
Abstract
Multiple sclerosis (MS) is a chronic autoimmune disease that degenerates the central nervous system (CNS). B cells exacerbate the progression of CNS lesions in MS by producing auto-antibodies, pro-inflammatory cytokines, and presenting auto-antigens to activated T cells. Long non-coding RNAs (lncRNAs) play a crucial role in complex biological processes and their stability in body fluids combined with their tissue specificity make these biomolecules promising biomarker candidates for MS diagnosis. In the current study, we investigated memory B cell-specific lncRNAs located, on average, less than 50 kb from differentially expressed protein-coding genes in MS patients compared to healthy individuals. Moreover, we included in our selection criteria lncRNA transcripts predicted to interact with microRNAs with established involvement in MS. To assess the expression levels of lncRNAs and their adjacent protein-coding genes, quantitative reverse transcription PCR was performed on peripheral blood mononuclear cells samples of 50 MS patients compared to 25 controls. Our results showed that in relapsing MS patients, compared to remitting MS patients and healthy controls, lncRNA RP11-530C5.1 was up-regulated while AL928742.12 was down-regulated. Pearson’s correlation tests showed positive correlations between the expression levels of RP11-530C5.1 and AL928742.12 with PAWR and IGHA2, respectively. The results of the ROC curve test demonstrated the potential biomarker roles of AL928742.12 and RP11-530C5.1. We conclude that these lncRNAs are potential markers for detection of relapsing MS patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Almost 2.5 million people around the world suffer from multiple sclerosis (MS), a chronic inflammatory disease of the central nervous system (CNS). MS is characterized by axon demyelination of the brain and spinal cord (Dendrou et al. 2015). Genetic and environmental factors influence the onset and development of MS (Olsson et al. 2017). Although it has been proposed that MS is caused by the action of CD4+ T cells (McFarland and Martin 2007), recent evidence revealed that memory B cells also play an important role in the pathogenesis of MS (Corcione et al. 2004; Cepok et al. 2005; Baker et al. 2017). B cells participate in the progress of the CNS lesions by producing auto-antibodies and secreting pro-inflammatory cytokines and also by presenting auto-antigens to activated T cells (Krumbholz et al. 2012; Fillatreau et al. 2002; Bar-Or et al. 2010; Tintore et al. 2008; Ignacio et al. 2010; Berger et al. 2003; Sellebjerg et al. 2009). Depletion of memory B cells is a high-efficacy treatment for relapsing MS, while increasing memory B cells exacerbates MS (Baker et al. 2017). Clinical trials with Rituximab, a monoclonal antibody against CD20, was an effective treatment for MS progression through the targeting of memory B cells (Bar-Or et al. 2008; Duddy et al. 2007).
Long non-coding RNAs (lncRNA) are defined as transcripts with more than 200 nucleotides in length without a significant open reading frame to encode protein (Ulitsky and Bartel 2013; Liao et al. 2011). lncRNAs play important roles in multiple biological processes through the regulation of gene expression (Fatica and Bozzoni 2014; Lee 2012). As disease biomarker candidates they have advantages over protein-coding genes due to their relative stability in body fluids and the relative ease of their detection by highly sensitive and specific PCR methods (Geisler and Coller 2013; Tong and Lo 2006).
Around 50% of protein-coding genes are correlated with the co-expression of a lncRNA located less than 50 kb away. Moreover, this correlation does not depend on the orientation of transcription of lncRNA genes and co-expressed protein-coding genes (Spurlock III et al. 2015).
There are several evidances indicating modulation of miRNAs levels in autoimmune diseases, thus regulation of such miRNAs may prevent development of autoimmune diseases, as reported for MS pathogenesis (Tufekci et al. 2011). LncRNAs could exert their functions through interacting with such miRNAs (Paraskevopoulou et al. 2012; Li et al. 2013). The influence of lncRNAs and microRNAs (miRNAs) on each other is rapidly emerging in recent studies. In some cases, the stability of a lncRNA is reduced by interacting with specific miRNAsIn other cases, lncRNAs act as decoys for miRNAs, suppressing miRNA repression of target messenger RNAs (mRNA). Other lncRNAs compete with miRNAs for interaction with the shared target and thereby influence mRNA expression. Additionally, some lncRNAs repress target mRNAs by producing miRNAs (Chen et al. 2012; Yoon et al. 2014).
