
Abstract
Systemic autoimmune diseases (SADs) encompass a wide spectrum of clinical signs as a reflection of their complex physiopathology. A variety of mechanisms related with the innate immune system are in the origin of the loss of self-tolerance in these diseases, and for most of them, the myeloid leukocytes are key actors. Monocytes, macrophages, dendritic cells, and neutrophils are first-line immune effectors located in the interface between innate and adaptive immunity. They are crucial in the organization of the local and systemic responses to damage-associated molecular patterns (DAMPs) and determine the intensity, orientation, and duration of the local immune response through the expression of chemokines, costimulatory or protolerogenic factors. In this review, we summarize the current knowledge about the role of the main myeloid populations in the induction and maintenance of systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), primary antiphospholipid antibody syndrome (PAPS), systemic sclerosis (SSc), and Sjögren’s syndrome (SjS), based on the data from both mouse preclinical models and patients. According to these data, our challenge in the next few years is to better dissect the fine mechanisms underlying the pathological role of myeloid cells in these diseases in order to define specific cell subsets or proteins that can be potential targets for drug development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Features of Systemic Autoimmune Diseases
The mammalian immune system is made of an intricate set of cellular, chemical, and soluble protein mechanisms specialized in the protection of the organism from infections and tumors through the recognition and neutralization of pathogens or aberrant cells, without attacking the body’s own structures. In steady state, the immune system is tolerized to the antigens and the structures expressed by the body’s own cells and thus does not respond to elements that are expressed in endogenous tissues. All those peacefully coexist in a state known as self-tolerance. If the self-tolerance equilibrium is persistently unbalanced, this can ultimately result in development of autoimmune diseases, which can be described as the result of a sustained and persistent immune response against self-constituents. A variety of mechanisms have been described for the breakdown of tolerance supported by experimental models: failure in the deletion of autoreactive lymphocytes, central and peripheral tolerance malfunction, abnormal presentation of autoantigens, molecular mimicry, epitope spreading, or polyclonal lymphocyte activation (reviewed in [1]). A common feature of all autoimmune diseases is the presence of autoantibodies and/or self-reactive lymphocytes, chronic inflammation, and tissue destruction [2].
Nowadays, it is accepted that autoimmune reactions are part of the physiological functioning of the healthy immune system. In serum of normal individuals, the natural self-reactive antibodies are found at low concentrations and antigen avidity [3]. It is likely that natural autoantibodies are used by the organism to facilitate the clearance of senescent cells and cell-free autoantigens, and consequently, prevent the activation of cognate autoimmune responses. However, in the serum of patients with autoimmune diseases, high concentrations of IgG-switched autoantibodies are detected, which show high avidity for the antigen and somatic hypermutations of the variable region. These autoimmune response-associated autoantibodies are the product of a T-helper cell-dependent activation of B cells, which, in conditions of prolonged contact with the antigen, leads to clonal selection [4].
Regarding the target organs, autoimmune diseases can be classified into two groups: organ-specific and systemic autoimmune diseases (SADs). In SADs, the pathogenic antigens are widely expressed in the body, and therefore, many organs and tissues are targeted by the activated immune system [5]. The ubiquity of the autoantigens and the systemic nature of the resulting disorders may be the cause of many common signs and symptoms that accompany various SADs [6]. This group includes systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), primary antiphospholipid antibody syndrome (PAPS), mixed connective tissue disease (MCTD), systemic sclerosis (SSc), and Sjögren’s syndrome (SjS). Numerous mouse models have been employed to determine the genetic, molecular, and cellular mechanisms involved in the pathogeny of SADs. These models can be useful not only to elucidate disease mechanisms, but also to identify new genes associated to disease, to test new therapies, or to validate therapeutic targets [7,8,9,10].
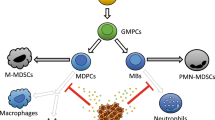
Initial studies focused on the role of the adaptive immune system since primary abnormalities of B and T lymphocyte functions in SLE and other SADs were considered for a long time the likely basis of the autoimmune condition [11]. Recent advances in the understanding of the innate immune system have changed this first paradigm. Thus, it has been increasingly recognized that several components of the innate immune system, which detect pathogen patterns, play a key role in self-antigen recognition in autoimmune diseases [12, 13]. Both components of the immune system, innate and adaptive, are then involved in the physiopathology of organ-specific and SADs [14]. The cellular components of the innate immune system include monocytes, macrophages, dendritic cells (DC), and neutrophils, all being myeloid phagocytes. Although their differentiation pathways are not fully understood, they all originate from a common myeloid progenitor (see Fig. 1). These cells are involved directly in the pathogenesis of SADs, as antigen-presenting cells as well as acting as accessory cells through the secretion of soluble mediators like cytokines or chemokines (see Fig. 2). They play key roles in immune surveillance, host defense, and tissue repair, and their activity and differentiation fate can be differentially modulated by the tissue microenvironment and the characteristics of the hematopoietic niche in steady state or pathogenic conditions [15]. Most of the main relevant roles of myeloid cells in the pathogenesis of autoimmune diseases have been revealed in mice. In this review, we discuss the role of the main myeloid populations in the pathogenesis of SADs in patients, as well as the knowledge obtained using preclinical models.

Main steps and players in myelopoiesis. Graph depicting the still evolving view of the myelopoiesis pathways in mice and humans

Myeloid cell involvement in SADs. Defective apoptotic cell clearance by myeloid phagocytes induces the accumulation of self-antigens in the tissues. DC take up these self-antigens and present them to autoreactive T and B lymphocytes in the presence of proinflammatory cytokines, inducing the active secretion of autoantibodies and the accumulation of pathogenic IC. Neutrophils recognize IC and, first, induce the release of ROS, cytokines, and proteases, and, second, die by NETosis. NET content activates pDC to secrete type I IFN, which in turn activates cDC and B lymphocytes, inducing antibody switching. Circulating monocytes migrate to the tissues and differentiate into tissue macrophages, which release ROS and mediate the differentiation of pathogenic resident cell types such as osteoclasts. Arrows, activated mechanism; whiskers, impaired mechanism
Dendritic Cells
Human DC are a heterogeneous population composed by different subsets with differing phenotypic and functional features [16]: (i) plasmacytoid DC (pDC) can be differentiated from both lymphoid and myeloid precursors [17], producing huge amounts of type I interferon (IFN) after activation [18], and (ii) conventional DC (cDC) of myeloid origin. In steady state, human myeloid DC progenitors in the bone marrow (Fig. 1) originate two main specialized subpopulations characterized by the expression of BDCA-3 (CD141+) or, alternatively, BDCA-1 (CD1c+) [19]. During inflammation, an additional DC population of inflammatory DC (infDC) is generated in the tissues after differentiation of newly recruited monocytes [20]. Mouse DC present high homology with human DC [21]. Murine steady-state DC subsets can also be divided into pDC and cDC. cDC derive from a common precommitted precursor, the pre-cDC, that is dependent on FLT3L (Fig. 1) [22]. Murine cDC can be classified in CD8− and CD8+. Human cDC share key functional properties with their mouse counterpart, such as constitutive expression of MHC class II molecules, ability to process antigens and stimulate naïve T cells, as well as molecular signatures [23, 24]. cDC can be further divided into lymphoid organ-resident DC and migratory tissue DC. In this review, only classical myeloid DC (cDC or infDC) will be considered since pDC are beyond the scope of this review.
In healthy conditions, immature DC take up cell-derived self-antigens (autoantigens) in the tissues [25, 26]. DC present autoantigens to autoreactive T cells, but local immune suppressive factors induce tolerance through different mechanisms [27,28,29]. In the presence of danger signals, DC become mature and activated and initiate a maturation program optimizing their antigen presentation capabilities and their costimulatory activity [30, 31]. Mature DCs express high levels of MHC class I and II molecules, T lymphocyte costimulatory molecules and chemokine receptors, as well as an array of cytokines regulating adaptive immunity [32]. Depending on the nature of the environment, cDC can induce differentiation of Treg, Th1, Th2 or Th17 cells from naïve CD4+ T cells [20, 33]. cDC are also important for the promotion of the humoral responses [34] via the activation of specialized T follicular helper (TFH) cells or through the direct interaction with B cells [35, 36]. In addition, activated cDC produce high levels of B lymphocyte activation and survival factors, such as BAFF and APRIL, which have a key role in B lymphocyte differentiation and antibody production [37, 38]. The role of DC in inflammation and autoimmunity has been recently reviewed [39, 40].
DC in Mouse Models of SADs
Several animal models have shown that defects in different molecules acting as “eat me” or “find me” signals are involved in the SLE, RA, SSc, and SjS phenotypes, suggesting that a defect in apoptotic cell clearance by DC and other phagocytes can be driving the autoimmune condition (reviewed in [41]).
One of the main functions of DC is antigen presentation to T cells and the control of T cell differentiation. In general, DC ablation in mice leads to increased autoreactivity [42]. In the MRL-Fas/lpr mouse, the removal of DC drives a decrease in T cell expansion and in IgG and IgM autoantibody formation [43], but in inbred strains, DC depletion results in a normal development and number of total T and Treg cells [44]. These contradictory results can be explained by the background differences or by environmental factors, pointing out the relevance of the context on DC responses.
More specifically, gene ablation of different negative regulators of the immune activation in DC results in spontaneous autoimmune and/or inflammatory manifestations. For example, ablation of B lymphocyte-induced maturation protein-1 (blimp1) in DC induces an increased production of IL-6 and a preferential differentiation of follicular T helper cells (TFH) in vitro [45]. In a similar manner, at the same line, the deletion of the myeloid a20/tnfaip3 gene results in a polyclonal immune activation, spontaneous maturation of cDC, and an increase of cytokine production, accompanied by the development of an SLE-like disease [46]. A similar phenotype was observed after the depletion of the Src homology region 2 domain-containing phosphatase-1 (SHP1). The shp1-KO animals develop splenomegaly associated with more CD11c+ DC, increased numbers of Th1 cells, and an increased expression of CD86 and CCR7 in splenic cDC [47].
In RA, the main role proposed for cDC is the control of Treg differentiation. In mouse models of RA, injection of fully mature DC loaded with collagen prevents collagen-induced arthritis (CIA) after the induction of a Th2 shift [48]. Recently, it has been shown that the injection of immature cDC before collagen immunization suppresses the development of CIA by inducing a new subset of Tregs [49]. The maturation state of cDC has also been proposed to be important in RA development. Jaen et al. evaluated in the mouse CIA model the effect of cDC administration at different maturation states. LPS-stimulated DC are more effective than plasmid-stimulated DC in preventing the development of the disease and express indoleamine-pyrrole 2,3-dioxygenase (IDO), which may explain their better therapeutic effect. In both cases, the authors report an in vivo induction of Treg cells that appear first in lymph nodes and later in the spleen [50]. Immature DC (iDC) can expand and activate a novel regulatory population of CD49b+ T cells, with high immunosuppressive potential able to mediate protection against a systemic autoimmune disease [49]. The route of injection is also crucial in these experiments because the local injection of collagen-loaded mature cDC induces a local increase in the severity of arthritis [51]; however, cDC injected intravenously are able to prevent CIA [48].
