
Abstract
Substantial evidence indicates that the human ether-a-go-go-related gene potassium channel (hERG, Kv11.1, KCNH2) is overexpressed in human glioblastoma multiforme (GBM) specimens and plays an essential role in the malignant proliferation of glioma cells. However, its upstream regulator in glioma cells is not fully elucidated. The present study was designed to determine whether the expression of hERG gene is regulated by miR-133b or miR-34a, thereby contributing to the anti-proliferation effect of arsenic trioxide (ATO) in U251 human glioma cells. Real-time polymerase chain reactions (qRT-PCR) and Western blot results demonstrated that hERG mRNA and protein levels were dramatically upregulated in clinical GBM specimens. Conversely, both miR-133b and miR-34a were markedly downregulated in clinical GBM specimens by qRT-PCR. The hERG gene was a direct target of miR-133b and miR-34a by bioinformatics analyses and luciferase reporter assays. Moreover, ATO, which is an emerging chemotherapy drug for glioma disease, remarkably elevated the level of miR-133b, but not miR-34a in U251 glioma cells. The level of miR-133b upstream transactivator serum response factor (SRF) was also suppressed by ATO. The transfection of anti-miR-133b oligonucleotide (AMO-133b) remarkably prevented the decrease of hERG protein by 5 μM ATO treatment for 24 h in U251 cells, whereas anti-miR-34a oligonucleotide (AMO-34a) did not exhibit recuperated effect. Finally, the transient overexpression by miR-133b mimics and treatment with the hERG channel-specific blocker E4031 markedly facilitated the ATO inhibition of proliferation of and induced apoptosis in U251 cells, whereas AMO-miR-133b attenuated these changes. Our study provided the evidence for the pathological role of miR-133b and miR-34a in the development of GBM and thus expanded our understanding of the hERG gene expression and ATO chemotherapeutic roles of miRNAs. Targeting miR-133b/hERG pathway may be a new strategy for chemotherapy of malignant gliomas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gliomas are the most common brain tumors. High-grade gliomas (WHO grades III and IV) are notoriously difficult to treat, and recurrences arise in virtually every case (Patil et al. 2013; Wang and Jiang 2013). Obviously, it is necessary to elucidate the critical molecular mechanism underlying malignant tumor progression and resistance to treatment, with the aim of developing chemotherapies and biological therapies based on more effective clinical therapies.
The human ether-a-go-go-related (hERG1) channel (KCNH2 or Kv11.1, according to the most recent nomenclature) is a voltage-dependent K+ channel that has an essential role in cardiac action potential repolarization (Chu et al. 2012). Interestingly, the hERG channel is also overexpressed in a variety of cancer cells, including in human glioblastoma multiforme (GBM) specimens (Bai et al. 2013; Masi et al. 2005). The hERG channels have been implicated in signaling pathways promoting cell proliferation and inhibiting apoptosis (Asher et al. 2010). However, its upstream molecular regulator remains largely unknown in the development of glioma disease.
MicroRNAs (miRNAs) are endogenous ∼22 nucleotide (nt) non-coding RNAs that anneal to inexact complementary sequences in the 3’ UTRs of target mRNAs of protein-coding genes to specify translational repression or/and mRNA cleavage (Koshkin et al. 2013). Increasing numbers of miRNAs abnormally expressed in gliomas have been identified by miRNA expression profiling methods (Silber et al. 2008). Among these miRNAs, miRNA-133b and miR-34a were expressed in astrocytes and were downregulated in human GBM specimens (Li et al. 2009; Luan et al. 2010; Silber et al. 2008). Using a bioinformatics analysis, two putative target sites for miR-133b or miR-34a were shown in hERG 3’ UTRs, suggesting the hERG channel is a potential target for miR-133b or miR-34a. Moreover, arsenic trioxide (ATO) therapy has recently been found to be highly effective in chemotherapy for glioma patients and induces apoptosis in glioma cells (Cohen et al. 2013; Grimm et al. 2012; Kanzawa et al. 2005) and inhibited hERG gene expression in neonatal cardiomyocytes (Chu et al. 2012; Ficker et al. 2004), but the exact mechanism has not been fully clarified.
On the basis of these premises, we investigated the expression of miR-133b, miR-34a, and hERG channels in human GBM specimens and determined whether hERG is a direct target of miR-133b or miR-34a and evaluated their roles in the arsenic-induced inhibitory effect on proliferation of U251 glioma cells.