In the current study, we analyzed a subset of lncRNAs that are specifically expressed in memory B cells at less than 50 kb distance away from differentially expressed genes in peripheral blood mononuclear cells (PBMC) of relapsing-remitting multiple sclerosis (RRMS) patients.
Materials and Methods
The approaches used to identify candidate lncRNAs are illustrated in Fig. 1 and described in more detail below.

In silico workflow of the study. Flow chart showing the methodological approaches used to identify lncRNAs linked to MS in B cells. In the first step, memory B cell lineage-specific lncRNAs retrieved from whole-genome RNA-seq data and chromosomal locations and transcripts of these lncRNAs were extracted from Ensembl GRCh37. a We obtained differentially expressed genes with adjusted p values < 0.05 and log fold change > ±1 from GSE21942 GEO dataset, comparing MS to healthy individuals. Finally, using python programming language, selected lncRNAs with less than 50 kb distance from protein-coding genes were selected as appropriate candidate lncRNAs for this study. b The sequences of lncRNA transcripts were entered into the LncDisease software and one of the lncRNAs found effective with miRNAs of MS
LncRNAs Selection
In this study 48 memory B cell lineage-specific lncRNAs were retrieved from whole-genome RNA-seq data (Ranzani et al. 2015). In the next step, the chromosomal locations and the identified transcripts for these lncRNAs were mapped using Ensembl (genome assembly GRCh37) for further analyses.
LncRNA Adjacent to Differentially Expressed Genes in MS
We used the dataset from the Gene Expression Omnibus (GEO) database and GEO series (GSE) by accession number: GSE21942 (Platform: GPL570, Affymetrix Human Genome U133 Plus 2.0 Array; 29 samples) to extract genes expressed differentially in MS patients compared to healthy individuals. Next, GEO2R analyzer was used to compare two groups of samples, control and patient, to identify differentially expressed genes. Genes with expression levels with adjusted p values < 0.05 and absolute log fold change greater than 1 were selected. Finally, using Python programming language (v3.6.0), selected lncRNAs that are located on the same chromosome with relevant protein-coding genes were identified, and then those lncRNAs with less than 50 kb distance from the protein-coding genes were considered appropriate candidate lncRNAs for this study.
LncRNA–miRNA Interactions and Associated Diseases
To identify the possible interaction between lncRNAs and miRNAs, LncDisease software (a sequence-based bioinformatics tool) was used to identify the effective transcripts of the lncRNA associated with MS through interaction with miRNAs involved in MS disease (Wang et al. 2016). LncDisease utilizes TargetScan and miRanda criteria to perform analysis as well as the HMDD database, and human miRNA-disease associations, to reach the final results.
Ethical Issue
The human subject protocol used for this study was approved by an Institutional Review Board of the Royan Institute (Project ID. No. 91000573). All study procedures were carried out in accordance with the approved guidelines.
Human Subjects
The present case–control study was designed at the Royan Institute of Isfahan. Written informed consent was obtained from each individual. The datasets analyzed during the current study are available in the GSE21942 dataset in the GEO database, and all data generated during this study are included in this article and its supplementary information files. Patients were clinically diagnosed with multiple sclerosis by a neurologist through clinical and laboratory parameters based on the revised McDonald criteria (Polman et al. 2011). In this study, PBMCs were isolated from 50 RRMS patients and 25 healthy controls. A total of 25 MS patients was in remitting phase and under regular treatment with interferon beta-1α (CinnoVex®) and another 25 cases were in the relapsing phase. Age- and sex-matched healthy controls with no history of autoimmune diseases or malignancies and no acute or chronic infections were sampled. Disability was graded using the Expanded Disability Status Scale (EDSS), method of calculating disability in MS patients (Kurtzke 1983).
