
Abstract
Long non-coding RNAs (lncRNAs) play critical roles in regulation of immunological pathways. Consequently, their expression profile represents new biomarkers for susceptibility and progression of immunological disorders. However, their role in chronic inflammatory diseases such as multiple sclerosis (MS) remained unknown. Here, we assessed the expression of lncRNAs MALAT1 and HOTAIRM1 as well as their target genes in peripheral blood of MS patients to show their possible roles in disease initiation and progression. In this study, 50 patients with relapsing-remitting MS and 50 healthy matched controls were enrolled. Comparative Ct method via TaqMan assay was used to quantify transcript levels of MALAT1, HOTAIRM1, AGO2, CSTF2, CPSF7, and WDR33. Our analysis depicted significant differences in lncRNAs and their target genes expression levels. AGO2 expression was significantly elevated in MS patients (P < 0.001) whereas, CSTF2 expiration was considerably down-regulated (P < 0.042). These findings suggest that AGO2 and CSTF2 can be considered as potential theoretical biomarkers for MS and can be helpful for diagnosis and prognosis of responding patients to interferon.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a heterogeneous autoimmune disease with poor diagnosis and higher prevalence in women (Eftekharian et al. 2016). Previous studies on MS pathogenesis depict that the immune system dysregulation is associated with changes in transcriptome and gene expression. About 85% of affected patients have relapsing-remitting multiple sclerosis (RR-MS) (Hatami et al. 2018). Inflammatory reactions trigger immune cells to cause localized demyelinating of nerve fibers. The unpredictable episodes of relapses or exacerbations lead to worsening of neurologic function or deterioration of existing symptoms (Compston and Coles 2008). Identification of genetic markers would facilitate prediction of disease prognosis during the early stages of the disease and monitoring both disease course and response to therapy.
Long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) belong to non-coding RNAs (ncRNAs) family which is involved in many cellular processes (Eftekharian et al. 2017). miRNAs mediate regulation of other classes of ncRNAs and also harmonize the expression of protein coding genes by targeting their transcripts to AGO2-containing complexes in the cytoplasm. Mechanistically, it is proposed that lncRNAs perform “sponge-like” effect on various miRNAs, which subsequently inhibit miRNA-mediated functions. RNA-seq analyses have shown that the alteration in lncRNAs expression contribute to diverse neurological disorders (Wu et al. 2013). The lncRNA lung adenocarcinoma transcript 1 (MALAT1), also referred to as non-coding nuclear-enriched abundant transcript 2 (NEAT2), was initially reported to contribute in metastasis in lung cancer (Gutschner et al. 2013; Li et al. 2015). MALAT1 is up-regulated in many solid tumors and is associated with cancer metastasis and recurrence (Gutschner et al. 2013). HOXA transcript antisense RNA, Myeloid-Specific 1 (HOTAIRM1) as an lncRNAs is located between HOXA1 and HOXA2 genes, in the HOX gene cluster. The gene is expressed in myeloid lineage cells and is involved in myeloid transcriptional regulation. Transcription of HOTAIRM1 is induced by retinoic acid and transcripts participating in myelopoiesis regulation. HOTAIRM1 also plays an essential role in the regulation of autophagy pathways in myeloid cells differentiation through the degradation of PML-RARA oncoprotein. HOTAIRM1 makes a miRNA sponge in a pathway which contains miR-20a/106b, miR-125b, and their targets AGO2, ULK1, E2F1, and DRAM2 (Chen et al. 2017). Recent studies confirmed the critical role of HOTAIRM1 in controlling the development of other immune cell-types such as innate lymphoid cells (ILC), monocyte-macrophages, T cell subsets (Th1, Th2, Th17, and Treg), and B cells (Atianand and Fitzgerald 2014).
Functional studies have indicated that alteration in the expression of Cleavage Stimulatory Factor-64 kDa (CSTF2), which binds to a GU/U-rich cis-element downstream to the specific polyA site (PAS), affect pre-mRNA processing. The transcriptome-wide study has also demonstrated that the expression level of CSTF2 has a positive correlation with the tendency of global mRNA 3′-UTR shortening (Yao et al. 2012). It was revealed that the CSTF2 expression was up-regulated upon T cell receptor (TCR) stimulation. On the other hand, in native T cells, the expression level of CSTF2 is lower and thus proximal PASs are not efficiently exploited for gene regulation. However, when the TCR was stimulated, the CSTF2 level was overexpressed and accordingly the usage of the proximal PAS increased (Chuvpilo et al. 1999). The protein encoded by CPSF7 is one of the six necessary factors for correct end-termination in 3′ UTRs in precursor mRNAs. This protein acts as a hetrotetramer, each containing a dimmer of 25 kDa subunits connected to 59 and 68 kDa subunits. The complex protein is composed of numerous factors which are involved in the editing and splicing of newly produced mRNAs like U2, and also in regulation of large complex activity associated with polyadenilation and its sub units like CSTF2.