Material and Methods
Clinical Glioma Specimens
Patient tissue samples were obtained from the Second Affiliated Hospital of Harbin Medical University (Harbin, China). All patients provided informed consent prior to collection of specimens according to institutional guidelines. All GBM tissue specimens were obtained from untreated patients undergoing primary surgical treatment. Normal tissues were obtained far from the center of the cancer in surgical specimens. Tissue samples were snap-frozen in the operating room immediately after surgery and the samples were sent to pathology for diagnosis by a board-certified neuropathologist. The protocol was approved by the Institutional Review Board, and informed consent was obtained from the patients or their guardians.
The Profile of miRNAs Targeting to hERG Gene by Bioinformatics Analysis
Considering that miRNA targets predicted by multiple algorithms might be more reliable, the miRNA-target interactions appearing in at least two of the nine databases (TargetScan, miRanda, PicTar, miRBase, DIANA-microT, PITA, miRNAMap, miRTarBase, and miRecords) were included in our analysis. The miRNA expression profile of glioblastomas (GBM), including 27 GBM samples and 10 controls, in batch 8 were downloaded from the Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/). The t test was used to detect differentially expressed miRNAs in GBM compared with the control group (p < 0.05). Potassium channel pathway was downloaded from http://www.genenames.org/genefamilies/VGIC#CACN1. The hypergeometric model was employed to test whether potassium channel pathways was significantly enriched with the targets of miRNAs (p < 0.05). Hierarchical clustering of the expression values of differentially expressed miRNAs was performed with R software (http://www.r-project.org/) using the metric of Euclidean distance and complete linkage. The network visualization was implemented in the Cytoscape software (http://cytoscape.org/).
Cell Culture and Transfection
The human glioblastoma cell line U251 and neuroblastoma cell line SH-SY5Y were obtained from the China Academia Sinica Cell Repository (Shanghai, China). The cells were maintained in RPMI 1640 (Gibco, USA) supplemented with 15 % fetal bovine serum (Gibco, USA), 2 mM glutamine (Sigma, USA), 100 units of penicillin/ml (Sigma, USA), and 100 μg of streptomycin/ml (Sigma, USA), which was incubated at 37 °C with 5 % CO2. Once the cells were 80 % confluent, they were starved in RPMI 1640 with 1 % FBS for 24 h and maintained in this low-serum condition for the course of all treatments. 2′OMe-miR-133b (5′-uuugguccccuucaaccagcua-3′), negative control oligonucleotide (5′-caguacuuuuguguaguacaa-3′), anti-miRNA oligonucleotides specific to miR-133b (AMO-133b) (5′-tagctggttgaaggggaccaaa-3′), 2′OMe-miR-34a (5′-uggcagugucuuagcugguugu-3′), negative control oligonucleotide (5′-uucuccgaacgugucacgutt-3′), and anti-miRNA oligonucleotides specific to miR-34a (AMO-34a) (5′-acaaccagc taagacactgcca-3′) were synthesized by GenePharma (Shanghai, China). The above oligonucleotides were transfected into U251 cells (200 nmol per well) using Lipofectamine2000 reagent (Invitrogen, USA).
Cell Viability Assay
Cells were seeded in a 96-well plate with the same starting cell number per well (1 × 104 cells/well) and allowed to attach for 24 h. After treatment, the media in each well were removed and replaced with 100 μl of 5 mg/ml 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, USA), and then, the plate was further incubated at 37 °C for 3 h. All of the remaining supernatant was then removed, and 100 μl of DMSO was added to each well and mixed thoroughly to dissolve the formed crystal formazan. After 10 min of incubation to ensure all crystal formazan were dissolved, the cell viability was detected by measuring the absorbance of each well at 570 nm. The relative cell viability was calculated as the absorbance percentage of the treatment group to the control group.
TUNEL Assay
Apoptotic cells were detected in situ with a Cell Death Detection Kit. POD analysis was performed with a commercially available kit for immunohistochemical detection and quantification of apoptosis (programmed cell death) at the single cell level, as based on labeling of DNA strand breaks according to the manufacturer’s instructions. After staining, the coverslips were observed with a confocal microscope (Olympus, FV-100). The ratio of apoptotic (TUNEL-positive) cells to total (DAPI-stained nuclei) was calculated (n = 5). Measurements were performed using Scion Image software (Beta 4.03; Scion Corporation, MD). All of the measurements were performed in a double-blind manner by two independent researchers.
Electron Microscopy
To perform electron microscopy analysis, the U251 cells were fixed in 2.5 % glutaraldehyde in PBS for 2 h at 4 °C and then postfixed in 1 % osmium tetroxide. After dehydration in a series of graded ethanol baths (30–100 %) and in propylene oxide, the cells were embedded in Epon. Cell sections (80–200 nm) were obtained using a Reichert Ultracut E microtome and stained with uranyl acetate. Grids were examined with a Jeol 1200 EXII electron microscope (Tokyo, Japan).