PBMC Preparation
Peripheral blood was collected from all subjects in tubes containing Ethylenediaminetetraacetic acid (EDTA) to prevent coagulation. Human PBMCs were isolated on a ficoll-hypaque lymphocyte separation medium (STEMCELL Technologies, USA) density gradient.
RNA Extraction and cDNA Synthesis
Total RNA was isolated with Trizol® reagent (Invitrogen, USA) following the manufacturer’s instructions. The RNA quality and quantity were assessed using a NanoDrop Spectrophotometer (Nanodrop 1000, Thermo Scientific, USA) and electrophoresis performed on 1% agarose gels. Next, in order to remove DNA contaminations, RNA was treated with deoxyribonuclease I (DNase I) (Thermo Scientific, USA). Total RNA (1 μg) was used for the biosynthesis of complementary DNA (cDNA) following recommended procedures from the manufacturer (Thermo Scientific, USA). cDNAs were stored at − 80 °C until use.
Quantitative Reverse Transcription PCR (RT-qPCR)
Quantitative reverse transcription PCR (RT-qPCR) was carried out with the StepOne™ RT-qPCR System (Applied Biosystems, USA). RT-qPCR amplifications were performed in triplicate. The RT-qPCR process was carried out in a final content volume of 10 μL and included SYBR Premix Ex Taq II (TaKaRa, Japan) and specific primer pairs for each lncRNA and coding gene. In order to normalize gene expression in healthy controls, relapsing and remitting patients, UBC and YWHAZ mRNAs were used as reference genes (Oturai et al. 2016). The list of primer sequences used for PCR are summarized in Table 1.
Statistical Analysis
The statistical analyses were carried out using SPSS 17 software (SPSS, Chicago, IL, USA) and Graph Pad Prism (version 6; Graph Pad software). Data normality was checked using the Shapiro–Wilk test and differences between groups were assessed by One-way ANOVA followed by pairwise comparisons and using Tukey’s correction. The correlation between lncRNAs and coding genes were assessed using Pearson’s correlation. Statistical significance was considered as p values less than 0.05. Receiver Operating Characteristic (ROC) curve analysis was used to determine the discriminatory power of identified biomarkers between the patients and controls.
Results
LncRNAs Selection
From the available RNA-Seq data, 48 lncRNAs were identified that were lineage-specific to memory B cells and were annotated to chromosomes using the GRCh37 genome assembly in Ensembl. Next, we considered differentially expressed protein-coding genes which had been identified in the PBMC of MS patients compared to healthy controls (497; Supplementary Table 1) and were retrieved from GEO database. We examined the proximity of this set of differentially expressed genes to the 48 selected lncRNAs and identified 2 lncRNAs located less than 50 kb distance away from the respective protein-coding genes. AL928742.12 was 10 kb away from immunoglobulin heavy constant alpha 2 (IGHA2) and RP11-530C5.1 has overlap with PRKC apoptosis WT1 regulator (PAWR), indicating the potential cis-regulatory relationships of these neighboring genes (Figs. 2 and 3). To select the appropriate candidates among the three annotated transcripts associated with AL928742.12, respective exonic sequences (in FASTA format) were entered into the ‘LncRNAs Input’ panel of the LncDisease software. AL928742.12-001 was scored as an MS-associated transcript according to predicted miRNA-lncRNA interactions. In the case of RP11-530C5.1, there was only one annotated transcript.

Physical gene proximities of LncRNAs with expressed protein-coding genes from array data. Proximities of differentially expressed protein-coding genes relative to lncRNAs in MS patients were determined using python programming language. mRNAs retrieved from GEO were inserted to STRING db and then combined with the selected lncRNAs and visualized using Cytoscape. Identified protein-coding gene–lncRNAs pairs located within the distance parameters (50 kbp) are indicated with thick lines

Chromosomal organization of protein-coding genes and lncRNA clusters. Genomic data were obtained from the UCSC genome browser. Protein-coding genes are in blue, and lncRNA genes in green. The region on the respective chromosomes displayed in detail is indicated with a red bar in each chromosome idiogram
Demographic and Clinical Characteristics of Enrolled Samples
MS patients and healthy individuals enrolled in this study completed questionnaires. Subject information that was included in the final analyses is shown in Table 2 and additional information on all individuals is provided in supplementary Table 2. Statistical analyses showed no significant differences between samples in each group with regard to sex and age.