Another key role of cDC is the production of B cell survival factors (see Fig. 2). Type I IFN increases the production of BAFF and APRIL by cDC, which are involved in the survival of autoreactive B cells, and this contributes to B cell differentiation and Ig class switching, which are important for generating pathogenic autoantibodies in SLE [52,53,54]. Deletion of the type I IFN receptor (ifnar) results in an ameliorated disease in different lupus-prone strains such as NZB [55], C57BL/lpr [56], (B6.Nba2 3 NZW)F1 [57], NZM2328 [58], and MRL/lrp [59]. More specifically, irak1 depletion in B6.Sle1 results in lower numbers of B cell blasts and activated CD4+ T cells [60]. In B6.Sle3, deletion of irak1 results in significantly reduced levels of anti-ssDNA and anti-double-stranded DNA (dsDNA) antibodies and a milder kidney pathology [60]. A similar approach was used by others and the deletion of stat4 in NZW derived mice (NZW2328) results in reduced levels of IFNγ but decreased survival rates and accelerated nephritis, even though levels of antibodies were low [61].
DC in SAD Patients
Flow cytometry and histologic analyses of DC subsets have shown a trend toward a reduced number of circulating DC in SAD patients, associated with an increase in the inflamed tissues [62,63,64,65,66]. Mature infDC have also been shown to be infiltrated in the synovial fluid of RA patients [20]. Mature cDC accumulate in the perivascular region of RA patients’ synovium, in association with T and B lymphocyte aggregates. These infiltrating cDC express the CCL20 receptor CCR6, which mediates the attraction of DC and Th17 cells to the tissues [67]. This finding suggests that a local maturation process is mediating the sequestration of DC in the leukocyte aggregates in the inflamed tissue in RA patients. In a similar manner, immune cell infiltrates of minor salivary glands of SjS patients contain typically macrophages and cDC [68].
Mature DC polarize naïve T lymphocytes into Th1, Th2, Treg, or Th17 through the secretion of different sets of cytokines. The accumulation of danger signals in the inflamed tissue stimulates and drives the DC to immunogenic or tolerogenic profiles. The release of cytokines that prime an improper autoantigen presentation leads to dysregulated autoreactive T and B lymphocytes that contribute to the physiopathology of autoimmune disorders. Mature cDC producing high amounts of Il-12 and IL-23 have been reported in the infiltrates of synovial tissues of RA patients, suggesting that these cells have a role in the polarization of pathogenic T lymphocytes [62, 66, 67]. infDC from synovial fluids of RA patients induce the secretion of IL-17 in naïve CD4+ T lymphocytes through the secretion of TGFβ, IL-1β, IL-6, and IL-23 [20]. In RA, IL-17 induces chondrocytes to secrete cartilage-degrading factors, and interferes with the synthesis of the cartilage matrix through the production of nitric oxide [69], or deregulating the RANK-RANKL (receptor activator of nuclear factor kappa-B ligand) pathway of osteoclast survival and differentiation [70].
As discussed before, suboptimal clearance of dying cells results in the accumulation of cell debris in the tissues and the ensuing local release of inflammatory signals [71]. Accordingly, a gene polymorphism in the mfge8 (milk fat globule-EGF factor 8 protein) gene, involved in phagocytosis mediating the interaction between phagocytes and dying cells [72], has been associated with SLE risk [73]. After antigen uptake in the tissues, mature DC migrate to the lymph nodes to activate antigen-specific T and B lymphocytes, in a process mediated by the CCR7 receptor and the chemokines CCL19 and CCL21 [74, 75]. Memory T cell activation by cDC can also occur within the inflamed tissues [76] in structured de novo formations with a follicular organization called ectopic lymphoid structures, where B cell activation leading to local production of pathogenic autoantibodies can be induced [77, 78]. The accumulation of cDC in autoimmune sites can be a consequence of the increased expression of chemokine receptors or their specific ligands in the tissue, or alternatively due to their defective migration to the draining lymph nodes from the inflamed tissue. Both mechanisms have been described in different SADs such as RA, SLE, and SjS [67, 79,80,81,82].
Monocytes and Macrophages
Monocytes have a critical role in innate immunity, not only as precursors of tissue macrophages and infDC but also through their function as phagocytes, antigen-presenting cells, and cytokine producers. Monocytes are produced in the bone marrow from hematopoietic stem cell precursors (see Fig. 1) and circulate in the periphery before migrating into the tissues, where they differentiate into different types of macrophages and DC [83]. Three major subsets of human circulating monocytes can be recognized by their surface phenotype: the classical CD14++CD16− monocytes, the nonclassical CD14+CD16++ monocytes, and an intermediate CD14+CD16+ population, considered as a transitional state between conventional and nonclassical monocytes [84]. After microbial stimulation, nonclassical monocytes are highly activated, become strong antigen-presenting cells, and produce high amounts of proinflammatory cytokines.
Macrophages are the main resident leukocytes in most tissues, differentiated in specialized phenotypes, e. g., Kupffer cells in the liver and microglial cells in the brain. Their numbers increase massively in inflammation and autoimmune diseases where they influence the normal cell turnover and tissue remodeling, facilitating the repair of injured sites [85]. Macrophages are known for their phenotypic heterogeneity, polarization, and plasticity. When the macrophages are recruited into the tissues, they become polarized, and generally, they can be classified as M1 macrophages, which are proinflammatory, and M2 macrophages, which are regulatory [86].
Monocytes and Macrophages in Mouse Models of SADs
In the autoimmune condition, the pathogenic roles of monocytes and macrophages are mainly due to alterations of immune complex (IC) recognition and clearance, nucleic acid recognition via toll-like receptors (TLR) signaling, and IFN signaling. Monocytes and macrophages, as well as other effector cells in the immune system, express cell-surface receptors specific for the Fc region of IgG (FcγR). They show differences in affinity according to the IgG subclass [87] and have a relevant role in autoimmune diseases [88]. The deposition in the kidney of autoantibodies in the form of IC and their interaction with FcγR is thought to trigger the local inflammatory response typical of SLE, leading to glomerulonephritis. Lostor-reduced expression of FcγRIIB results in development of lupus-like symptoms in the nonautoimmune C57BL/6 strain, with presence of autoantibodies and autoimmune glomerulonephritis, but this effect seems to be strain dependent [89]. In the NZB/NZW F1 mouse strain, direct activation of FcR in monocytes/macrophages is sufficient to initiate the response to glomerular IC deposit [90]. Mice deficient in FcγR do not develop proteinuria and inflammatory responses; however, the deposits of IgG and C3 are still present in the glomerulae [91,92,93,94]. In an interesting paper, Marino et al. identified a peptide able to bind to immunoglobulins and to interfere with FcγR recognition. Administration of this peptide to MRL/lpr mice results in a remarkable increase in the survival rate. Treated mice show lower IC deposition accompanied by a significant reduction in proteinuria [95]. These data demonstrate the relevance of FcR in controlling the kidney failure present in SLE, and blocking this receptor could be an attractive alternative to treat renal failure in the disease.
Another mechanism involved in lupus nephritis is the recruitment of monocytes and neutrophils mediated by type I IFN. In an experimental model of autoantibody-induced nephritis, the production of type I IFN by resident populations in the kidney seems to be responsible for tissue damage caused by deposition of autoantibodies. Increased levels of type I IFN aggravated the renal disease, whereas inhibiting IFN-I activity results in milder symptoms [96]. In the pristane-induced lupus model, a novel population of Ly6Chigh macrophages has been described as the main producer of type I IFN independently of DC activation [97]. Ly6Chigh monocytes from the bone marrow go into the circulation and then to the peritoneal cavity where they accumulate. A striking correlation between the numbers of Ly6Chigh monocytes and the production of autoantibodies is also observed. Monocyte depletion results in a decrease in type I IFN and IFN-induced gene expression, though the systemic depletion of DC has little effect. The expression of TNFα also diminished upon CD11b+Ly6Chigh monocyte depletion, whereas the expression of IL-12 does not change significantly. These results support the possibility of production of type I IFN by immature monocytes independently of DC in lupus [97].
To elucidate the role of complement in this model, the same authors designed new experiments in two different knockouts: C1qa (BALB/C and C57BL/6 strains) and C3 (BALB/c). Surprisingly, C1qa−/− mice develop lower titers of circulating autoantibodies and milder arthritis compared with the controls. Two months after pristane injection, a decrease in the number of CD11b+Ly6Chigh monocytes in peritoneal exudates was detected in C1qa−/− mice; conversely, the number of the circulating population was higher. In vitro, peritoneal macrophages from C1qa−/− BALB/c mice injected with pristane produce less CCL3, CCL2, CXCL1, and IL-6 after TLR7 stimulation in vitro, but after stimulation of TLR3, TLR4, and TLR9, the levels of cytokines/chemokines are similar to WT animals. Deletion of other complement components, such as C3, does not affect the chemokine/cytokine production in the same conditions [98]. Based on these data, we can conclude that C1qa has an important role in the recruitment of circulating monocytes to the peritoneum in the pristine-induced lupus model.
Elevated levels of cytokines and chemokines in tissues also contribute to SLE development and can lead to renal leukocyte infiltration and tissue damage. The presence of leukocytes in renal infiltration is usually associated with poor prognosis in SLE. During experimental lupus nephritis, F4/80hi cells expressing high levels of CD11b, CD80, CD86, MMP2, MMP14, Ikkε, CXCL13, and IL-10 are a major renal source of proinflammatory cytokines and chemokines [99]. In NZB/W mice, nephritis onset is associated with a specific renal macrophage/DC signature. Renal F4/80hi/CD11cint macrophages are located throughout the interstitium, whereas F4/80lo/CD11chi DC accumulate in perivascular lymphoid aggregates. CD11b+/CD11chi/F4/80lo cells appear in large numbers in lymphoid aggregates during nephritis [99] and disappear upon remission. A new type of renal F4/80hi/CD11cint macrophage has been described in the kidney with a Gr1lo/Ly6Clo/VLA4lo/MHCIIhi/CD43lo/CD62Llo phenotype different from that described for inflammatory macrophages [100].
High levels of expression of two ligands for CCR1, CCL3, and CCL5, in association with mononuclear phagocytes and T cell infiltration, have been reported in NZB/W mice, as well as in other models of SLE and in human lupus nephritis [101,102,103]. In mouse models of lupus nephropathy, the expression of CCR1 on myeloid and some subsets of T cells seems to guide them to inflamed target organs such as the kidney. In NZB/W mice, CCR1 inhibition ameliorates the progression of lupus nephritis [104]. MRL(lpr/lpr) mice treated with the CCR1 antagonist BX471 show a reduced renal expression of CCL2, CCL3, CCL4, and CCL5 and the chemokine receptors CCR1, CCR2, and CCR5, together with reduced kidney fibrosis. However, this treatment has no effect on the levels of serum anti-dsDNA autoantibodies, proteinuria, or glomerular injury [105]. Short-term treatment with the orally available CCR1 antagonist BL5923 resulted in lower numbers of T cells and macrophages in the kidney infiltrates [104]. At longer times, CCR1 antagonist administration results in a minor kidney accumulation of effector/memory CD4+ T cells, Ly6C+ monocytes, and both M1 and M2 macrophages in MRL-lpr mice. The tissue damage is reduced resulting in a delayed proteinuria and increased survival [105]. In transference experiments done in the NZB/WF1 model, it has been reported that splenic T, B, and myeloid cells from nephritic mice migrated into noninflamed syngenic kidneys [102]. When the transfer was done to chronically inflamed kidneys, the process was improved, suggesting that this process could be autoregulated with a loop between kidney signaling and circulating leukocytes.
