richichi C, Ortensi B, Del Bene M, Fornasari L, Beznoussenko G, Mironov A et al (2015) Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 6(31):31413–31427. https://doi.org/10.18632/oncotarget.5105
Setti M, Savalli N, Osti D, Richichi C, Angelini M, Brescia P, Fornasari L, Carro MS et al (2013) Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J Natl Cancer Inst 105(21):1644–1655. https://doi.org/10.1093/jnci/djt278
Lui VC, Lung SS, JK P, Hung KN, Leung GK (2010) Invasion of human glioma cells is regulated by multiple chloride channels including ClC-3. Anticancer Res 30(11):4515–4524
Sahlender DA, Savtchouk I, Volterra A (2014) What do we know about gliotransmitter release from astrocytes? Philos Trans R Soc Lond Ser B Biol Sci 369(1654):20130592. https://doi.org/10.1098/rstb.2013.0592
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr., Carter BS, Krichevsky AM, Breakefield XO (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10 (12):1470–1476. doi:https://doi.org/10.1038/ncb1800
Bortner CD, Cidlowski JA (2007) Cell shrinkage and monovalent cation fluxes: role in apoptosis. Arch Biochem Biophys 462(2):176–188. https://doi.org/10.1016/j.abb.2007.01.020
Strange K (2004) Cellular volume homeostasis. Adv Physiol Educ 28(1–4):155–159. https://doi.org/10.1152/advan.00034.2004
Algharabil J, Kintner DB, Wang Q, Begum G, Clark PA, Yang SS, Lin SH, Kahle KT et al (2012) Inhibition of Na(+)-K(+)-2Cl(−) cotransporter isoform 1 accelerates temozolomide-mediated apoptosis in glioblastoma cancer cells. Cell Physiol Biochem : Int J Exp Cell Physiol Biochem Pharmacol 30(1):33–48. https://doi.org/10.1159/000339047
Cong D, Zhu W, Shi Y, Pointer KB, Clark PA, Shen H, Kuo JS, Hu S et al (2014) Upregulation of NHE1 protein expression enables glioblastoma cells to escape TMZ-mediated toxicity via increased H(+) extrusion, cell migration and survival. Carcinogenesis 35(9):2014–2024. https://doi.org/10.1093/carcin/bgu089
Beskina O, Miller A, Mazzocco-Spezzia A, Pulina MV, Golovina VA (2007) Mechanisms of interleukin-1beta-induced Ca2+ signals in mouse cortical astrocytes: roles of store- and receptor-operated Ca2+ entry. Am J Physiol Cell Physiol 293(3):C1103–C1111. https://doi.org/10.1152/ajpcell.00249.2007
Zhu W, Carney KE, Pigott VM, Falgoust LM, Clark PA, Kuo JS, Sun D (2016) Glioma-mediated microglial activation promotes glioma proliferation and migration: roles of Na+/H+ exchanger isoform 1. Carcinogenesis 37(9):839–851. https://doi.org/10.1093/carcin/bgw068
Ostrow LW, Suchyna TM, Sachs F (2011) Stretch induced endothelin-1 secretion by adult rat astrocytes involves calcium influx via stretch-activated ion channels (SACs). Biochem Biophys Res Commun 410(1):81–86. https://doi.org/10.1016/j.bbrc.2011.05.109
Li JH, Zhao ST, Wu CY, Cao X, Peng MR, Li SJ, Liu XA, Gao TM (2013) Store-operated Ca2+ channels blockers inhibit lipopolysaccharide induced astrocyte activation. Neurochem Res 38(10):2216–2226. https://doi.org/10.1007/s11064-013-1130-0
Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, Li P, Li M et al (2015) Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527(7576):100–104. https://doi.org/10.1038/nature15376
Funding
This work was supported in part by NIH grant R01NS75995 (DS).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Guan, X., Hasan, M.N., Maniar, S. et al. Reactive Astrocytes in Glioblastoma Multiforme. Mol Neurobiol 55, 6927–6938 (2018). https://doi.org/10.1007/s12035-018-0880-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-0880-8