LncRNAs Expression Levels in RRMS Compared to the Healthy Controls
After measuring differentially expressed lncRNAs in each group, statistical analyses showed a significant up-regulation of RP11-530C5.1 in relapsing MS patients compared to the remitting patients (p value = 0.046) and healthy controls (p value = 0.002). In contrast, there was a significant decrease of AL928742.12 expression in relapsing MS patients compared to the controls (p value < 0.001) (Fig. 4).

Expression level analyses of AL928742.12 and RP11-530C5.1 lncRNAs in relapsing–remitting and control samples. a Scatter-plot of the expression level of AL928742.12 and b Scatter-plot of the expression level of RP11-530C5.1 in MS and control samples was measured by RT-qPCR and values are given as the mean normalized expression relative to UBC and YWHAZ. (*p < 0.05, **p < 0.01 and*** p < 0.001)
Evaluating Expression Levels of Coding Genes (IGHA2 and PAWR)
In the next step, we measured the expression levels of protein-coding genes, PAWR and IGHA2, which were located less than 50 kb distance away from the differentially expressed lncRNAs. No statistically significant changes in PAWR and IGHA2 expression levels were detected when we compared RRMS patients to healthy controls (Fig. 5).

Expression of IGHA2 and PAWR in MS patients compared to the controls. Scatter-plots of the differential expression level of aPAWR and bIGHA2 in relapsing, remitting, and control samples. Values are given as mean normalized expression relative to UBC and YWHAZ. (*p < 0.05, **p < 0.01 and*** p < 0.001)
Correlation Coefficient Between lncRNA and Coding Genes
Pearson’s correlation tests showed positive correlations between the expression levels of RP11-530C5.1 and PAWR (Pearson’s correlation = 0.269, p value = 0.047). Likewise, there was a significant correlation between AL928742.12 and IGHA2 (Pearson’s correlation = 0.476, p value < 0.001) (Fig. 6).

Correlation analyses between lncRNAs and protein-coding genes in MS. Pearson correlation of expression between a AL928742.12 and IGHA2 and b RP11-530C5.1 and PAWR
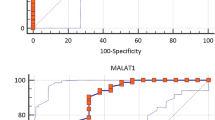
ROC Curve Analysis Results
The results of ROC curve tests showed positive biomarker potential for AL928742.12 (AUC = 0.723, p value = 0.006), as well as the RP11-530C5.1 (AUC = 0.825, p value < 0.0001) (Fig. 7). These findings indicate that these two lncRNAs have the potential to serve as diagnostic biomarkers to distinguish healthy controls from relapse phase MS patients.

ROC curve analysis indicates lncRNAs as likely biomarkers for MS. Discriminatory power of the individual lncRNAs, a AL928742.12 and b RP11-530C5.1, as biomarkers for the diagnosis of MS patients and controls
Discussion
Several specific lncRNAs were recently shown to be deregulated in the PBMCs of MS patients, leading to a proposed role for specific lncRNAs in the progression of MS pathogenesis (Zhang and Cao 2016; Teimuri et al. 2018; Hosseini et al. 2019). LncRNAs have important roles in regulating gene expression and abnormal expression of lncRNAs has recently been linked to the pathogenesis and progression of multiple diseases (DiStefano 2018). Recent studies also demonstrated control of the immune system by lncRNAs (Zhang and Cao 2016; Heward and Lindsay 2014). B lymphocytes have a key role in the normal immune response by secreting antibodies in humoral immunity. Therapies that target memory B cells have become an important focus in MS disease research (Baker et al. 2017).
In this study, we aimed to identify candidate memory B cell-specific lncRNAs involved in MS pathogenesis. To accomplish this, we selected lncRNAs specifically expressed in memory B cell lineage that were also located less than 50 kb distance away from genes differentially expressed in the PBMC of MS patients. Because these differentially expressed genes might be involved in MS pathogenesis, their close physical association with differentially expressed lncRNAs suggests an associated involvement of the lncRNA with MS as well. We evaluated the expression levels of the identified lncRNAs and their neighboring mRNAs in relapsing and remitting phase MS patients compared to healthy individuals.