The deficiency on interferon regulatory factor 4 (IRF4), a transcription factor required for M2 macrophage polarization [106], also inhibits TLR signaling through its binding to MyD88 [107]. As a consequence, irf4 deficiency enhances the activation of antigen-presenting cells and the production of NF-κB-dependent proinflammatory cytokines in the lupus-prone B6lrp mice but protects the animal against IC deposition in the kidney and the resulting glomerulonephritis [108].
Macrophages are also a relevant population in the control of other SADs as RA in human and mouse models. In this disease, the most relevant role of the macrophages is the production of cytokines that control osteoclast activity (Fig. 2). Th1 cytokines, such as IL-12 and IFNγ, and Th2 cytokines, such as IL-4 and IL-10, are inhibitory for osteoclastogenesis [109,110,111,112]. It is also relevant in the production of NO by synoviocytes and macrophages that induces degeneration of chondrocytes. Other cytokines such as colony-stimulating factor 1 (CSF-1) and its receptor, CSF-1R, play an important role in regulating tissue-resident macrophages and osteoclasts. The expression of CSF1-R increases during CMP differentiation to macrophages (Fig. 1) and CSF-1R downstream signaling regulates macrophage survival, proliferation, differentiation, and chemotaxis [113]. In two different RA models, such as the CIA model and the passive serum transfer, the blockade of CSF-1R abrogated cartilage damage, bone erosion, and systemic bone loss. In both cases, this effect was associated with depletion of osteoclasts. A significant reduction in inflammation was also observed that was accompanied by the absence of synovial macrophages and a reduction of the number of splenic monocytes, pointing out the relevant role of CSF-1R in controlling these populations in RA [114].
SjS animal models show a predominance of CD4+ T lymphocytes infiltrated into lachrymal and salivary glands. The presence of macrophages in the infiltrates has been detected but their pathogenic role is not well defined yet, even though it is known that they are important players [10, 115]. In NOD mice, macrophages and DC initiate the infiltration into the salivary glands that will develop into a lymphocytic focus. Therein, M1 and M2 macrophages are detected together with B and T cells [116]. The role of macrophages in SjS pathogenesis has been investigated by Zhou et al. using a knockout mouse model for the autoimmune regulator (AIRE). These knockout mice present a multiorgan autoimmune disease, including an exocrinopathy affecting the salivary and lacrimal glands [117, 118]. In the absence of AIRE, F4/80+ macrophages accumulate in the cornea. Subconjunctival injection of clodronate liposomes depletes macrophages locally with no effect on CD11c+ DC and improves corneal epitheliopathy, hyperplasia, and stromal fibrosis. In AIRE KO mice, macrophages appear to function locally, downstream of CD4+ T cell activation and infiltration. Clodronate systemic administration does not improve the ocular epitheliopathy but results in an improvement in tear secretion and decreased damage to lachrymal glands [119]. Thus, even if CD4+ T cells are the main population in the infiltrates and primary effectors in the development of the pathogenesis, macrophages seem to have a relevant role in the development of the complete SjS phenotype. In the NOD/B10-H2 b strain after prophylactic treatment with cobra venom factor (the complement-activating protein from cobra venom that functionally resembles C3b), animals failed to develop salivary dysfunction and showed reduced levels of leukocyte infiltration, reduction of antinuclear autoantibodies, and major alterations in the B lymphocyte profiles [120]. The role of complement has also been studied in the C57BL/6.NOD-Aec1Aec2 SjS mouse model. In this case, the deletion of C3 resulted in a decrease in clinical signs. C3 KO animals presented reduced acinar cell apoptosis, reduced levels of caspase-3, lack of leukocyte infiltration of submandibular glands, and reduced synthesis of pathogenic autoantibodies. The glandular architecture and retention/secretion of saliva were normal [121]. RNA expression microarray studies have been carried out in lachrymal glands of NOD mice comparing them with age-matched BALB/c mice. The results showed an upregulation of cathepsins and proinflammatory factors including TNFα, IL-6 and IL-1β [122]. In C57BL/6.NOD-Aec1Aec2 mice, caspase-11, expressed primarily in macrophages and DCs, was significantly upregulated at 8 weeks of age, but not caspase-9 [123]. The upregulation of caspase-11 in the submandibular gland before disease onset is apparently associated with the enhanced transcriptional activity of the signal transducer and activator of transcription 1 (STAT1) gene [124]. In general, the presence of elevated levels of proinflammatory cytokines in the submandibular gland enhances IFNγ production by epithelial cells, resulting in further activation of macrophages [124].
It is also worthy to mention the role of macrophages in SSc. Macrophages are a potent source of reactive oxygen species (ROS). ROS have multiple effects including DNA oxidative damage and unbalanced oxidative stress, which has been implicated in the pathogenesis of scleroderma. There are increased numbers of macrophages at the early stages of fibrosis, and they release proinflammatory and fibrogenic mediators, such as TGFβ and PDGF [125]. A high number of macrophages have been detected in the skin of Scl-GVHD [126] and bleomycin-induced mouse models [127]. In the bleomycin model, TGFβ is produced by fibroblasts and infiltrating cells that are predominantly comprised of macrophages at the sclerotic stage [128]. CCL2 and its receptor CCR2 are upregulated in dermal fibroblasts and inflammatory cells from both SSc bleomycin-treated mice as it happens in patients [129]. In the CCL2-deficient mice, skin fibrosis was diminished even after the bleomycin treatment [130]. In the Scl-GVHD model, populations of monocytes/macrophages (CD11b+/2F8+) and CD3+ T cells of donor origin are the main components of the skin lesion. There are also high levels of CCL2, IFN-inducible chemokines, VEGF, and adhesion molecules in the skin [131].
Monocytes and Macrophages in SAD Patients
One of the major functions of blood monocytes is the elimination of opsonized microorganisms and apoptotic debris by phagocytosis or receptor-specific endocytosis through pattern-recognition receptors (PRR) [132]. Among them, C1q mediates the recognition of a wide variety of plasma proteins and pathogen molecules [133, 134], ensuring uneventful removal. It has been shown that the deficiency in the C1q protein is associated with a risk of developing SLE and RA [135, 136], suggesting that the role of C1q in the clearance of microbial elements and self debris could be crucial for preventing the induction of pathogenic autoantibodies [137]. Circulating monocytes from SjS patients release spontaneously higher amounts of the two B lymphocyte-stimulating cytokines IL-6 and BAFF [138] and show high levels of phosphorylation of STAT5, correlating with serum IgG levels and anti-SSB/La autoantibody titers [139]. However, they show an impaired capacity of phagocytosis of apoptotic cells [140].
A significant role for monocyte activation in PAPS-mediated thrombogenesis has been suggested. From a proteomics analysis of monocytes from PAPS patients with thrombosis, a differential expression of annexin I and annexin II, as well as RhoA, Nedd8, and Hsp60 proteins, has been observed [141]. Circulating antibodies and autoantibodies from PAPS patients activate monocytes through TLR2 and CD14, inducing the expression of ROS and the secretion of tissue factors [142, 143]. They also induce the overexpression of TLR8 and its translocation from the endoplasmic reticulum to the endosomal compartment, sensitizing monocytes to TLR8 ligands [144].
SSc patients have a higher proportion of CD14+ monocytes in the blood, showing an activated phenotype [145]. These activated monocytes overexpress both CD169, a macrophage marker induced by type I IFN, and CD204, a marker for activated profibrotic M2 macrophages [146]. Moreover, LPS stimulation of SSc circulating monocytes increases CD163 expression compared to monocytes from control individuals [147].
SLE macrophages are unable to clear efficiently apoptotic cells and show an altered proinflammatory status characterized by an overproduction of inflammatory cytokines, such as type I IFN, TNFα, and IL-6 [148, 149]. They show enhanced antigen presentation capacity and are primed for activation, leading to a skew toward autoimmunity [150]. In this inflammatory context, SLE monocytes and macrophages present self-antigens to autoreactive T lymphocytes instead of inducing peripheral tolerance after phagocytosis of apoptotic cells [148]. Interestingly, CD68+ mononuclear phagocyte infiltration in the kidneys of lupus nephritis patients is associated with poor prognosis [151,152,153].
Macrophages produce many proinflammatory cytokines and chemokines in the synovial tissue of RA patients, contributing to cartilage and bone destruction [154]. Indeed, infiltrating macrophage numbers constitute a biomarker for disease severity, as well as a predictor of the response to therapy [155]. It has been reported that there is a positive correlation between the number of infiltrated macrophages and the degree of joint erosion [156]. The polarization of RA synovial tissue macrophages depends on the stage of the rheumatic inflammation. Actually, patients with highly active RA show a prevalence of the M1 phenotype. On the contrary, macrophages of RA patients with low disease score or in clinical remission show an M2 phenotype [157], and furthermore, glucocorticoid treatment induces an M2 state [158]. The infiltration of macrophages into the labial salivary glands of SjS patients correlates with the biopsy focus score [159]. Additionally, patients with SjS show higher expression of IL-18 in the infiltrated macrophages, with a positive correlation with salivary gland enlargement [68].
Skin infiltrates of SSc patients are composed mainly of T lymphocytes and macrophages. Among them, infiltrated CD163+ macrophages seem to be the main source of CCL19, a chemokine strongly correlated with vascular markers, suggesting a role of CCL19 in the recruitment of macrophages to the inflamed SSc skin [160]. In terms of phenotype, it has been reported that there is overexpression of TLR4, CD14, and MD2 in the skin of diffuse SSc patients. The expression of these genes correlates with progressive skin disease [161], suggesting that these markers can be used for the monitoring of skin disease progression. CD14 is mainly expressed by macrophages, although it can also be expressed at lower levels by cDC and neutrophils. In addition, dermal macrophages of SSc patients acquire a profibrotic phenotype after stimulation with IL-13 [162, 163]. Microarray analysis of lung samples of patients of SSc-associated interstitial lung disease shows a unique gene signature compared with other similar lung diseases. Many genes of this specific signature correspond to alveolar macrophage activation and fibrosis [164, 165]. Studies of expression profiles of bronchoalveolar lavages of SSc patients with lung inflammation describe the induction of markers of alveolar macrophage activation [166], together with a consistent increase in the expression of CCL18 transcripts [167]. This result is in agreement with the high levels of serum CCL18 in SSc patients, related with lung involvement [168,169,170]. In addition, it has been shown that circulating monocytes and alveolar macrophages of SSc patients of interstitial lung disease responded more intensely to LPS stimulation [147, 166].