Quantification of hERG mRNA, miRNA-133b, and miR-34a Levels
Total RNA isolated by TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) was treated with a Turbo DNA free kit (Ambion, Austin, TX, USA) to eliminate genomic DNA contamination. The hERG1 mRNA was detected with a specific forward primer (5′-CTGGCCATGAAGTTCAAGACCACAC-3′) and reverse primer (5′-AGGACTCCCTTTGCTTTGGATGTG-3′). Quantitative real-time PCR (qRT-PCR) was used to assess the expression of miR-133b and miR-34a with kits from Applied Biosystems (Foster City, CA, USA). In each sample, we calculated a ΔCt (target–reference). The fold-change between the ATO-treated samples and a normal control for miR-133b and miR-34a were calculated with the 2-ΔΔCt method, in which ΔΔCT = ΔCT (target–reference)–ΔCT (target–reference) (in untreated samples). qRT-PCR was repeated in triplicate for each sample, and an average 2-ΔΔCt value along with its SD was calculated for each sample relative to the normal control for expression of miR-133b and miR-34a. U6 and GeNorm were used as internal reference genes to which the expression of miR-133b and miR-34a were normalized.
Luciferase Reporter Assay
The hERG 3′-UTR containing the conserved miR-133b or miR-34a binding site was synthesized by Invitrogen and amplified by PCR. The PCR fragment was cloned downstream of the luciferase gene between the SacI and HindIII sites in pMIR-Report (Promega). Then, 0.1 μg of the luciferase reporters containing the 3′-UTR were co-transfected with miR-133b or miR-34a mimics into U251 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). As an internal control, 10 ng of Renilla luciferase reporter was also included. Then, 48 h after transfection, the cells were collected, and dual luciferase activities were measured with a luminometer according to the manufacturer’s instructions.
Western Blotting
U251 cell lysates (100 μg/lane) were loaded onto a 10 % gradient gel for SDS-polyacrylamide gel electrophoresis (SDS-PAGE). After SDS-PAGE, the gels were blotted onto an Immunobilon-P nylon membrane. After blocking, the blots were incubated overnight with the appropriate primary antibody at 4 °C. A rabbit polyclonal anti-hERG antibody and corresponding antigen peptide were purchased from the Alomon company (1:500 dilution; Alomone labs, Jerusalem, Israel). Anti-serum response factor (SRF) antibody was purchased from Sigma-Aldrich (1:500 dilution; Sigma-Aldrich Co., LLC, USA). The goat anti-rabbit secondary antibody was from Invitrogen (Carlsbad, CA, USA). Western blot bands were quantified using Odyssey v1.2 software by measuring the band intensity (area × OD) for each group, with normalizing to GAPDH (anti-GAPDH antibody from Kangcheng, Shanghai, China), which acted as an internal control for the Western blot assay.
Statistical Analysis
Data are presented as the mean ± SEM. Statistical comparison was performed using Student’s t test and analysis of variance (ANOVA), with a value of p < 0.05 considered significant.
Results
Overexpression of hERG mRNA and Protein in GBM Clinical Specimens
The overexpression of hERG gene plays an important role in glioma cell proliferation (Asher et al. 2010). Thus, we detected the level of hERG mRNA and protein expression in specimens of grade IV astrocytoma GBM. The pre-incubation with the peptide antigen was used as negative control. Consistent with a previous study (Asher et al. 2010), a significantly increased level of hERG mRNA (Fig. 1a) and protein (Fig. 1b) was found in GBM specimens compared with nontumor tissues. Interestingly, the level of hERG protein was enhanced ∼3.45-fold (n = 5), whereas the level of hERG mRNA was only increased ∼1.35-fold in GBM specimens compared with nontumor tissues (Fig. 1), suggesting that the hERG gene was post-transcriptionally repressed in GBM by unknown mechanisms, such as miRNAs.