CPSF7 and other factors such as CPSF73, CPSF100, Symplekin, and WDR33 cooperate in detection of poly(A) signal region and formation of the complex (Schönemann et al. 2014). WD repeats contain 40 amino acid regions typically include glycine, histidine, tryptophan, and asparagine which may facilitate the formation of heterotrimeric or multi-protein compounds. WDR33 protein as a member of WD proteins is involved in a variety of cellular processes, including cell cycle progression, cell signal transduction, apoptosis, and nucleus protein regulation. This gene plays an important role in cell differentiation or DNA recombination and has several transcripts produced through alternative splicing.
In the present study, we evaluated the expression of MALAT1, HOTAIRM1, AGO2, CSTF2, CPSF7, and WDR33 in patients with RR-MS compared to normal control.
Materials and Methods
A total of 50 unrelated patients in the relapsing stage of RR-MS disease and 50 healthy control subjects were recruited to participate in this study at Department of Medical Genetics, Shahid Beheshti University of Medical Sciences Tehran, Iran, during 2-year study period (2016–2018). (Table 1). Exclusion criteria for control group were pregnancy, steroid therapy, infection within 1 month prior to sample collection, existence of other autoimmune diseases, malignancies, and chronic infectious diseases. The selection criteria for RR-MS individuals included clinical or radiological relapse within 1 month from sample collection and no history of smoking. The patients were confirmed to have McDonald criteria for MS, as revised in 2010, and stable phase of the disease (Polman et al. 2011). The study groups were prospectively matched for the same geographical region, sex, age (mean age 35.3 ± 2.1), and body mass index (BMI) (Table 1). Individuals in case group were classified in three different groups based on their age ranges (< 30, 30–40, > 40). They were treated with interferon (IFN)-β therapy for at least 2 years (Cinno Vex had been injected subcutaneously at doses of 20 μg three-times a week) (Taheri et al. 2017; Rezazadeh et al. 2018). Informed consent was obtained from each participant and the study was approved by the institutional ethics committee (No. 3275).
Total RNA was extracted from each blood sample by the Geneall Hybrid-RTM blood RNA extraction Kit (cat No. 305-101) according to the manufacturer’s instructions. Total RNA was treated with DNase I for 30 min at 42 °C. The mRNA was reverse-transcribed into complementary DNA (cDNA) by using Geneall cDNA synthesis kit as described by the manufacturer’s protocol. The expression level of lncRNAs was measured by qRT-PCR using TaqMan® assay in the Corbett Rotor gene 6000 machine (Corbett Life Science). The reaction system of PCR was performed in duplicate containing 2× concentrated Master Mix, 2 μL of template cDNA, and 100 nM of primers in a final volume of 25 μL. The sequence of probes and primers has been presented in Table 2. Hypoxanthine guanine phosphoribosyl transferase (HPRT) was applied as a reference gene based on its stable expression within different sample groups (Vandesompele et al. 2002). The initial denaturation step at 95 °Cfor 10 min was followed by 40 cycles at 95 °C for 15 s, and 60 °C for 45 s. Relative expressions of genes were calculated with 2-ΔΔCt method, using HPRT as an internal control.
To identify the mRNAs as “cis-regulated target genes” in up- or downstream of the given lncRNAs and the correlation between lncRNA and mRNA expressions some databases were reviewed including LNCipedia, ChIPBASE, DIANA tools, GENECODE, ENCODE, lncRNA, disease databases, lncrnadb, FANTOM, lnCeDB, STRING, and HGNC.
The lncRNAs and co-expression genes such as chromatin regulators and transcription factors (TFs) were identified by using ENCODE and other online databases. The “TF–lncRNAs” two element networks were generated in the first step. After the primary evaluation merged network has been proposed.