Analysis of microarray data retrieved from gene expression profiling in PBMCs from relapsing–remitting MS patients demonstrates that PAWR is among the top differentially expressed genes in B cell (Comabella et al. 2015). PAWR is a pro-apoptotic gene and a recent study showed that B cells derived from patients with RRMS induce apoptosis in oligodendrocytes and neurons via unknown secreted factors (Lisak et al. 2017). IGHA2, on the other hand, encodes the constant region of immunoglobulin heavy chains. Immunoglobulins serve as receptors that initiate B lymphocyte differentiation into antibody-secreting plasma cells. Secreted immunoglobulins mediate the effector phase of humoral immunity, which blocks antigen binding to these receptors (McHeyzer-Williams et al. 2012; Schroeder and Cavacini 2010).
RP11-530C5.1 was significantly higher in relapsing MS patients compared to remitting phase patients and healthy controls. The correlation between RP11-530C5.1 and PAWR expression suggests a cis-regulatory role for RP11-530C5.1 on PAWR in memory B cells.
Recently, it was shown that AL928742.12 is down-regulated in inflammatory bowel disease (Mirza et al. 2015). Our results indicated that AL928742.12 was also significantly down-regulated in relapsing MS patients compared to the healthy controls. AL928742.12 expression was also significantly correlated with IGHA2 expression.
LncRNAs may have correlation with their adjacent genes and exert a positive or negative effect on expression of these genes at both transcriptional and post-transcriptional levels. Such regulation is important in development, differentiation, or even progress of human disease (Wilusz et al. 2009; Taft et al. 2010; Yap et al. 2010).
Conclusion
In this study, candidate lncRNAs involved in MS disease were identified from B cell-specific lncRNAs that were identified through adjacent differentially expressed genes in the PBMC of MS patients compared to the healthy controls, and also through interactions with miRNAs involved in MS. Here, we propose that deregulated lncRNAs identified from these associations could provide a valuable resource for studies to discern the important lncRNAs in diseases involving specific cell types and their associations with, and potential regulation of, nearby protein-coding genes.
Abbreviations
- cDNA:
-
Complementary DNA
- CNS:
-
Central nervous system
- DNase I:
-
Deoxyribonuclease I
- EDSS:
-
Expanded disability status scale
- GEO:
-
Gene expression omnibus
- GSE:
-
GEO series
- IGHA2 :
-
Immunoglobulin heavy constant alpha 2
- EDTA:
-
Ethylenediaminetetraacetic acid
- lncRNA:
-
Long non-coding RNA
- miRNA:
-
microRNA
- MS:
-
Multiple sclerosis
- PAWR :
-
PRKC apoptosis WT1 regulator
- PBMC:
-
Peripheral blood mononuclear cell
- RT-qPCR:
-
Quantitative reverse transcription PCR
- ROC curve:
-
Receiver operating characteristic
- RRMS:
-
Relapsing-remitting multiple sclerosis
References
Baker, D., Marta, M., Pryce, G., Giovannoni, G., & Schmierer, K. (2017). Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine,16, 41–50.
Bar-Or, A., Calabresi, P. A., Arnold, D., Markowitz, C., Shafer, S., Kasper, L. H., et al. (2008). Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Annals of Neurology,63(3), 395–400.
Bar-Or, A., Fawaz, L., Fan, B., Darlington, P. J., Rieger, A., Ghorayeb, C., et al. (2010). Abnormal B-cell cytokine responses a trigger of T-cell–mediated disease in MS? Annals of Neurology,67(4), 452–461.
Berger, T., Rubner, P., Schautzer, F., Egg, R., Ulmer, H., Mayringer, I., et al. (2003). Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. New England Journal of Medicine,349(2), 139–145.
Cepok, S., Rosche, B., Grummel, V., Vogel, F., Zhou, D., Sayn, J., et al. (2005). Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain,128(7), 1667–1676.