The changes of hERG mRNA and protein in clinical GBM specimens. a A modest upregulation of hERG transcripts in clinical GBM specimens according to qRT-PCR and b a significant upregulation of hERG in clinical GBM specimens according to Western blot analysis. Antigen peptide was used as negative control (2 μg peptide per 1 μg antibody). Values are shown as the mean ± SEM. n = 5 for each group, *p < 0.05 vs. control. Ctl control human sample, GBM human glioblastoma multiforme specimen
miR-133b and miR-34a Targeting hERG Gene are Downregulated in GBM Clinical Specimen
According to the miRNA-target interaction information in 9 databases, 54 miRNAs are predicted to target potassium channel gene hERG. By analyzing the level 3 miRNA expression profile of GBM from the Cancer Genome Atlas (TCGA), there are 232 significantly differentially expressed miRNAs in 27 GBM samples compared with 10 controls (p < 0.05, t test) (Fig. 2a). Fifteen differentially expressed miRNAs, among which six miRNAs are downregulated and nine miRNAs are upregulated, are overlapped with the miRNAs that regulate hERG (Fig. 2b). The miR-133b is predicted to target 1628 genes, which are significantly enriched with the potassium channel genes (p = 0.012). Also, the targets of miR-34a are significantly enriched with the potassium channel genes (p = 0.018). And, both miR-133b and miR-34a are predicted to target the potassium channel gene hERG. We predicted the miR-133b was significantly downregulated in GBM (Fig. 2c, p = 1.71E-10) and miR-34a was significantly downregulated in GBM (Fig. 2c, p = 6.04E-11). Finally, we detected the expression of miR-133b and miR-34a in the normal brain, GBM specimens, glioma cell lines U251, and neuroblastoma SH-SY5Y cells by qRT-PCR. By comparison of miR-133b and miR-34a expression levels, it was found that both of them were markedly reduced in GBM specimens and U251 cells (Fig. 2d). However, only miR-34a expression was decreased in neuroblastoma SH-SY5Y cells, suggesting miR-133b decreased in GBM specimens was mainly due to enriched astrocyte proliferation. Thus, we hypothesize that the downregulation of miR-133b and miR-34a could induce the increasing of hERG, which is a potential target of miR-133b and miR-34a, to participate in the progression of GBM.

Downregulation of the miR-133b and miR-34a in clinical GBM specimens. a Heatmap of differentially expressed miRNAs between GBM and controls. Hierarchical clustering analysis and heatmap for miRNA expression were performed using R software. Rows represent miRNAs and columns represent samples in heatmap. Red represents highly expressed and green means low expression value. b miRNA-hERG interaction network. Rounded rectangles represent miRNAs and circles represent genes. The significantly downregulated (upregulated) miRNAs in GBM compared with controls are marked with green (red). c Prediction the miR-133b and miR-34a expression in GBM and control samples. d Downregulation of miR-133b and miR-34a in clinical GBM specimens according to qRT-PCR. Values are shown as the mean ± SEM. n = 5 for each group, *p < 0.05 vs. control
hERG is a Direct Target of miR-133b and miR-34a
After scrutinizing the sequences between miR-133b, miR-34a, and hERG 3′-UTR, we found that the seed sites of miR-133b and miR-34a, respectively, matched the hERG 3′-UTR (Fig. 3a), suggesting that hERG may be one of the targets of miR-133b and miR-34a. Then, transfection of miR-133b or miR-34a into U251 cells and the Western blotting results confirmed our assumption. As depicted in Fig. 3b, hERG expression was significantly downregulated in U251 cells with transfected miR-133b or miR-34a, whereas the expression level of hERG could be, respectively, restored in the cells co-transfected with miR-133b and antisense-miR-133b (AMO-133b) or miR-34a and antisense-miR-34a (AMO-34a). To evaluate whether miR-133b and miR-34a could interfere with hERG 3′-UTR, we, respectively, utilized the pMIR-report construct containing hERG binding sites for miR-133b and miR-34a. Notably, qRT-PCR demonstrated that miR-133b had no effect on the expression of hERG mRNA (Fig. 3c) compared to miR-34a, indicating that miR-133b post-transcriptionally regulates hERG. As illustrated in Fig. 3d, overexpression of miR-133b or miR-34a (20 nmol/L) with the luciferase expression constructs carrying the target fragments caused a significant reduction of luciferase activities compared with that of the negative control (NC), whereas the reduction of luciferase activities was efficiently reversed by AMO-133b or AMO-34a (10 nmol/L), suggesting hERG is a direct target of miR-133b and miR-34a.