SPSS software version 18 (statistical package for social sciences Inc., Chicago, IL, USA) was used to analyze the data. Independent t test was applied to compare expressions of genes between two groups. Pearson correlation coefficient was calculated for evaluation of the correlation between variables. P values less than 0.05 were considered as statistically significant. Spearman rank order correlation test was performed to evaluate the possible correlation between relative expression levels of genes and clinical variables.
Results
The Expression Levels of lncRNAs and Their Target Genes
The expression levels of MALAT1, HOTAIRM1, AGO2, CSTF2, CPSF7, and WDR33 have been compared between MS patients and control group based on their age and sex. The results are presented in Table 3. As shown in Table 3, the expression of MALAT1 gene was significantly different between two groups participants who were under the age of 30 (p = 0.04). The mRNA levels of AGO2 was considerably up-regulated in MS patients compared to healthy subjects (p < 0.001) (Table 3). The results of statistical analysis showed no difference in the HOTAIRM1 expression between participants (Table 3). There was significant decrease in expression level of CSTF2 (REx = 0.7533, p = 0.042) between two groups especially when comparing female patients with female controls (Table 3). Compared with healthy controls, our patients demonstrated an increase in expression of CPSF7 and WDR33 levels (REx = 3.2, p = 0.02 and REx = 2.05, p = 0.02 respectively).
Co-expression Analysis
Co-expression analysis between expression of genes in healthy controls and MS patients revealed a significant correlation between MALAT1 and AGO2 (r = 0.747, p < 0.0001), whereas HOTAIRM1 and CSTF2 showed no remarkable correlation (r = 0.158, p = 0.117) (Table 3). We also detected direct and moderate correlation between MALAT1 and WDR33 expressions (r = 0.489, p < 0.0001).
Correlation Between Expression Levels and Clinical Characteristics of Patients
Data analysis showed a negative correlation between MALAT1, HOTAIRM1, and AGO2 expressions and age at disease onset. The positive correlation between AGO2 and WDR33 expressions and disease duration, and between HOTAIRM1 expression and age were significant.
Receiver Operating Characteristic (ROC) Curve Analysis
We assessed diagnostic power of all genes in MS through calculation of area under curve (AUC) in ROC curves. Based on the AUC values, MALAT1 and AGO2 genes had the highest diagnostic power (AUC = 0.882) (Fig. 1). The AUC values for HOTAIRM1, CPSF7, WDR33, and CSTF2 were 0.8, 0.74, 0.639, and 0.62 respectively.

ROC curve analysis for AGO2 (a) and MALAT1 (b)
Discussion
lncRNAs are a novel class of transcripts that are pervasively transcribe in the genome. Recent studies have approved that lncRNAs dysregulation are associated with human diseases including neurodegenerative disorders, but their expression have not been exhaustively investigated in MS. In recent years, many efforts have been done to identify genes which are involved in the MS initiation and severity. In this work, we highlighted the interaction of MALAT1 and HOTAIRM1 as lncRNAs and their target genes including AGO2, CSTF2, CPSF7, and WDR33 with MS pathogenesis and progression.
LncRNAs act as competing endogenous RNAs against miRNAs via sponge mechanism. miRNAs could inhibit lncRNAs expression through AGO2-mediated pathway (Leucci et al. 2013). MALAT1 as an active member of lncRNAs is mostly expressed in hippocampal neurons and particularly in RR-MS disease (Fenoglio et al. 2016). It seems that MALAT1 promotes the inflammatory response in microglia via MyD88/IRAK1/TRAF6 signaling pathway (Wang and Zhou 2018). Also, it contributes in the inflammatory response through modulating miR-199b/IKKβ/NF-κB signaling and promotes the production of proinflammatory cytokines including TNF-α and IL-1β by down-regulating of miR-199b (Zhou et al. 2016). Although MALAT1 regulation mechanism by miRNAs has been remained unknown, it has been shown that miR-101 and miR-217 could suppress this lncRNA. This posttranscriptional regulation may lead to inhibition of cell growth, invasion, and metastasis (Wang et al. 2015). MALAT1 also modulates expression of a subset of coding genes which are involved in regulation of synapsis. It is proposed that MALAT1-CREB complex conserve phosphorylated condition of CREB via inhibiting of PP2A-mediated dephosphorylation, which leads to continuous CREB signaling activation (Yao et al. 2016).