Chen, G., Wang, Z., Wang, D., Qiu, C., Liu, M., Chen, X., et al. (2012). LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Research,41(D1), D983–D986.
Comabella, M., Cantó, E., Nurtdinov, R., Río, J., Villar, L. M., Picón, C., et al. (2015). MRI phenotypes with high neurodegeneration are associated with peripheral blood B-cell changes. Human Molecular Genetics,25(2), 308–316.
Corcione, A., Casazza, S., Ferretti, E., Giunti, D., Zappia, E., Pistorio, A., et al. (2004). Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proceedings of the National academy of Sciences of the United States of America,101(30), 11064–11069.
Dendrou, C. A., Fugger, L., & Friese, M. A. (2015). Immunopathology of multiple sclerosis. Nature Reviews Immunology,15(9), 545.
DiStefano, J. K. (2018). The emerging role of long noncoding RNAs in human disease. Methods in Molecular Biology, 1706, 91–110.
Duddy, M., Niino, M., Adatia, F., Hebert, S., Freedman, M., Atkins, H., et al. (2007). Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. The Journal of Immunology,178(10), 6092–6099.
Fatica, A., & Bozzoni, I. (2014). Long non-coding RNAs: New players in cell differentiation and development. Nature Reviews Genetics,15(1), 7.
Fillatreau, S., Sweenie, C. H., McGeachy, M. J., Gray, D., & Anderton, S. M. (2002). B cells regulate autoimmunity by provision of IL-10. Nature Immunology,3(10), 944.
Geisler, S., & Coller, J. (2013). RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nature Reviews Molecular Cell Biology,14(11), 699.
Heward, J. A., & Lindsay, M. A. (2014). Long non-coding RNAs in the regulation of the immune response. Trends in Immunology,35(9), 408–419.
Hosseini, A., Teimuri, S., Ehsani, M., Rasa, S. M. M., Etemadifar, M., Nasr Esfahani, M. H., et al. (2019). LncRNAs associated with multiple sclerosis expressed in the Th1 cell lineage. Journal of Cellular Physiology,1, 1. https://doi.org/10.1002/jcp.28779.
Ignacio, R. J., Liliana, P., & Edgardo, C. (2010). Oligoclonal bands and MRI in clinically isolated syndromes: Predicting conversion time to multiple sclerosis. Journal of Neurology,257(7), 1188–1191.
Krumbholz, M., Derfuss, T., Hohlfeld, R., & Meinl, E. (2012). B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nature Reviews Neurology,8(11), 613.
Kurtzke, J. F. (1983). Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology,33(11), 1444.
Lee, J. T. (2012). Epigenetic regulation by long noncoding RNAs. Science,338(6113), 1435–1439.
Li, J.-H., Liu, S., Zhou, H., Qu, L.-H., & Yang, J.-H. (2013). starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Research,42(D1), D92–D97.
Liao, Q., Liu, C., Yuan, X., Kang, S., Miao, R., Xiao, H., et al. (2011). Large-scale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Research,39(9), 3864–3878.
Lisak, R. P., Nedelkoska, L., Benjamins, J. A., Schalk, D., Bealmear, B., Touil, H., et al. (2017). B cells from patients with multiple sclerosis induce cell death via apoptosis in neurons in vitro. Journal of Neuroimmunology,309, 88–99.
McFarland, H. F., & Martin, R. (2007). Multiple sclerosis: A complicated picture of autoimmunity. Nature Immunology,8(9), 913.
McHeyzer-Williams, M., Okitsu, S., Wang, N., & McHeyzer-Williams, L. (2012). Molecular programming of B cell memory. Nature Reviews Immunology,12(1), 24.
Mirza, A. H., Berthelsen, C. H., Seemann, S. E., Pan, X., Frederiksen, K. S., Vilien, M., et al. (2015). Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Medicine,7(1), 39.
Olsson, T., Barcellos, L. F., & Alfredsson, L. (2017). Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nature Reviews Neurology,13(1), 25.