hERG gene is direct target for miR-133b and miR-34a. a The sequences showed the two binding sites of miRNA: mRNA complementary between miR-133b, miR-34a, and human hERG gene. b Compared with the control, transfection of miR-133b or miR-34a resulted in a significant decrease of hERG protein. Co-application of miR-133b with AMO-133b or co-application of miR-34a with AMO-34a alleviated the decrease of hERG protein, whereas the NC displayed no effects. c qRT-PCR showed that miR-34a markedly inhibited hERG mRNA expression, whereas miR-133b had no effect on the expression of hERG mRNA after transfection of miR-133b into U251 cells. d Compared with the control, transfection of miR-133b with the luciferase reporter gene linked to the wild-type 3′-UTR of hERG resulted in a significant decrease of luciferase activity. Co-application of miR-133b with AMO-133b or co-application of miR-34a with AMO-34a alleviated the reduction of luciferase activity, whereas NC displayed no effects. n = 3 represents three independent experiment under each condition. Data are shown as the mean ± SEM. *p < 0.05 vs. control, # p < 0.05 vs. miR-133b or miR-34a correspondingly. Ctl control, NC transfection of negative control, miR-133b transfection of miR-133b, +AMO-133b co-transfection of miR-133b and its inhibitor (AMO-133b), miR-34a transfection of miR-34a, +AMO-34a co-transfection of miR-34a and its inhibitor (AMO-34a)
ATO Upregulates Expression of miR-133b and Downregulates hERG Protein in Cultured U251 Cells
Recent reports indicate that ATO is beneficial for improving chemotherapy for patients with glioma (Cohen et al. 2013; Grimm et al. 2012). Given the established pharmacological effects of miR-133b and miR-34a with glioma, we proposed that ATO might produce its beneficial actions by affecting miR-133b or miR-34a expression. We compared the expression levels of miR-133b, miR-34a, and hERG protein in the absence and presence of ATO. As illustrated in Fig. 4a, the miR-133b level was found to be ∼3.5-fold higher in the ATO-treated U251 cells (10 μM) than that in the untreated control cells. However, ATO had no effect on the expression level of miR-34a. Accordingly, 5 μM ATO caused an approximately 52 % percent decrease of hERG protein (Fig. 4b), which could be targeted by miR-133b and miR-34a. Transfection of AMO-133b could abolish the ATO-induced hERG protein downregulation in U251 cells (Fig. 4b), whereas AMO-34a have no reversed effect (Fig. 4c), indicating miR-133b probably mediated the ATO chemotherapy for glioma cells. To further elucidate the molecular mechanism of ATO increase of miR-133b, we assayed a transcriptional factor of miR-133b, serum response factor (SRF), which can promote miR-133b expression and regulate the expression of genes and cellular response (Ivey et al. 2008; Villar et al. 2011). As we expected, SRF expression was increased by 2.6-fold in U251 cells following treatment with 10 μM ATO compared with control cells (Fig. 4d), suggesting miR-133b is specifically regulated by ATO.

Effects of ATO on miR-133b, miR-34a, hERG, and SRF expression in U251 glioma cells. Cells were treated with 5 or 10 μM ATO as indicated for 24 h or were pre-transfected with AMO-133b or AMO-34a 24 h before ATO exposure. a The miR-133b and miR-34a levels were examined using qRT-PCR. b The role of AMO-133b in ATO-induced downregulation of hERG protein. c The role of AMO-34a in ATO-induced downregulation of hERG protein. d The effect of ATO on the expression of SRF. Proteins were determined by Western blot analysis. Values are shown as the mean ± SEM. n = 5 for each group, *p < 0.05 vs. control
MiR-133b Contributes to ATO-Induced Apoptosis in U251 Cells
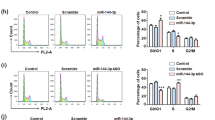
We hypothesized that miR-133b suppression of the hERG channel contributes to inhibition of cell proliferation and the apoptotic process in U251 cells with ATO treatment. Therefore, we first performed MTT assay to determine whether miR-133b was involved in the process of ATO-inhibited U251 cell proliferation. As shown in Fig. 5a, the transfection of miR-133b and pretreatment with E4031, a specific hERG channel blocker, facilitated the ATO-induced decrease of cell viability, whereas AMO-133b prevented the ATO-induced decrease of cell viability. TUNEL staining (Fig. 5b) indicated that under control conditions, it is hard to detect nuclear staining in U251 cells. In contrast, a significantly increased amount of stained nuclei, indicating apoptosis, occurred in U251 cells treated with 10 μM ATO. To determine if miR-133b is involved in ATO-induced apoptosis in these cells by targeting hERG, we assessed similar experiments as described above in the presence of miR-133b, AMO-133b, and E4031. The abundance of apoptotic cells was increased dramatically after 24 h of treatment with ATO. MiR-133b transfection or E4031 pretreatment promoted ATO-induced cell apoptosis. In contrast, transfection of AMO-133b dramatically prevented an increase of ATO-induced cell apoptosis in U251 cells (Fig. 5b). We also examined the micro-morphological changes induced by ATO using an electron microscope at an original magnification of ×5000, as an alternative indication of apoptosis. As shown in Fig. 5c, the micro-structure of the cells under control conditions appears to be normal. However, in the cells treated with ATO, there were a considerable number of cells that exhibited early nuclear morphological changes during apoptosis, including the disappearance of microvilli and chromosomal DNA condensation. Furthermore, some of the late stage apoptotic changes occurred in ATO-treated cells transfected with miR-133b or incubated with E4031, such as mitochondrial swelling and formation of separated apoptotic bodies. In contrast, the changes in cellular micro-structure induced by ATO were, at least in part, prevented by transfection with AMO-133b (Fig. 5c). The results obtained from TUNEL assays and electron microscopy examinations are consistent with the notion that ATO indeed induces apoptosis, and these results suggest that miR-133b plays an important role in ATO-induced apoptosis in cultured U251 cells by targeting the hERG channel.