Neutrophils
Neutrophils have key roles in the control of infectious agents through their capability to quickly migrate from the circulation to the infected tissues in response to regulatory or chemotactic signals [171]. Once they arrive at these sites, they turn into “primed” neutrophils, recognizing and destroying the invading pathogens using a wide variety of degrading enzymes contained in their granules, in addition to their ability to generate ROS [172]. Using these arms, neutrophils have the highest killing activity among the immune cells. Primed neutrophils extend their lifespan and promote inflammation using chemokines and cytokines that attract other actors of the immune system, and regulating almost every element of the inflammatory response [173]. When the infection is resolved, they die by apoptosis [174] or NETosis, through the release toward the extracellular milieu of granule-derived protein-decorated chromatin forming neutrophil extracellular traps (NETs) [175, 176]. High titers of autoantibodies against dsDNA, histones, or anti-citrullinated protein antibodies (ACPA) are hallmarks of SAD patients. Since the proteins associated with the DNA in the NETs include citrullinated histones and proteins with altered immunogenicity after posttranslational modifications such as oxidations, neutrophils can also be a source of autoantigens through degranulation or NETosis [177,178,179].
In some conditions, neutrophils can also infiltrate tissues and become improperly activated in sterile tissues via deposed IC [180], and secrete the content of their granules, attacking host tissues if local detoxification pathways become overburdened. As a consequence, the connective tissues are dissolved and normal cells destroyed [181]. Besides their direct tissue damage induction, neutrophil-derived regulatory factors also organize a sterile inflammatory response [182]. Neutrophil-secreted cytokines have been shown to contribute to the deregulation of the immune responses in several SADs [173]. Other neutrophil functions have been shown to be deregulated in several SADs as shown in Table 1 and detailed below.
Neutrophils in Mouse Models of SADs
Strong evidence about the important role of neutrophils in SLE pathogenesis comes from in vivo depletion experiments [184]. The depletion of the neutrophil population in lupus-prone autoimmune B6.Faslpr/JTnfrsf17−/− mice, deficient in a BAFF receptor, results in a reduction in autoantibody titers, serum IFNα and BAFF, T cell activation, as well as high numbers of splenic germinal center B cells and plasma cells. In this strain, high production of BAFF by neutrophils may help to drive the selection and survival of autoimmune B cell clones that produce self-reactive antibodies, such as anti-dsDNA antibodies [184]. Interaction between BAFF, T cells, and IFNγ has also been proposed in the Lyn-deficient autoimmune mouse model. Lyn−/− mice present a ∼30–50% reduction in mature B cell numbers, and lyn −/− myeloid cells are hyperresponsive to engagement of surface integrins, showing increased secondary granule release [205]. Scapini et al. have described a population of hyperactivated myeloid cells in these animals that produces high levels of BAFF, that activates T cells to release high levels of IFNγ. Administration of anti-BAFF monoclonal antibody reduced disease development in Lyn−/− mice and a similar effect was observed with the genetic deletion of IFNγ [185].
Other important mechanism by which neutrophils drive autoimmune responses is through the release of ROS, proteases, and proinflammatory cytokines (Fig. 2). In the MRL/lpr mouse, blockade of mitochondrial ROS production has recently been reported to be sufficient to block NETosis in vitro, reducing disease severity and type I IFN responses [186]. The KO of the NADPH oxidase nox results in increased lupus disease symptoms in this particular strain [206]. However, the role of NOX in immune responses is not all clear since in patients with chronic granulomatous disease and lack of functional NOX, a proinflammatory phenotype has been observed [207].
The relevance of NET formation in lupus disease has been tested recently in animal models. Injection of netting neutrophil cell lines to wild-type mice does not result in the development of lupus disease. Although it is unclear if those injections could mimic the in vivo NET formation and prime the activation of TLR signaling, IgG and IgM antibody levels were increased [187]. Another key process in NET formation in vivo is the citrullination of histones by peptidyl arginine deiminase 4 (PAD4). Neutrophils from MRL/lpr and NZ2328 mice demonstrate accelerated NET formation compared with controls and an accelerated NET formation [188, 189]. In MRL/lpr inhibition of PAD1, PAD2, and PAD4 using the Cl-amidine inhibitor markedly improves endothelial function and reduces proteinuria and IC deposition in the kidney while protecting against skin disease [188]. In NZM, the same treatment inhibits NET formation in vivo and significantly alters circulating autoantibody profiles and complement levels while reducing glomerular IgG deposition [189].
In RA, neutrophils also have a critical role in the initiation and maintenance of the disease. Neutrophils are abundant in murine autoimmune arthritis and contribute to the pathogenesis through the release of cytotoxic products and immunoregulatory mediators. In addition, neutrophils may promote autoimmunity by formation of NETs and the associated promotion of anticitrullinated protein/peptide antibodies [182, 208]. Interestingly, neutrophil-depleted mice are completely resistant to the disease-inducing effects of K/BxN serum transfer [209]. In CIA models, it has also been proven that they have a critical role in initiating and maintaining the inflammatory responses. In a similar way to what happens in patients, there is also a prominence of neutrophil recruitment in RA models [209,210,211]. It has been demonstrated that neutrophils participate in their own recruitment in murine arthritis through C5aR and FcγR signaling [212]. Once in the joints, neutrophil activation by IC promotes IL-1β production, which stimulates synovial cells to produce chemokines, amplifying the neutrophil recruitment into the joints [212]. Neutrophils infiltrating the synovial membranes and joints in rats with arthritis upregulate cathelicidins [213], antibacterial peptides with potent proinflammatory and immunomodulatory activities [214].
Recent evidence indicates that the inflammatory loops initiated by the molecules externalized in NETs may be key in arthritis development [215]. As it was mentioned before, citrullination of histones by PAD4 is a key step in NET formation. PAD4 mRNA, absent from healthy synovium, is transcribed and translated by neutrophils infiltrating synovial tissue during inflammation. As a consequence, several synovial proteins are citrullinated in this compartment [216]. Of interest, the PAD inhibitor CI-amidine mitigates collagen-induced arthritis and decreases the clinical disease score [190]. However, no abrogation of disease severity was observed in the PAD4 knockout mice using the K/BxN serum transfer model of arthritis [217].
Granulocyte-colony stimulating factor (G-CSF) has also been found to be a key player in arthritis models, participating in the interactions between hematopoietic cells through the control of myeloid cell numbers and activation [218]. Neutrophil depletion or reduction of their G-CSF production also inhibits disease development in the CIA arthritis model [191]. This cytokine is also required for neutrophil recruitment in the K/BxN serum transfer arthritis model [192]. In this line of research, antibody blockade or knockout of key neutrophil signaling receptors, such as CXCR1 and CXCR2, ameliorates disease signs in an antigen-induced model [193, 194]. A similar effect of reduction in disease progression was observed in the CIA model with ablation of C5aR [195], involved in neutrophil recruitment [212]. In the K/BxN serum transfer-induced arthritis model, an important role of FcγR has also been demonstrated. Expression of human FcγRIIa on neutrophils in mice that lacked their own results in the restoration of susceptibility to K/BxN serum induced RA, neutrophil recruitment, synovitis, and bone destruction [219]. Other molecules involved in the recruitment of pathogenic neutrophils are L-selectin [196], IFNγ, [197, 198], and IL-17 [199]. Neutrophil production of IL-17 has also been pointed to as an amplifier of arthritis in the K/BxN model [200]. Neutrophil activation following recognition of early IC in the joint may also lead to changes in vascular permeability, which further promotes IgG deposition [220]. The production of other effectors, such as ROS, is also important for the induction of the disease. In summary, we can conclude that decreased disease activity and joint destruction directly correlates with lower influx of neutrophils to joints and less neutrophil activity.
Production of ROS by neutrophils seems to also be a concern in SSc pathogenesis [203]. Repeated injections of hypochlorous acid, a product of neutrophil burst, induced skin and lung fibrosis as well as anti-DNA topoisomerase 1 (Scl70) antibody production, mimicking the diffuse form of SSc in patients and proving the relevant role of ROS in SSc [203, 204].
Neutrophils in SAD Patients
Neutropenia is found in a significant proportion of SLE patients as reported in [221, 222]. Circulating neutrophils of SLE patients display abnormal features, such as impaired phagocytic activity [223] and lower recognition by the C1q-mediated apoptotic cell clearance [224]. The lower production of ROS by circulating neutrophils from SLE patients with more severe symptoms indicates that these cells are not primed but show a skewed phenotype [225]. On the other hand, enriched numbers of low density granulocytes (LDG), with an activated phenotype but morphologically similar to immature cells, are characteristic in the blood of these patients [226, 227]. The number of circulating LDG correlates with dsDNA-specific autoantibody titers and disease severity [228]. The enhancement of NETosis activity in LDG and neutrophils from SLE patients is well established. It has been hypothesized that neutrophil death induces the type I IFN production by pDC characteristic of SLE [229], facilitating the uptake of extracellular DNA by pDC and their activation [178]. IFN and IC trigger the activation of neutrophils, inducing again their NETosis in a self-amplifying process [230]. Several studies have reported the finding of neutrophils in the kidney biopsies of lupus nephritis patients [230,231,232,233], and tissue NETosis has been correlated with higher titers of anti-dsDNA autoantibodies [230]. Altogether, these data support the notion of neutrophils having a key role in the pathogenesis of SLE [234].
In healthy individuals, blood neutrophils need to be primed in order to migrate to the tissues and become active. In RA patients, circulating and infiltrated neutrophils have a longer lifespan [235] and show an activated phenotype [236], together with activation of the NF-κB pathway [237], increase of their chemotactic capacity [238,239,240], high phagocytic activity [241], and enhanced ROS production [242] in response to IC [243]. This phenotype participates actively in the damage of the synovial joints [182]. The process consisting in the adherence of activated neutrophils to IC in the synovial fluid, causing degranulation and liberation of ROS and collagenases [244], has been called “frustrated phagocytosis” [245]. The phenotype of synovial neutrophils of RA patients is quite similar to tissue macrophages, in terms of secretion of a wide variety of proinflammatory cytokines and chemokines [174], thus facilitating the delay in the neutrophil apoptosis induction. Recent reports suggest a role of neutrophil NETosis in the joint damage in RA [182], since anti-citrullinated protein antibodies (ACPA) are characteristic of erosive RA, and NETs contain citrullinated histones [179]. Spontaneous NETosis of neutrophils in culture is higher in RA compared with controls, and they have more nuclear citrullinated histone H3 [246]. Interestingly, antibodies specific for citrullinated vimentin are associated with the severity of RA [247]. Moreover, neutrophils from healthy donors bearing the T allele of the RA risk-associated gene ptpn22(C1858T) have a high migration capacity, superior ROS production, and enhanced NET release [248], indicating that the neutrophils could be acting in the very first steps of the pathogenic processes of the disease.
The information about the role of neutrophils in the physiopathology of other SADs is less complete compared to SLE and RA. For instance, there are some pieces of evidence pointing to a role of NETosis in the pathogenesis of PAPS. Similar to SLE, the sera of PAPS patients show a decrease in the NET-degrading activity [249]. In line with this finding, the sera of these patients also have high levels of cell-free DNA and NET components, and the circulating neutrophils have high spontaneous NETosis activity [250]. A LDG population has been also described in the blood of PAPS patients [251]. Other neutrophil functions, such as ROS generation, are skewed in neutrophils of SSc patients [252, 253].