In the present study, the expression level of MALAT1 in RR-MS patients was not significantly increased compared with the control group. The results of Spearman test revealed that there is a reverse correlation between the expression of MALAT1 and age, whereas the expression of this gene decreased with disease duration. This gene may be considered as a trigger for MS disease and also may cooperate with some factors in disease development but further experiment needed to confirm this.
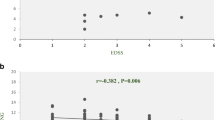
Previous studies showed that AGO2 has regulatory functions in inflammatory pathways and subsequently in inflammatory diseases such as rheumatoid arthritis and osteoarthritis (Pearson and Jones 2016; Yao et al. 2016). The role of MALAT1 identified in abnormalities of capillary systems. However, its role in neurodegenerative abnormalities including MS has not been discovered yet. Our network analysis demonstrated that AGO2 may have interaction with MALAT1. The data showed AGO2 expression level was increased up to 2.6-fold in RR-MS patients compared to controls. This increase was more than 3 times in male patients while in female patients it was 1.9 times. Interestingly, the increase in expression level of this gene was about 13.7 times in affected males with age of 30–40 years. At the same age range, in the female patients, this rate was increased to 7-fold. The expression of AGO2 showed direct and moderate correlation with EDSS. These outcomes suggest that increasing in the expression level of AGO2 may associate with worsening of clinical conditions. However, we found that inverse correlation between AGO2 expression and disease duration. Evidences suggest that miRNAs regulate protein coding genes by leading their mRNAs to LARC complex. On the one hand, lncRNAs may compete with the miRNAs inside the cell. On the other hand, miRNAs can suppress the expression of lncRNAs via AGO2. It seems this process for MALAT1 happens at the transcriptional level (Wang et al. 2015). In a very interesting way, miR-9 has critical role in degradation of MALAT1 through the AGO2 pathway in the nucleus (Leucci et al. 2013). These findings depict the existence of a new direct regulatory link between the two most important classes of non-coding RNAs, miRNAs, and lncRNAs. In line with these observations, our results indicate that MALAT1 and AGO2 have a very strong correlation (r = 0.694, p < 0.0001), suggesting that with increase in the expression level of AGO2, the possible inhibitory effect of miRNAs such as miR-9 on the MALAT1 is compensated by higher expression of MALAT1.
HOTAIRM1 is expressed in myeloid cells and can play an important role in regulation of myeloid transplantation. Many effects of lncRNAs are done by employing histone modifying complexes, such as Polycomb suppressor complexes (PRC2), to induce silencing of target genes. Consequently, transcription may be active or suppressed depending on the three-dimensional complexes formed on the chromatin. Interestingly, some lncRNAs, such as HOTAIRM1, interact with a number of different chromatin proteins, such as CSTF2. This lncRNA seems to act like a molecular sponge to collect some targets such as CSTF2, AGO2, and mir-3960. In the current study, the results showed that HOTAIRM1 expression in female patients aged between 30 and 40 years old was up-regulated compared with the corresponding control group. We also detected an increase in the expression of this gene by increasing the level of EDSS. Additionally, there was a remarkable correlation between the expression levels of HOTAIRM1 and AGO2 which proposes that AGO2 expression level is upgraded to compensate the sponge activity of HOTAIRM1.
The CSTF2 gene encodes a nuclear protein with RRM domain which is essential for exclusive binding to a specific position in target RNAs. The encoded protein is a cleavage stimulation factor which attaches to the GU-rich elements at the 3′ UTR of the specific mRNAs. According to previous data, interaction between CSTF2 and TNF-α was anticipated to be involved in the process of immunologic diseases. CSTF2 protein is also involved in the inflammatory processes in the development of neurodegenerative diseases (Curinha et al. 2014). It seems that the onset of neurodegenerative diseases is due to the collapse of the natural interactions between the inflammatory proteins and CSTF2. Transcripts of some lncRNAs, such as HOTAIRM1, are polyadenylated and also participate in the process of creating this poly-adenyl tail. The CSTF2 gene also has interactions with GAS5, WDR33, and NR3C1. Expression profiling in rheumatoid arthritis has shown that changes in expression level of CSTF2 leads to autoimmune responses. The gene also has expression in both B and T cells and is involved in immune responses and expression regulation. Therefore, CSTF2 can be considered as an appropriate candidate for the evaluation of the pathogenesis of autoimmune disorders such as MS disease (Nunez-Iglesias et al. 2010). In this study, the CSTF2 expression level in MS patients was significantly decreased. Such decrease was particularly prominent in female patient aged more than over 40 which is in line with the different pathways involved in the pathogenesis of MS in males and females.