miR-133b contributes to the arsenic-induced decrease of cell viability and apoptosis in U251 glioma cells. Cells were transfected with various constructs as indicated for 24 h or were pretreated with hERG inhibitor E4031 1 h before ATO exposure. a The effect of ATO on cell viabilities in the U251 cells under the indicated condition was determined by MTT assay. TUNEL staining (b) and transmission electron microscopy (c) were performed to estimate cell apoptosis. Cell viabilities and percentage of apoptotic cells were obtained from four independent experiments (n = 4). The values are shown as the mean ± SEM. *p < 0.05 vs. control; # p < 0.05 vs. ATO group. Magnification of the transmission electron microscope was set at × 5000
Discussion
hERG channels are specifically overexpressed in high-grade A, particularly in primary GBM, and are implicated in the accelerated proliferation and inhibition of apoptosis of cancer cells (Masi et al. 2005). Here, we revealed differential changes of hERG protein and transcript expression in GBM specimens. Similarly, hERG expression at the transcriptional and post-transcriptional levels diverge in diabetic hearts, with the protein levels being reduced by some 60 %, whereas the mRNA levels display no significant change (Zhang et al. 2006). MiRNAs can control the expression levels of target genes. Thus, overexpression of hERG protein in human glioma disease might be due to dysregulation of miRNAs. Recently, Lin H et al. revealed that miR-34, a known transcriptional target of p53, is an important negative regulator of hERG by directly repressing hERG gene in SH-SY5Y cells (Lin et al. 2011). A report by Bai Y et al. also demonstrated that miR-296-3p could modulate the sensitivity of glioma cells to some anticancer drugs, possibly through regulating hERG expression (Bai et al. 2013). However, because a given target might similarly be targeted by multiple miRNAs, it is still essential to further identify a key miRNA in the regulation of hERG expression in GBM disease. Recent studies indicate that miR-133b is expressed in astrocytes and exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to astrocytes (Xin et al. 2012, 2013). Importantly, Silber et al. found, in an array study, a significant decrease of miR-133b in clinical GBM specimens (Silber et al. 2008). To build on the correlative evidence, we conducted luciferase experiments and found miR-133b directly repressed hERG at the post-transcriptional level. Here, our study provides an explanation for the observed discrepancy between the changes of hERG expression at the protein and mRNA levels in clinical GBM.
Recently, a few clinical surveys revealed that ATO is a newly emerging drug for GBM disease (Cohen et al. 2013; Kanzawa et al. 2005). In the present study, our results indicate that ATO inhibited hERG protein expression perhaps due to the upregulation of the SRF-dependent miR-133b level but not a miR-34a, resulting in apoptosis in U251 cells, which provides a new understanding of the mechanism of how ATO kills cancer cells. Consistently, Shan et al. reported that ATO is able to upregulate SRF-dependent miR-133 expression in cardiomyocytes underlying ATO-induced cardiac electrical disorders (Shan et al. 2013), whereas Wu et al. detected the microRNAs expression profile in acute promyelocytic leukemia (APL) cells by ATO treatment and revealed that ATO has no effect on miR-34a expression in APL cells (Wu et al. 2012). We also found miR-133b is directly involved in the anti-proliferative effect and induction of apoptosis in U251 cells by ATO treatment. It has been demonstrated that miR-133b targets both pro-apoptotic and anti-apoptotic proteins and regulates the apoptotic process in various cell types (Liu et al. 2012; Patron et al. 2012; Qin et al. 2012). Patron J et al. also revealed that transfection with miR-133b rendered resistant HeLa cells sensitive to tumor necrosis factor-alpha (TNF-α)-induced cell death (Patron et al. 2012). Indeed, cell type- and environment-specific effects on apoptosis are suggested by reports of hERG activity promoting apoptosis. For example, selectively blocking the hERG channel promotes cell apoptosis in ovarian cancer cells (Asher et al. 2011), HL-1 cells, and prostatic cancer cells (Gonzalez-Juanatey et al. 2003; Zhao et al. 2005). This evidence suggests miR-133b possibly improves arsenic-induced apoptosis in U251 glioma cells by targeting the hERG channel. Thus far, several methods have been employed for enhancing tumor cell response to arsenic chemotherapy. Primon M found that cathepsin L silencing improves ATO-mediated in vitro cytotoxicity and apoptosis in glioblastoma U87 cells (Primon et al. 2013). Interestingly, He J et al. found that the combination of ATO and miR-203 presents therapeutic potential for chronic myelogenous leukemia (He et al. 2013). In the present study, overexpression of miR-133b also enhanced the efficiency of ATO-suppressed proliferation in U251 glioma cells. Therefore, exploiting the synergistic effects between miR-133b and ATO may be an effective clinical strategy for glioma chemotherapy, and miR-133b may be a potential drug target in glioma cells.