Concluding Remarks
As summarized in Table 1, scientific evidence pointing to a key role of the myeloid cells in the pathogenesis of SADs is abundant and strong. Dendritic cell alterations in immune diseases include presentation of self-antigens to autoreactive T cells, increased secretion of proinflammatory cytokines, and promotion of autoantibody production in B cells. DC also act through the control of T cell differentiation and activation. Monocytes and macrophages are important cytokine producers able to control migration of other populations to the inflamed tissues and remodeling processes such as condrogenesis and osteoclast activity. They respond to the IC deposit and produce proinflammatory cytokines and chemokines participating in tissue damage in SADs. Finally, neutrophils release ROS, proteases, and proinflammatory cytokines that act as danger signals. Netting neutrophils release intracellular modified antigens promoting the induction of pathogenic autoantibodies.
According to these data, our challenge in the next few years is to better dissect the immunopathological mechanisms underlying these disturbances in order to define specific cell subsets or proteins that can be potential targets for drug development.
cDC conventional dendritic cells, CIA collagen-induced arthritis, DC dendritic cells, IC immune complex, infDC inflammatory dendritic cells, IFN interferon, pDC plasmacytoid dendritic cells, RA rheumatoid arthritis, ROS reactive oxygen species, SADs systemic autoimmune diseases, SjS Sjögren’s syndrome, SLE systemic lupus erythematosus, SSc systemic sclerosis
References
Selmi C (2016) Autoimmunity in 2015. Clinical Reviews in Allergy & Immunology 51(1):110–119. doi:10.1007/s12016-016-8576-1
Carroll M (2001) Innate immunity in the etiopathology of autoimmunity. Nat Immunol 2(12):1089–1090. doi:10.1038/ni1201-1089
Wu T, Ding H, Han J, Arriens C, Wei C, Han W, Pedroza C, Jiang S, Anolik J, Petri M, Sanz I, Saxena R, Mohan C (2016) Antibody-array-based proteomic screening of serum markers in systemic lupus erythematosus: a discovery study. J Proteome Res 15(7):2102–2114. doi:10.1021/acs.jproteome.5b00905
Suurmond J, Diamond B (2015) Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125(6):2194–2202. doi:10.1172/JCI78084
Gregersen PK, Behrens TW (2006) Genetics of autoimmune diseases—disorders of immune homeostasis. Nat Rev Genet 7 (12):917–928. doi:10.1038/nrg1944
Marrack P, Kappler J, Kotzin BL (2001) Autoimmune disease: why and where it occurs. Nat Med 7(8):899–905. doi:10.1038/9093590935
Wooley PH (2004) The usefulness and the limitations of animal models in identifying targets for therapy in arthritis. Best Pract Res Clin Rheumatol 18(1):47–58. doi:10.1016/j.berh.2003.09.007
Perry D, Sang A, Yin Y, Zheng YY, Morel L (2011) Murine models of systemic lupus erythematosus. J Biomed Biotechnol 2011:271694. doi:10.1155/2011/271694
Beyer C, Schett G, Distler O, Distler JH (2010) Animal models of systemic sclerosis: prospects and limitations. Arthritis Rheum 62(10):2831–2844. doi:10.1002/art.27647
Donate A, Voigt A, Nguyen CQ (2014) The value of animal models to study immunopathology of primary human Sjogren’s syndrome symptoms. Expert Rev Clin Immunol 10(4):469–481. doi:10.1586/1744666X.2014.883920
Hanh B (2005) Harrison’s principles of internal medicine. Systemic lupus erythematosus, New York City
Marshak-Rothstein A (2006) Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 6(11):823–835. doi:10.1038/nri1957
Theofilopoulos AN, Baccala R, Beutler B, Kono DH (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23:307–336
Waldner H (2009) The role of innate immune responses in autoimmune disease development. Autoimmun Rev (8, 5):400–404. doi:10.1016/j.autrev.2008.12.019
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12(4):253–268. doi:10.1038/nri3175
Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S (2014) Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14(8):571–578. doi:10.1038/nri3712
Swiecki M, Colonna M (2015) The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15(8):471–485. doi:10.1038/nri3865
Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ (1999) The nature of the principal type 1 interferon-producing cells in human blood. Science 284(5421):1835–1837
Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M (2010) The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8{alpha}+ dendritic cells. J Exp Med 207(6):1283–1292
Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, Dalod M, Soumelis V, Amigorena S (2013) Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 38(2):336–348. doi:10.1016/j.immuni.2012.10.018
Crozat K, Guiton R, Guilliams M, Henri S, Baranek T, Schwartz-Cornil I, Malissen B, Dalod M (2010) Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunol Rev 234(1):177–198. doi:10.1111/j.0105-2896.2009.00868.x
Waskow C, Liu K, Darrasse-Jeze G, Guermonprez P, Ginhoux F, Merad M, Shengelia T, Yao K, Nussenzweig M (2008) The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol 9(6):676–683. doi:10.1038/ni.1615
Steinman RM, Idoyaga J (2010) Features of the dendritic cell lineage. Immunol Rev 234(1):5–17. doi:10.1111/j.0105-2896.2009.00888.x
Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, Hashimoto D, Chow A, Price J, Greter M, Bogunovic M, Bellemare-Pelletier A, Frenette PS, Randolph GJ, Turley SJ, Merad M, Immunological Genome C (2012) Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol 13(9):888–899. doi:10.1038/ni.2370
Steinman RM, Inaba K, Turley S, Pierre P, Mellman I (1999) Antigen capture, processing, and presentation by dendritic cells: recent cell biological studies. Hum Immunol 60(7):562–567
Maranon C, Desoutter JF, Hoeffel G, Cohen W, Hanau D, Hosmalin A (2004) Dendritic cells cross-present HIV antigens from live as well as apoptotic infected CD4+ T lymphocytes. Proc Natl Acad Sci U S A101(16):6092–6097
Steinman RM, Hawiger D, Liu K, Bonifaz L, Bonnyay D, Mahnke K, Iyoda T, Ravetch J, Dhodapkar M, Inaba K, Nussenzweig M (2003) Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci 987:15–25
Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM (2002) Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med 196(8):1091–1097
Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, Wang Z, Watkins SC, Falo LD Jr, Thomson AW (2003) Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood 101(2):611–620. doi:10.1182/blood-2002-06-1769
Matzinger P (2002) The danger model: a renewed sense of self. Science 296(5566):301–305. doi:10.1126/science.1071059
Valente M, Baey C, Louche P, Dutertre CA, Vimeux L, Maranon C, Hosmalin A, Feuillet V (2014) Apoptotic cell capture by DCs induces unexpectedly robust autologous CD4+ T-cell responses. Eur J Immunol 44(8):2274–2286. doi:10.1002/eji.201344191
Reis e Sousa C (2006) Dendritic cells in a mature age. Nat Rev Immunol 6(6):476–483. doi:10.1038/nri1845
Steinman RM, Hawiger D, Nussenzweig MC (2003) Tolerogenic dendritic cells. Annu Rev Immunol 21:685–711
Ueno H, Schmitt N, Palucka AK, Banchereau J (2010) Dendritic cells and humoral immunity in humans. Immunol Cell Biol 88(4):376–380. doi:10.1038/icb.2010.28
Jego G, Pascual V, Palucka AK, Banchereau J (2005) Dendritic cells control B cell growth and differentiation. Curr Dir Autoimmun 8:124–139. doi:10.1159/000082101
Qi H, Egen JG, Huang AY, Germain RN (2006) Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science 312(5780):1672–1676. doi:10.1126/science.1125703
MacLennan I, Vinuesa C (2002) Dendritic cells, BAFF, and APRIL: innate players in adaptive antibody responses. Immunity 17(3):235–238
Kalled SL, Ambrose C, Hsu YM (2005) The biochemistry and biology of BAFF, APRIL and their receptors. Curr Dir Autoimmun 8:206–242. doi:10.1159/000082105
Segura E, Amigorena S (2013) Inflammatory dendritic cells in mice and humans. Trends Immunol 34(9):440–445. doi:10.1016/j.it.2013.06.001
Ganguly D, Haak S, Sisirak V, Reizis B (2013) The role of dendritic cells in autoimmunity. Nat Rev Immunol 13(8):566–577. doi:10.1038/nri3477
Green DR, Oguin TH, Martinez J (2016) The clearance of dying cells: table for two. Cell Death Differ 23(6):915–926. doi:10.1038/cdd.2015.172
Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D (2009) Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 206(3):549–559. doi:10.1084/jem.20082394
Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ (2010) Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity 33(6):967–978. doi:10.1016/j.immuni.2010.11.025
Birnberg T, Bar-On L, Sapoznikov A, Caton ML, Cervantes-Barragan L, Makia D, Krauthgamer R, Brenner O, Ludewig B, Brockschnieder D, Riethmacher D, Reizis B, Jung S (2008) Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity 29(6):986–997. doi:10.1016/j.immuni.2008.10.012
Kim SJ, Zou YR, Goldstein J, Reizis B, Diamond B (2011) Tolerogenic function of Blimp-1 in dendritic cells. J Exp Med 208(11):2193–2199. doi:10.1084/jem.20110658
Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, van Praet J, Branco-Madeira F, Janssens S, Reizis B, Elewaut D, Beyaert R, Hammad H, Lambrecht BN (2011) The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 35(1):82–96. doi:10.1016/j.immuni.2011.05.013
Kaneko T, Saito Y, Kotani T, Okazawa H, Iwamura H, Sato-Hashimoto M, Kanazawa Y, Takahashi S, Hiromura K, Kusakari S, Kaneko Y, Murata Y, Ohnishi H, Nojima Y, Takagishi K, Matozaki T (2012) Dendritic cell-specific ablation of the protein tyrosine phosphatase Shp1 promotes Th1 cell differentiation and induces autoimmunity. J Immunol 188(11):5397–5407. doi:10.4049/jimmunol.1103210
van Duivenvoorde LM, Louis-Plence P, Apparailly F, van der Voort EI, Huizinga TW, Jorgensen C, Toes RE (2004) Antigen-specific immunomodulation of collagen-induced arthritis with tumor necrosis factor-stimulated dendritic cells. Arthritis Rheum 50(10):3354–3364. doi:10.1002/art.20513
Charbonnier LM, van Duivenvoorde LM, Apparailly F, Cantos C, Han WG, Noel D, Duperray C, Huizinga TW, Toes RE, Jorgensen C, Louis-Plence P (2006) Immature dendritic cells suppress collagen-induced arthritis by in vivo expansion of CD49b+ regulatory T cells. J Immunol 177(6):3806–3813
Jaen O, Rulle S, Bessis N, Zago A, Boissier MC, Falgarone G (2009) Dendritic cells modulated by innate immunity improve collagen-induced arthritis and induce regulatory T cells in vivo. Immunology 126(1):35–44. doi:10.1111/j.1365-2567.2008.02875.x