Taken together, the expression pattern of these genes were distinct in females and males. Such finding is in line with the previously reported effects of gender in genetic, immunological, and clinical aspects of MS disease (Harbo et al. 2013).
The Spearman test showed statistically significant correlation between HOTAIRM1 and MALAT1, but the correlation between HOTAIRM1 and CPSF7 as a second target for this lncRNA was not remarkable. Our study showed a 3.2-fold increase in expression of CPSF7 in patients compared to control participants. The rate of increase in expression of CPSF7 has gradually slowed down with increased age. Expression levels of CSTF2 and CPSF7 genes were directly correlated and showed a simultaneous increase in expression.
The WDR33 expression in patients was 2-fold upper than controls. Relative expression of this gene in male patients aged between 30 and 40 was 5.9-fold higher compared to control group. The expression of this gene had reverse correlation with duration of the MS disease. The WDR33 protein is directly connected to the AAUAAA signal, the branch point of the poly A, and the CPSF family factors interfere with this reaction (Chan et al. 2014). One of the factors within this complex is CPSF7 which is involved in the polyadenylation process. The performed studies have shown that inhibiting the polyadenylation reaction leads to decrease in induction of genes involved in the inflammation process and one of the contributing factors is CPSF7 (Kondrashov et al. 2012).
In conclusion, our findings propose a novel direct regulatory link between two important classes of ncRNAs, miRNAs, and lncRNAs, and coding genes. Besides, this work demonstrates robust dysregulation of several lncRNAs associated genes that they play significant roles in cellular mechanisms and regulation in MS patients compared with controls. Finally, we demonstrated high diagnostic power of MALAT1 and AGO2 in MS disease. However, our study has a limitation regarding small sample size. The logic of this project might then be applied to set up future studies with the purpose of discovering potential biomarkers for MS and response to therapy.
References
Atianand MK, Fitzgerald KA (2014) Long non-coding RNAs and control of gene expression in the immune system. Trends Mol Med 20:623–631
Chan SL, Huppertz I, Yao C, Weng L, Moresco JJ, Yates JR 3rd, Ule J, Manley JL, Shi Y (2014) CPSF30 and Wdr33 directly bind to AAUAAA in mammalian mRNA 3' processing. Genes Dev 28:2370–2380
Chen Z-H, Wang W-T, Huang W, Fang K, Sun Y-M, Liu S-R, Luo X-Q, Chen Y-Q (2017) The lncRNA HOTAIRM1 regulates the degradation of PML-RARA oncoprotein and myeloid cell differentiation by enhancing the autophagy pathway. Cell Death Differ 24:212–224
Chuvpilo S, Zimmer M, Kerstan A, Glöckner J, Avots A, Escher C, Fischer C, Inashkina I, Jankevics E, Berberich-Siebelt F (1999) Alternative polyadenylation events contribute to the induction of NF-ATc in effector T cells. Immunity 10:261–269
Compston A, Coles A (2008) Multiple sclerosis. Lancet 372:1502–1517
Curinha A, Oliveira Braz S, Pereira-Castro I, Cruz A, Moreira A (2014) Implications of polyadenylation in health and disease. Nucleus 5:508–519
Eftekharian MM, Ghannad MS, Taheri M, Roshanaei G, Mazdeh M, Musavi M, Hormoz MB (2016) Frequency of viral infections and environmental factors in multiple sclerosis. Hum Antibodies 24:17–23
Eftekharian MM, Ghafouri-Fard S, Soudyab M, Omrani MD, Rahimi M, Sayad A, Komaki A, Mazdeh M, Taheri M (2017) Expression analysis of long non-coding RNAs in the blood of multiple sclerosis patients. J Mol Neurosci 63:333–341
Fenoglio C, Calvi A, Serpente M, De Riz M, Comi C, Lecchi E, Pietroboni A, Arcaro M, Cioffi S, Oldoni E (2016) Long non coding RNA (LncRNAs) expression analysis in patients with multiple sclerosis: potential biomarkers of disease susceptibility and progression. Mult Scler J. SAGE Publications Ltd 1 Olivers Yard, 55 City Road, London EC1Y 1SP, England, p 271