In summary, we observed downregulation of miR-133b, miR-34a and overexpression of hERG gene in human GBM specimens and further demonstrated that the hERG gene is a direct target of both miR-133b and miR-34a. The overexpression of miR-133b may be potentially exploited for arsenic chemotherapy of GBM in the future.
References
Asher V, Sowter H, Shaw R, Bali A, Khan R (2010) Eag and HERG potassium channels as novel therapeutic targets in cancer. World J Surg Oncol 8:113
Asher V, Warren A, Shaw R, Sowter H, Bali A, Khan R (2011) The role of Eag and HERG channels in cell proliferation and apoptotic cell death in SK-OV-3 ovarian cancer cell line. Cancer Cell Int 11:6
Bai Y, Liao H, Liu T, Zeng X, Xiao F, Luo L, Guo H, Guo L (2013) MiR-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-a-go-go (EAG1). Eur J Cancer 49:710–724
Chu W, Li C, Qu X, Zhao D, Wang X, Yu X, Cai F, Liang H, Zhang Y, Zhao X, Li B, Qiao G, Dong D, Lu Y, Du Z, Yang B (2012) Arsenic-induced interstitial myocardial fibrosis reveals a new insight into drug-induced long QT syndrome. Cardiovasc Res 96:90–98
Cohen KJ, Gibbs IC, Fisher PG, Hayashi RJ, Macy ME, Gore L (2013) A phase I trial of arsenic trioxide chemoradiotherapy for infiltrating astrocytomas of childhood. Neuro Oncol 15:783–787
Ficker E, Kuryshev YA, Dennis AT, Obejero-Paz C, Wang L, Hawryluk P, Wible BA, Brown AM (2004) Mechanisms of arsenic-induced prolongation of cardiac repolarization. Mol Pharmacol 66:33–44
Gonzalez-Juanatey JR, Iglesias MJ, Alcaide C, Pineiro R, Lago F (2003) Doxazosin induces apoptosis in cardiomyocytes cultured in vitro by a mechanism that is independent of alpha1-adrenergic blockade. Circulation 107:127–131
Grimm SA, Marymont M, Chandler JP, Muro K, Newman SB, Levy RM, Jovanovic B, McCarthy K, Raizer JJ (2012) Phase I study of arsenic trioxide and temozolomide in combination with radiation therapy in patients with malignant gliomas. J Neurooncol 110:237–243
He JH, Li YM, Li YG, Xie XY, Wang L, Chun SY, Cheng WJ (2013) hsa-miR-203 enhances the sensitivity of leukemia cells to arsenic trioxide. Exp Ther Med 5:1315–1321
Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, Hsiao EC, Schwartz RJ, Conklin BR, Bernstein HS, Srivastava D (2008) MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2:219–229
Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S (2005) Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 24:980–991
Koshkin PA, Chistiakov DA, Chekhonin VP (2013) Role of microRNAs in mechanisms of glioblastoma resistance to radio- and chemotherapy. Biochemistry (Mosc) 78:325–334
Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, Marcinkiewicz L, Jiang J, Yang Y, Schmittgen TD, Lopes B, Schiff D, Purow B, Abounader R (2009) MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res 69:7569–7576
Lin H, Li Z, Chen C, Luo X, Xiao J, Dong D, Lu Y, Yang B, Wang Z (2011) Transcriptional and post-transcriptional mechanisms for oncogenic overexpression of ether a go-go K + channel. PLoS One 6:e20362
Liu L, Shao X, Gao W, Zhang Z, Liu P, Wang R, Huang P, Yin Y, Shu Y (2012) MicroRNA-133b inhibits the growth of non-small-cell lung cancer by targeting the epidermal growth factor receptor. FEBS J 279:3800–3812
Luan S, Sun L, Huang F (2010) MicroRNA-34a: a novel tumor suppressor in p53-mutant glioma cell line U251. Arch Med Res 41:67–74
Masi A, Becchetti A, Restano-Cassulini R, Polvani S, Hofmann G, Buccoliero AM, Paglierani M, Pollo B, Taddei GL, Gallina P, Di Lorenzo N, Franceschetti S, Wanke E, Arcangeli A (2005) hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br J Cancer 93:781–792