Oturai, D. B., Søndergaard, H., Börnsen, L., Sellebjerg, F., & Romme Christensen, J. (2016). Identification of suitable reference genes for peripheral blood mononuclear cell subset studies in multiple sclerosis. Scandinavian Journal of Immunology,83(1), 72–80.
Paraskevopoulou, M. D., Georgakilas, G., Kostoulas, N., Reczko, M., Maragkakis, M., Dalamagas, T. M., et al. (2012). DIANA-LncBase: Experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Research,41(D1), D239–D245.
Polman, C. H., Reingold, S. C., Banwell, B., Clanet, M., Cohen, J. A., Filippi, M., et al. (2011). Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Annals of Neurology,69(2), 292–302.
Ranzani, V., Rossetti, G., Panzeri, I., Arrigoni, A., Bonnal, R. J., Curti, S., et al. (2015). The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nature Immunology,16(3), 318.
Schroeder, H. W., & Cavacini, L. (2010). Structure and function of immunoglobulins. Journal of Allergy and Clinical Immunology,125(2), S41–S52.
Sellebjerg, F., Börnsen, L., Khademi, M., Krakauer, M., Olsson, T., Frederiksen, J., et al. (2009). Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology,73(23), 2003–2010.
Spurlock, C. F., III, Tossberg, J. T., Guo, Y., Collier, S. P., Crooke, P. S., III, & Aune, T. M. (2015). Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nature Communications,6, 6932.
Taft, R. J., Pang, K. C., Mercer, T. R., Dinger, M., & Mattick, J. S. (2010). Non-coding RNAs: Regulators of disease. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland,220(2), 126–139.
Teimuri, S., Hosseini, A., Rezaenasab, A., Ghaedi, K., Ghoveud, E., Etemadifar, M., et al. (2018). Integrative analysis of lncRNAs in Th17 cell lineage to discover new potential biomarkers and therapeutic targets in autoimmune diseases. Molecular Therapy-Nucleic Acids,12, 393–404.
Tintore, M., Rovira, A., Rio, J., Tur, C., Pelayo, R., Nos, C., et al. (2008). Do oligoclonal bands add information to MRI in first attacks of multiple sclerosis? Neurology,70(13 Part 2), 1079–1083.
Tong, Y.-K., & Lo, Y. D. (2006). Diagnostic developments involving cell-free (circulating) nucleic acids. Clinica Chimica Acta,363(1–2), 187–196.
Tufekci, K. U., Oner, M. G., Genc, S., & Genc, K. (2011). MicroRNAs and multiple sclerosis. Autoimmune Diseases,1, 1. https://doi.org/10.4061/2011/807426.
Ulitsky, I., & Bartel, D. P. (2013). lincRNAs: Genomics, evolution, and mechanisms. Cell,154(1), 26–46.
Wang, J., Ma, R., Ma, W., Chen, J., Yang, J., Xi, Y., et al. (2016). LncDisease: A sequence based bioinformatics tool for predicting lncRNA-disease associations. Nucleic Acids Research,44(9), e90. https://doi.org/10.1093/nar/gkw093.
Wilusz, J. E., Sunwoo, H., & Spector, D. L. (2009). Long noncoding RNAs: Functional surprises from the RNA world. Genes & Development,23(13), 1494–1504.
Yap, K. L., Li, S., Muñoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., et al. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Molecular Cell,38(5), 662–674.
Yoon, J.-H., Abdelmohsen, K., & Gorospe, M. (2014). Functional interactions among microRNAs and long noncoding RNAs. Seminars in Cell & Developmental Biology, 34, 9–14.
Zhang, Y., & Cao, X. (2016). Long noncoding RNAs in innate immunity. Cellular & Molecular Immunology,13(2), 138.
Acknowledgements
The authors declare that they have no competing interests. A part of this project was supported by the National Institute for Medical Research Development (NIMAD’s Project No. 942792) and also partly by Royan Institute for Biotechnology.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ghoveud, E., Teimuri, S., Vatandoost, J. et al. Potential Biomarker and Therapeutic LncRNAs in Multiple Sclerosis Through Targeting Memory B Cells. Neuromol Med 22, 111–120 (2020). https://doi.org/10.1007/s12017-019-08570-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-019-08570-6