Leung BP, Conacher M, Hunter D, McInnes IB, Liew FY, Brewer JM (2002) A novel dendritic cell-induced model of erosive inflammatory arthritis: distinct roles for dendritic cells in T cell activation and induction of local inflammation. J Immunol 169(12):7071–7077
Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J (2003) Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19(2):225–234
Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF (2001) Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14(4):461–470
Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, Rifkin IR (2007) Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol 178(11):6876–6885
Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulos AN (2003) Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med 197(6):777–788. doi:10.1084/jem.20021996
Braun D, Geraldes P, Demengeot J (2003) Type I interferon controls the onset and severity of autoimmune manifestations in lpr mice. J Autoimmun 20(1):15–25
Jorgensen TN, Roper E, Thurman JM, Marrack P, Kotzin BL (2007) Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 x NZW)F(1) mice. Genes Immun 8(8):653–662. doi:10.1038/sj.gene.6364430
Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Koss MN, Stohl W, Kovats S, Jacob CO (2009) Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol 183(9):6021–6029. doi:10.4049/jimmunol.0803872
Schwarting A, Wada T, Kinoshita K, Tesch G, Kelley VR (1998) IFN-gamma receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol 161(1):494–503
Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, Thomas JA, Reiff A, Myones BL, Ojwang JO, Kaufman KM, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Silverman E, Ziegler J, Kelly JA, Merrill JT, Harley JB, Ramsey-Goldman R, Vila LM, Bae SC, Vyse TJ, Gilkeson GS, Gaffney PM, Moser KL, Langefeld CD, Zidovetzki R, Mohan C (2009) Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A106(15):6256–6261. doi:10.1073/pnas.0901181106
Jacob CO, Zang S, Li L, Ciobanu V, Quismorio F, Mizutani A, Satoh M, Koss M (2003) Pivotal role of Stat4 and Stat6 in the pathogenesis of the lupus-like disease in the New Zealand mixed 2328 mice. J Immunol 171(3):1564–1571
Lebre MC, Jongbloed SL, Tas SW, Smeets TJ, McInnes IB, Tak PP (2008) Rheumatoid arthritis synovium contains two subsets of CD83-DC-LAMP- dendritic cells with distinct cytokine profiles. Am J Pathol 172(4):940–950. doi:10.2353/ajpath.2008.070703
Gill MA, Blanco P, Arce E, Pascual V, Banchereau J, Palucka AK (2002) Blood dendritic cells and DC-poietins in systemic lupus erythematosus. Hum Immunol 63(12):1172–1180
Jin O, Kavikondala S, Sun L, Fu R, Mok MY, Chan A, Yeung J, Lau CS (2008) Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus 17(7):654–662. doi:10.1177/0961203308089410
Migita K, Miyashita T, Maeda Y, Kimura H, Nakamura M, Yatsuhashi H, Ishibashi H, Eguchi K (2005) Reduced blood BDCA-2+ (lymphoid) and CD11c+ (myeloid) dendritic cells in systemic lupus erythematosus. Clin Exp Immunol 142(1):84–91. doi:10.1111/j.1365-2249.2005.02897.x
Jongbloed SL, Lebre MC, Fraser AR, Gracie JA, Sturrock RD, Tak PP, McInnes IB (2006) Enumeration and phenotypical analysis of distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthritis Res Ther 8(1):R15. doi:10.1186/ar1864
Page G, Miossec P (2004) Paired synovium and lymph nodes from rheumatoid arthritis patients differ in dendritic cell and chemokine expression. J Pathol 204(1):28–38. doi:10.1002/path.1607
Manoussakis MN, Boiu S, Korkolopoulou P, Kapsogeorgou EK, Kavantzas N, Ziakas P, Patsouris E, Moutsopoulos HM (2007) Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum 56 (12):3977–3988. doi:10.1002/art.23073
Benderdour M, Tardif G, Pelletier JP, Di Battista JA, Reboul P, Ranger P, Martel-Pelletier J (2002) Interleukin 17 (IL-17) induces collagenase-3 production in human osteoarthritic chondrocytes via AP-1 dependent activation: differential activation of AP-1 members by IL-17 and IL-1beta. J Rheumatol 29(6):1262–1272
Takayanagi H (2009) Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol 5(12):667–676. doi:10.1038/nrrheum.2009.217
Green DR, Ferguson T, Zitvogel L, Kroemer G (2009) Immunogenic and tolerogenic cell death. Nat Rev Immunol 9 (5):353–363. doi:10.1038/nri2545
Baghdadi M, Chiba S, Yamashina T, Yoshiyama H, Jinushi M (2012) MFG-E8 regulates the immunogenic potential of dendritic cells primed with necrotic cell-mediated inflammatory signals. PLoS One 7(6):e39607. doi:10.1371/journal.pone.0039607
Hu CY, Wu CS, Tsai HF, Chang SK, Tsai WI, Hsu PN (2009) Genetic polymorphism in milk fat globule-EGF factor 8 (MFG-E8) is associated with systemic lupus erythematosus in human. Lupus 18(8):676–681. doi:10.1177/0961203309103027
MartIn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F (2003) Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 198(4):615–621. doi:10.1084/jem.20030448
Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Forster R (2004) CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21(2):279–288. doi:10.1016/j.immuni.2004.06.014
Wakim LM, Waithman J, Van Rooijen N, Heath WR, Carbone FR (2008) Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 319(5860):198–202. doi:10.1126/science.1151869
Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, Spencer J, Pitzalis C (2009) Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med 6(1):e1. doi:10.1371/journal.pmed.0060001
Manzo A, Bombardieri M, Humby F, Pitzalis C (2010) Secondary and ectopic lymphoid tissue responses in rheumatoid arthritis: from inflammation to autoimmunity and tissue damage/remodeling. Immunol Rev 233(1):267–285. doi:10.1111/j.0105-2896.2009.00861.x
Tucci M, Ciavarella S, Strippoli S, Dammacco F, Silvestris F (2009) Oversecretion of cytokines and chemokines in lupus nephritis is regulated by intraparenchymal dendritic cells: a review. Ann N Y Acad Sci 1173:449–457. doi:10.1111/j.1749-6632.2009.04805.x
Page G, Lebecque S, Miossec P (2002) Anatomic localization of immature and mature dendritic cells in an ectopic lymphoid organ: correlation with selective chemokine expression in rheumatoid synovium. J Immunol 168(10):5333–5341
Noort AR, van Zoest KP, van Baarsen LG, Maracle CX, Helder B, Papazian N, Romera-Hernandez M, Tak PP, Cupedo T, Tas SW (2015) Tertiary lymphoid structures in rheumatoid arthritis: NF-kappaB-inducing kinase-positive endothelial cells as central players. Am J Pathol 185(7):1935–1943. doi:10.1016/j.ajpath.2015.03.012
Aziz KE, McCluskey PJ, Wakefield D (1997) Characterisation of follicular dendritic cells in labial salivary glands of patients with primary Sjogren syndrome: comparison with tonsillar lymphoid follicles. Ann Rheum Dis 56(2):140–143
Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ (2009) Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325(5940):612–616. doi:10.1126/science.1175202
Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB (2010) Nomenclature of monocytes and dendritic cells in blood. Blood 116(16):e74–e80. doi:10.1182/blood-2010-02-258558
Laria A, Lurati A, Marrazza M, Mazzocchi D, Re KA, Scarpellini M (2016) The macrophages in rheumatic diseases. J Inflamm Res 9:1–11. doi:10.2147/JIR.S82320
Cassetta L, Cassol E, Poli G (2011) Macrophage polarization in health and disease. Scientific World Journal 11:2391–2402. doi:10.1100/2011/213962
Ravetch JV, Bolland S (2001) IgG Fc receptors. Annu Rev Immunol 19:275–290. doi:10.1146/annurev.immunol.19.1.275
Takai T (2002) Roles of Fc receptors in autoimmunity. Nat Rev Immunol 2(8):580–592. doi:10.1038/nri856
Bolland S, Ravetch JV (2000) Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 13(2):277–285
Bergtold A, Gavhane A, D’Agati V, Madaio M, Clynes R (2006) FcR-bearing myeloid cells are responsible for triggering murine lupus nephritis. J Immunol 177(10):7287–7295
Clynes R, Dumitru C, Ravetch JV (1998) Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 279(5353):1052–1054
Suzuki Y, Shirato I, Okumura K, Ravetch JV, Takai T, Tomino Y, Ra C (1998) Distinct contribution of Fc receptors and angiotensin II-dependent pathways in anti-GBM glomerulonephritis. Kidney Int 54(4):1166–1174. doi:10.1046/j.1523-1755.1998.00108.x
Park SY, Ueda S, Ohno H, Hamano Y, Tanaka M, Shiratori T, Yamazaki T, Arase H, Arase N, Karasawa A, Sato S, Ledermann B, Kondo Y, Okumura K, Ra C, Saito T (1998) Resistance of Fc receptor-deficient mice to fatal glomerulonephritis. J Clin Invest 102(6):1229–1238. doi:10.1172/JCI3256
Wakayama H, Hasegawa Y, Kawabe T, Hara T, Matsuo S, Mizuno M, Takai T, Kikutani H, Shimokata K (2000) Abolition of anti-glomerular basement membrane antibody-mediated glomerulonephritis in FcRgamma-deficient mice. Eur J Immunol 30(4):1182–1190. doi:10.1002/(SICI)1521-4141(200004)30:4<1182::AID-IMMU1182>3.0.CO;2-H
Marino M, Ruvo M, De Falco S, Fassina G (2000) Prevention of systemic lupus erythematosus in MRL/lpr mice by administration of an immunoglobulin-binding peptide. Nat Biotechnol 18(7):735–739. doi:10.1038/77296
Fairhurst AM, Xie C, Fu Y, Wang A, Boudreaux C, Zhou XJ, Cibotti R, Coyle A, Connolly JE, Wakeland EK, Mohan C (2009) Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J Immunol 183(10):6831–6838. doi:10.4049/jimmunol.0900742
Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, van Rooijen N, Moldawer L, Satoh M, Reeves WH (2008) A novel type I IFN-producing cell subset in murine lupus. J Immunol 180(7):5101–5108
Carlucci F, Ishaque A, Ling GS, Szajna M, Sandison A, Donatien P, Cook HT, Botto M (2016) C1q modulates the response to TLR7 stimulation by pristane-primed macrophages: implications for pristane-induced lupus. J Immunol 196(4):1488–1494. doi:10.4049/jimmunol.1401009
Schiffer L, Bethunaickan R, Ramanujam M, Huang W, Schiffer M, Tao H, Madaio MP, Bottinger EP, Davidson A (2008) Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J Immunol 180(3):1938–1947
Bethunaickan R, Berthier CC, Ramanujam M, Sahu R, Zhang W, Sun Y, Bottinger EP, Ivashkiv L, Kretzler M, Davidson A (2011) A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J Immunol 186(8):4994–5003. doi:10.4049/jimmunol.1003010
Perez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, Mampaso F, Schlondorff D (2001) Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol 12(7):1369–1382