Patil SA, Hosni-Ahmed A, Jones TS, Patil R, Pfeffer LM, Miller DD (2013) Novel approaches to glioma drug design and drug screening. Expert Opin Drug Discov 8:1135–1151
Patron JP, Fendler A, Bild M, Jung U, Muller H, Arntzen MO, Piso C, Stephan C, Thiede B, Mollenkopf HJ, Jung K, Kaufmann SH, Schreiber J (2012) MiR-133b targets antiapoptotic genes and enhances death receptor-induced apoptosis. PLoS One 7:e35345
Primon M, Huszthy PC, Motaln H, Talasila KM, Torkar A, Bjerkvig R, Lah Turnsek T (2013) Cathepsin L silencing enhances arsenic trioxide mediated in vitro cytotoxicity and apoptosis in glioblastoma U87MG spheroids. Exp Cell Res 319:2637–2648
Qin W, Dong P, Ma C, Mitchelson K, Deng T, Zhang L, Sun Y, Feng X, Ding Y, Lu X, He J, Wen H, Cheng J (2012) MicroRNA-133b is a key promoter of cervical carcinoma development through the activation of the ERK and AKT1 pathways. Oncogene 31:4067–4075
Shan H, Zhang Y, Cai B, Chen X, Fan Y, Yang L, Chen X, Liang H, Zhang Y, Song X, Xu C, Lu Y, Yang B, Du Z (2013) Upregulation of microRNA-1 and microRNA-133 contributes to arsenic-induced cardiac electrical remodeling. Int J Cardiol 167:2798–2805
Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M, Vandenberg SR, Ginzinger DG, James CD, Costello JF, Bergers G, Weiss WA, Alvarez-Buylla A, Hodgson JG (2008) miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med 6:14
Villar AV, Merino D, Wenner M, Llano M, Cobo M, Montalvo C, Garcia R, Martin-Duran R, Hurle JM, Hurle MA, Nistal JF (2011) Myocardial gene expression of microRNA-133a and myosin heavy and light chains, in conjunction with clinical parameters, predict regression of left ventricular hypertrophy after valve replacement in patients with aortic stenosis. Heart 97:1132–1137
Wang Y, Jiang T (2013) Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett 331:139–146
Wu Y, Li XF, Yang JH, Liao XY, Chen YZ (2012) microRNAs expression profile in acute promyelocytic leukemia cell differentiation induced by all-trans retinoic acid and arsenic trioxide. Zhonghua Xue Ye Xue Za Zhi 33:546–551
Xin H, Li Y, Buller B, Katakowski M, Zhang Y, Wang X, Shang X, Zhang ZG, Chopp M (2012) Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 30:1556–1564
Xin H, Li Y, Liu Z, Wang X, Shang X, Cui Y, Gang Zhang Z, Chopp M (2013) Mir-133b Promotes Neural Plasticity and Functional Recovery after Treatment of Stroke with Multipotent Mesenchymal Stromal Cells in Rats Via Transfer of Exosome-Enriched Extracellular Particles. Stem Cells
Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, Zhang H, Bai Y, Yang B, Wang Z (2006) Restoring depressed HERG K + channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Heart Circ Physiol 291:H1446–H1455
Zhao H, Lai F, Nonn L, Brooks JD, Peehl DM (2005) Molecular targets of doxazosin in human prostatic stromal cells. Prostate 62:400–410
Acknowledgments
This study was supported by the National High Technology Research and Development Program of China (863 Program, 2012AA02A508) and the National Natural Science Foundation of China (81372700).
Conflict of Interest
The authors have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, J., Li, Y. & Jiang, C. MiR-133b Contributes to Arsenic-Induced Apoptosis in U251 Glioma Cells by Targeting the hERG Channel. J Mol Neurosci 55, 985–994 (2015). https://doi.org/10.1007/s12031-014-0455-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-014-0455-8