Gutschner T, Hämmerle M, Eißmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Groß M (2013) The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 73:1180–1189
Harbo HF, Gold R, Tintore M (2013) Sex and gender issues in multiple sclerosis. Ther Adv Neurol Disord 6:237–248
Hatami M, Salmani T, Arsang-Jang S, Davood Omrani M, Mazdeh M, Ghafouri-Fard S, Sayad A, Taheri M (2018) STAT5a and STAT6 gene expression levels in multiple sclerosis patients. Cytokine 106:108–113
Kondrashov A, Meijer HA, Barthet-Barateig A, Parker HN, Khurshid A, Tessier S, Sicard M, Knox AJ, Pang L, De Moor CH (2012) Inhibition of polyadenylation reduces inflammatory gene induction. Rna 18:2236–2250
Leucci E, Patella F, Waage J, Holmstrøm K, Lindow M, Porse B, Kauppinen S, Lund AH (2013) microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci Rep 3:2535
Li C, Chen J, Zhang K, Feng B, Wang R, Chen L (2015) Progress and prospects of long noncoding RNAs (lncRNAs) in hepatocellular carcinoma. Cell Physiol Biochem 36:423–434
Nunez-Iglesias J, Liu CC, Morgan TE, Finch CE, Zhou XJ (2010) Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One 5:e8898
Pearson MJ, Jones SW (2016) Long noncoding RNAs in the regulation of inflammatory pathways in rheumatoid arthritis and osteoarthritis. Arthritis Rheumatol 68:2575–2583
Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L (2011) Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69:292–302
Rezazadeh M, Gharesouran J, Moradi M, Noroozi R, Omrani MD, Taheri M, Ghafouri-Fard S (2018) Association study of ANRIL genetic variants and multiple sclerosis. J Mol Neurosci:1–6
Schönemann L, Kühn U, Martin G, Schäfer P, Gruber AR, Keller W, Zavolan M, Wahle E (2014) Reconstitution of CPSF active in polyadenylation: recognition of the polyadenylation signal by WDR33. Genes Dev 28:2381–2393
Taheri M, Ghafouri-Fard S, Solgi G, Sayad A, Mazdeh M, Omrani MD (2017) Determination of cytokine levels in multiple sclerosis patients and their relevance with patients’ response to Cinnovex. Cytokine 96:138–143
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034. 1
Wang L-Q, Zhou H-J (2018) LncRNA MALAT1 promotes high glucose-induced inflammatory response of microglial cells via provoking MyD88/IRAK1/TRAF6 signaling. Sci Rep 8:8346
Wang X, Li M, Wang Z, Han S, Tang X, Ge Y, Zhou L, Zhou C, Yuan Q, Yang M (2015) Silencing of long noncoding RNA MALAT1 by miR-101 and miR-217 inhibits proliferation, migration, and invasion of esophageal squamous cell carcinoma cells. J Biol Chem 290:3925–3935
Wu P, Zuo X, Deng H, Liu X, Liu L, Ji A (2013) Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull 97:69–80
Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, Shi Y (2012) Transcriptome-wide analyses of CstF64–RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci 109:18773–18778
Yao J, Wang XQ, Li YJ, Shan K, Yang H, Yao MD, Liu C, Li XM, Shen Y, Liu JY (2016) Long non-coding RNA MALAT1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol Med e201505725
Zhou HJ, Wang LQ, Xu QS, Fan ZX, Zhu Y, Jiang H, Zheng XJ, Ma YH, Zhan RY (2016) Downregulation of miR-199b promotes the acute spinal cord injury through IKKbeta-NF-kappaB signaling pathway activating microglial cells. Exp Cell Res 349:60–67
Acknowledgments
We thank our patients for participating in this study. This article has been extracted from thesis written by Jalal Gharesouran in Department of Medical Genetics, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran (registration no; 3).
Funding Sources
This work was financially supported by a grant allocated by the Deputy of Research, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gharesouran, J., Taheri, M., Sayad, A. et al. A Novel Regulatory Function of Long Non-coding RNAs at Different Levels of Gene Expression in Multiple Sclerosis. J Mol Neurosci 67, 434–440 (2019). https://doi.org/10.1007/s12031-018-1248-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-018-1248-2