Adalid-Peralta L, Mathian A, Tran T, Delbos L, Durand-Gasselin I, Berrebi D, Peuchmaur M, Couderc J, Emilie D, Koutouzov S (2008) Leukocytes and the kidney contribute to interstitial inflammation in lupus nephritis. Kidney Int 73(2):172–180. doi:10.1038/sj.ki.5002625
Sobel ES, Morel L, Baert R, Mohan C, Schiffenbauer J, Wakeland EK (2002) Genetic dissection of systemic lupus erythematosus pathogenesis: evidence for functional expression of Sle3/5 by non-T cells. J Immunol 169(7):4025–4032
Bignon A, Gaudin F, Hemon P, Tharinger H, Mayol K, Walzer T, Loetscher P, Peuchmaur M, Berrebi D, Balabanian K (2014) CCR1 inhibition ameliorates the progression of lupus nephritis in NZB/W mice. J Immunol 192(3):886–896. doi:10.4049/jimmunol.1300123
Anders HJ, Belemezova E, Eis V, Segerer S, Vielhauer V, Perez de Lema G, Kretzler M, Cohen CD, Frink M, Horuk R, Hudkins KL, Alpers CE, Mampaso F, Schlondorff D (2004) Late onset of treatment with a chemokine receptor CCR1 antagonist prevents progression of lupus nephritis in MRL-Fas(lpr) mice. J Am Soc Nephrol 15(6):1504–1513
Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S (2010) The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11 (10):936–944. doi:10.1038/ni.1920
Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, Matsuyama T, Taniguchi T, Honda K (2005) Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A102(44):15989–15994. doi:10.1073/pnas.0508327102
Lech M, Weidenbusch M, Kulkarni OP, Ryu M, Darisipudi MN, Susanti HE, Mittruecker HW, Mak TW, Anders HJ (2011) IRF4 deficiency abrogates lupus nephritis despite enhancing systemic cytokine production. J Am Soc Nephrol 22(8):1443–1452. doi:10.1681/ASN.2010121260
Hong MH, Williams H, Jin CH, Pike JW (2000) The inhibitory effect of interleukin-10 on mouse osteoclast formation involves novel tyrosine-phosphorylated proteins. J Bone Miner Res 15(5):911–918. doi:10.1359/jbmr.2000.15.5.911
HorwoodNJ EJ, Martin TJ, Gillespie MT (2001) IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J Immunol 166(8):4915–4921
Abu-AmerY (2001) IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB. J Clin Invest 107 (11):1375–1385. doi:10.1172/JCI10530
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T (2000) T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408(6812):600–605. doi:10.1038/35046102
Stanley ER, Chitu V (2014) CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol 6(6). doi:10.1101/cshperspect.a021857
Toh ML, Bonnefoy JY, Accart N, Cochin S, Pohle S, Haegel H, De Meyer M, Zemmour C, Preville X, Guillen C, Thioudellet C, Ancian P, Lux A, Sehnert B, Nimmerjahn F, Voll RE, Schett G (2014) Bone- and cartilage-protective effects of a monoclonal antibody against colony-stimulating factor 1 receptor in experimental arthritis. Arthritis Rheumatol 66(11):2989–3000. doi:10.1002/art.38624
Zhou D, McNamara NA (2014) Macrophages: important players in primary Sjogren’s syndrome? Expert Rev Clin Immunol 10(4):513–520. doi:10.1586/1744666X.2014.900441
Baban B, Liu JY, Abdelsayed R, Mozaffari MS (2013) Reciprocal relation between GADD153 and Del-1 in regulation of salivary gland inflammation in Sjogren syndrome. Exp Mol Pathol 95(3):288–297. doi:10.1016/j.yexmp.2013.09.002
Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D (2002) Projection of an immunological self shadow within the thymus by the aire protein. Science 298(5597):1395–1401. doi:10.1126/science.1075958
De Voss JJ, Le Clair NP, Hou Y, Grewal NK, Johannes KP, Lu W, Yang T, Meagher C, Fong L, Strauss EC, Anderson MS (2010) An autoimmune response to odorant binding protein 1a is associated with dry eye in the Aire-deficient mouse. J Immunol 184(8):4236–4246. doi:10.4049/jimmunol.0902434
Zhou D, Chen YT, Chen F, Gallup M, Vijmasi T, Bahrami AF, Noble LB, van Rooijen N, McNamara NA (2012) Critical involvement of macrophage infiltration in the development of Sjogren’s syndrome-associated dry eye. Am J Pathol 181(3):753–760. doi:10.1016/j.ajpath.2012.05.014
Nguyen C, Cornelius J, Singson E, Killedar S, Cha S, Peck AB (2006) Role of complement and B lymphocytes in Sjogren’s syndrome-like autoimmune exocrinopathy of NOD.B10-H2b mice. Mol Immunol 43(9):1332–1339. doi:10.1016/j.molimm.2005.09.003
Nguyen CQ, Kim H, Cornelius JG, Peck AB (2007) Development of Sjogren’s syndrome in nonobese diabetic-derived autoimmune-prone C57BL/6.NOD-Aec1Aec2 mice is dependent on complement component-3. J Immunol 179(4):2318–2329
LiX WK, Edman M, Schenke-Layland K, MacVeigh-Aloni M, Janga SR, Schulz B, Hamm-Alvarez SF (2010) Increased expression of cathepsins and obesity-induced proinflammatory cytokines in lacrimal glands of male NOD mouse. Invest Ophthalmol Vis Sci 51(10):5019–5029. doi:10.1167/iovs.09-4523
Killedar SJ, Eckenrode SE, McIndoe RA, She JX, Nguyen CQ, Peck AB, Cha S (2006) Early pathogenic events associated with Sjogren’s syndrome (SjS)-like disease of the NOD mouse using microarray analysis. Lab Investig 86(12):1243–1260. doi:10.1038/labinvest.3700487
Bulosan M, Pauley KM, Yo K, Chan EK, Katz J, Peck AB, Cha S (2009) Inflammatory caspases are critical for enhanced cell death in the target tissue of Sjogren’s syndrome before disease onset. Immunol Cell Biol 87(1):81–90. doi:10.1038/icb.2008.70
Kovacs EJ (1991) Fibrogenic cytokines: the role of immune mediators in the development of scar tissue. Immunol Today 12(1):17–23. doi:10.1016/0167-5699(91)90107-5
Zhang Y, McCormick LL, Desai SR, Wu C, Gilliam AC (2002) Murine sclerodermatous graft-versus-host disease, a model for human scleroderma: cutaneous cytokines, chemokines, and immune cell activation. J Immunol 168(6):3088–3098
Yamamoto T, Takagawa S, Katayama I, Yamazaki K, Hamazaki Y, Shinkai H, Nishioka K (1999) Animal model of sclerotic skin. I: local injections of bleomycin induce sclerotic skin mimicking scleroderma. J Invest Dermatol 112(4):456–462. doi:10.1046/j.1523-1747.1999.00528.x
Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P (1989) Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med 170(3):655–663
Yamamoto T, Nishioka K (2003) Role of monocyte chemoattractant protein-1 and its receptor,CCR-2, in the pathogenesis of bleomycin-induced scleroderma. J Invest Dermatol 121(3):510–516. doi:10.1046/j.1523-1747.2003.12408.x
Ferreira AM, Takagawa S, Fresco R, Zhu X, Varga J, Di Pietro LA (2006) Diminished induction of skin fibrosis in mice with MCP-1 deficiency. J Invest Dermatol 126(8):1900–1908. doi:10.1038/sj.jid.5700302
Zhou L, Askew D, Wu C, Gilliam AC (2007) Cutaneous gene expression by DNA microarray in murine sclerodermatous graft-versus-host disease, a model for human scleroderma. J Invest Dermatol 127(2):281–292. doi:10.1038/sj.jid.5700517
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1(2):135–145. doi:10.1038/35100529
Ghebrehiwet B, Peerschke EI (2004) cC1q-R (calreticulin) and gC1q-R/p33: ubiquitously expressed multi-ligand binding cellular proteins involved in inflammation and infection. Mol Immunol 41(2–3):173–183. doi:10.1016/j.molimm.2004.03.014
Ghebrehiwet B, Hosszu KK, Valentino A, Peerschke EI (2012) The C1q family of proteins: insights into the emerging non-traditional functions. Front Immunol 3. doi:10.3389/fimmu.2012.00052
Walport MJ, Davies KA, Botto M (1998) C1q and systemic lupus erythematosus. Immunobiology 199(2):265–285. doi:10.1016/S0171-2985(98)80032-6
Liu CC, Navratil JS, Sabatine JM, Ahearn JM (2004) Apoptosis, complement and systemic lupus erythematosus: a mechanistic view. Curr Dir Autoimmun 7:49–86
Martin M, Blom AM (2016) Complement in removal of the dead—balancing inflammation. Immunol Rev 274(1):218–232. doi:10.1111/imr.12462
Yoshimoto K, Tanaka M, Kojima M, Setoyama Y, Kameda H, Suzuki K, Tsuzaka K, Ogawa Y, Tsubota K, Abe T, Takeuchi T (2011) Regulatory mechanisms for the production of BAFF and IL-6 are impaired in monocytes of patients of primary Sjogren’s syndrome. Arthritis Res Ther 13(5):R170. doi:10.1186/ar3493
Pertovaara M, Silvennoinen O, Isomaki P (2015) STAT-5 is activated constitutively in T cells, B cells and monocytes from patients with primary Sjogren’s syndrome. Clin Exp Immunol 181(1):29–38. doi:10.1111/cei.12614
Hauk V, Fraccaroli L, Grasso E, Eimon A, Ramhorst R, Hubscher O, Perez Leiros C (2014) Monocytes from Sjogren’s syndrome patients display increased vasoactive intestinal peptide receptor 2 expression and impaired apoptotic cell phagocytosis. Clin Exp Immunol 177(3):662–670. doi:10.1111/cei.12378
Lopez-Pedrera C, Cuadrado MJ, Herandez V, Buendia P, Aguirre MA, Barbarroja N, Torres LA, Villalba JM, Velasco F, Khamashta M (2008) Proteomic analysis in monocytes of antiphospholipid syndrome patients: deregulation of proteins related to the development of thrombosis. Arthritis Rheum 58(9):2835–2844. doi:10.1002/art.23756
Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, Bertolaccini ML, Khamashta MA, Rodriguez-Ariza A, Segui P, Collantes-Estevez E, Barbarroja N, Khraiwesh H, Gonzalez-Reyes JA, Villalba JM, Velasco F, Cuadrado MJ, Lopez-Pedrera C (2012) Mitochondrial dysfunction in antiphospholipid syndrome: implications in the pathogenesis of the disease and effects of coenzyme Q(10) treatment. Blood 119(24):5859–5870. doi:10.1182/blood-2011-12-400986
Satta N, Kruithof EK, Fickentscher C, Dunoyer-Geindre S, Boehlen F, Reber G, Burger D, de Moerloose P (2011) Toll-like receptor 2 mediates the activation of human monocytes and endothelial cells by antiphospholipid antibodies. Blood 117(20):5523–5531. doi:10.1182/blood-2010-11-316158
Shapira I, Andrade D, Allen SL, Salmon JE (2012) Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum 64(8):2719–2723. doi:10.1002/art.34440
Stifano G, Christmann RB (2016) Macrophage involvement in systemic sclerosis: do we need more evidence? Curr Rheumatol Rep 18(1):2. doi:10.1007/s11926-015-0554-8
Higashi-Kuwata N, Jinnin M, Makino T, Fukushima S, Inoue Y, Muchemwa FC, Yonemura Y, Komohara Y, Takeya M, Mitsuya H, Ihn H (2010) Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther 12(4):R128. doi:10.1186/ar3066
Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, Murray LA, Siner JM, Antin-Ozerkis DE, Montgomery RR, Reilkoff RA, Bucala RJ, Herzog EL (2010) Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Investig 90(6):812–823. doi:10.1038/labinvest.2010.73
Kavai M, Szegedi G (2007) Immune complex clearance by monocytes and macrophages in systemic lupus erythematosus. Autoimmun Rev (6, 7):497–502. doi:10.1016/j.autrev.2007.01.017
Sestak AL, Furnrohr BG, Harley JB, Merrill JT, Namjou B (2011) The genetics of systemic lupus erythematosus and implications for targeted therapy. Ann Rheum Dis 70(Suppl 1):i37–i43. doi:10.1136/ard.2010.138057
Byrne JC, Ni Gabhann J, Lazzari E, Mahony R, Smith S, Stacey K, Wynne C, Jefferies CA (2012) Genetics of SLE: functional relevance for monocytes/macrophages in disease. Clin Dev Immunol 2012:582352. doi:10.1155/2012/582352
Yang N, Isbel NM, Nikolic-Paterson DJ, Li Y, Ye R, Atkins RC, Lan HY (1998) Local macrophage proliferation in human glomerulonephritis. Kidney Int 54(1):143–151. doi:10.1046/j.1523-1755.1998.00978.x
Hill GS, Delahousse M, Nochy D, Remy P, Mignon F, Mery JP, Bariety J (2001) Predictive power of the second renal biopsy in lupus nephritis: significance of macrophages. Kidney Int 59(1):304–316. doi:10.1046/j.1523-1755.2001.00492.x
Hill GS, Delahousse M, Nochy D, Mandet C, Bariety J (2001) Proteinuria and tubulointerstitial lesions in lupus nephritis. Kidney Int 60(5):1893–1903. doi:10.1046/j.1523-1755.2001.00017.x
Horwood NJ (2016) Macrophage polarization and bone formation: a review. Clin Rev Allergy Immunol 51(1):79–86. doi:10.1007/s12016-015-8519-2
Gerlag DM, Tak PP (2008) Novel approaches for the treatment of rheumatoid arthritis: lessons from the evaluation of synovial biomarkers in clinical trials. Best Pract Res Clin Rheumatol 22(2):311–323. doi:10.1016/j.berh.2008.02.002
Mulherin D, Fitzgerald O, Bresnihan B (1996) Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum 39(1):115–124
Hamilton JA, Tak PP (2009) The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum 60(5):1210–1221. doi:10.1002/art.24505
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23(11):549–555
Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM (2010) Characteristics of the minor salivary gland infiltrates in Sjogren’s syndrome. J Autoimmun 34(4):400–407. doi:10.1016/j.jaut.2009.10.004
Mathes AL, Christmann RB, Stifano G, Affandi AJ, Radstake TR, Farina GA, Padilla C, McLaughlin S, Lafyatis R (2014) Global chemokine expression in systemic sclerosis (SSc): CCL19 expression correlates with vascular inflammation in SSc skin. Ann Rheum Dis 73(10):1864–1872. doi:10.1136/annrheumdis-2012-202814
Stifano G, Affandi AJ, Mathes AL, Rice LM, Nakerakanti S, Nazari B, Lee J, Christmann RB, Lafyatis R (2014) Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther 16(4):R136. doi:10.1186/ar4598
Seki E, DeMinicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF (2007) TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13(11):1324–1332. doi:10.1038/nm1663
Atamas SP, White B (2003) Cytokine regulation of pulmonary fibrosis in scleroderma. Cytokine Growth Factor Rev 14(6):537–550
Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA (2011) Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum 63(3):783–794. doi:10.1002/art.30159
Silver RM, Feghali-Bostwick CA (2014) Editorial: molecular insights into systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol 66(3):485–487. doi:10.1002/art.38287
Hamilton RF Jr, Parsley E, Holian A (2004) Alveolar macrophages from systemic sclerosis patients: evidence for IL-4-mediated phenotype changes. Am J Physiol Lung Cell Mol Physiol 286(6):L1202–L1209. doi:10.1152/ajplung.00351.2003
LuzinaI G, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B (2002) Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol 26(5):549–557. doi:10.1165/ajrcmb.26.5.4683
Prasse A, Pechkovsky DV, Toews GB, Schafer M, Eggeling S, Ludwig C, Germann M, Kollert F, Zissel G, Muller-Quernheim J (2007) CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum 56(5):1685–1693. doi:10.1002/art.22559
Schupp J, Becker M, Gunther J, Muller-Quernheim J, Riemekasten G, Prasse A (2014) Serum CCL18 is predictive for lung disease progression and mortality in systemic sclerosis. Eur Respir J 43(5):1530–1532. doi:10.1183/09031936.00131713
Tiev KP, Hua-Huy T, Kettaneh A, Gain M, Duong-Quy S, Toledano C, Cabane J, Dinh-Xuan AT (2011) Serum CC chemokine ligand-18 predicts lung disease worsening in systemic sclerosis. Eur Respir J 38(6):1355–1360. doi:10.1183/09031936.00004711
Nauseef WM, Borregaard N (2014) Neutrophils at work. Nat Immunol 15(7):602–611. doi:10.1038/ni.2921
Mayadas TN, Cullere X, Lowell CA (2014) The multifaceted functions of neutrophils. Annu Rev Pathol 9:181–218. doi:10.1146/annurev-pathol-020712-164023
Mantovani A, Cassatella MA, Costantini C, Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11(8):519–531. doi:10.1038/nri3024
Wright HL, Moots RJ, Bucknall RC, Edwards SW (2010) Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 49(9):1618–1631. doi:10.1093/rheumatology/keq045
Brinkmann V, Zychlinsky A (2012) Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol 198(5):773–783. doi:10.1083/jcb.201203170
Wright HL, Bucknall RC, Moots RJ, Edwards SW (2012) Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology (Oxford) 51(3):451–459. doi:10.1093/rheumatology/ker338
Simon D, Simon HU, Yousefi S (2013) Extracellular DNA traps in allergic, infectious, and autoimmune diseases. Allergy 68(4):409–416. doi:10.1111/all.12111
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 3(73):73ra19. doi:10.1126/scitranslmed.3001180
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, Thompson P, Chen P, Fox DA, Pennathur S, Kaplan MJ (2013) NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 5(178):178ra140. doi:10.1126/scitranslmed.3005580
Fossati G, Bucknall RC, Edwards SW (2002) Insoluble and soluble immune complexes activate neutrophils by distinct activation mechanisms: changes in functional responses induced by priming with cytokines. Ann Rheum Dis 61(1):13–19
Weiss SJ (1989) Tissue destruction by neutrophils. N Engl J Med 320(6):365–376. doi:10.1056/NEJM198902093200606
Wright HL, Moots RJ, Edwards SW (2014) The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol 10(10):593–601. doi:10.1038/nrrheum.2014.80
Zoja C, Liu XH, Donadelli R, Abbate M, Testa D, Corna D, Taraboletti G, Vecchi A, Dong QG, Rollins BJ, Bertani T, Remuzzi G (1997) Renal expression of monocyte chemoattractant protein-1 in lupus autoimmune mice. J Am Soc Nephrol 8(5):720–729
Coquery CM, Wade NS, Loo WM, Kinchen JM, Cox KM, Jiang C, Tung KS, Erickson LD (2014) Neutrophils contribute to excess serum BAFF levels and promote CD4+ T cell and B cell responses in lupus-prone mice. PLoS One 9(7):e102284. doi:10.1371/journal.pone.0102284
Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, CassatellaMA LCA (2010) Myeloid cells, BAFF, and IFN-gamma establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med 207(8):1757–1773. doi:10.1084/jem.20100086
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, DeRavin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22(2):146–153. doi:10.1038/nm.4027
Liu CL, Tangsombatvisit S, Rosenberg JM, Mandelbaum G, Gillespie EC, Gozani OP, Alizadeh AA, Utz PJ (2012) Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res Ther 14(1):R25. doi:10.1186/ar3707
Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ (2015) Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis 74(12):2199–2206. doi:10.1136/annrheumdis-2014-205365
Knight JS, Zhao W, Luo W, Subramanian V, O’Dell AA, Yalavarthi S, Hodgin JB, Eitzman DT, Thompson PR, Kaplan MJ (2013) Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 123(7):2981–2993. doi:10.1172/JCI67390
Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, Luo Y, Levitt B, Glogowska M, Chandra P, Kulik L, Robinson WH, Arend WP, Thompson PR, Holers VM (2011) N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol 186(7):4396–4404. doi:10.4049/jimmunol.1001620
Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, James WG, Metcalf D, Campbell IK, Wicks IP (2008) A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood 112(13):5193–5201. doi:10.1182/blood-2008-02-139535
Christensen AD, Haase C, Cook AD, Hamilton JA (2016) Granulocyte colony-stimulating factor (G-CSF) plays an important role in immune complex-mediated arthritis. Eur J Immunol 46(5):1235–1245. doi:10.1002/eji.201546185
Coelho FM, Pinho V, Amaral FA, Sachs D, Costa VV, Rodrigues DH, Vieira AT, Silva TA, Souza DG, Bertini R, Teixeira AL, Teixeira MM (2008) The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheum 58(8):2329–2337. doi:10.1002/art.23622
Grespan R, Fukada SY, Lemos HP, Vieira SM, Napimoga MH, Teixeira MM, Fraser AR, Liew FY, McInnes IB, Cunha FQ (2008) CXCR2-specific chemokines mediate leukotriene B4-dependent recruitment of neutrophils to inflamed joints in mice with antigen-induced arthritis. Arthritis Rheum 58(7):2030–2040. doi:10.1002/art.23597
Grant EP, Picarella D, Burwell T, Delaney T, Croci A, Avitahl N, Humbles AA, Gutierrez-Ramos JC, Briskin M, Gerard C, Coyle AJ (2002) Essential role for the C5a receptor in regulating the effector phase of synovial infiltration and joint destruction in experimental arthritis. J Exp Med 196(11):1461–1471
Sarraj B, Ludanyi K, Glant TT, Finnegan A, Mikecz K (2006) Expression of CD44 and L-selectin in the innate immune system is required for severe joint inflammation in the proteoglycan-induced murine model of rheumatoid arthritis. J Immunol 177(3):1932–1940
Grevers LC, van Lent PL, Koenders MI, Walgreen B, Sloetjes AW, Nimmerjahn F, Sjef Verbeek J, van den Berg WB (2009) Different amplifying mechanisms of interleukin-17 and interferon-gamma in Fcgamma receptor-mediated cartilage destruction in murine immune complex-mediated arthritis. Arthritis Rheum 60(2):396–407. doi:10.1002/art.24288
Williams AS, Richards PJ, Thomas E, Carty S, Nowell MA, Goodfellow RM, Dent CM, Williams BD, Jones SA, Topley N (2007) Interferon-gamma protects against the development of structural damage in experimental arthritis by regulating polymorphonuclear neutrophil influx into diseased joints. Arthritis Rheum 56(7):2244–2254. doi:10.1002/art.22732
Kelchtermans H, Schurgers E, Geboes L, Mitera T, Van Damme J, Van Snick J, Uyttenhove C, Matthys P (2009) Effector mechanisms of interleukin-17 in collagen-induced arthritis in the absence of interferon-gamma and counteraction by interferon-gamma. Arthritis Res Ther 11(4):R122. doi:10.1186/ar2787
Katayama M, Ohmura K, Yukawa N, Terao C, Hashimoto M, Yoshifuji H, Kawabata D, Fujii T, Iwakura Y, Mimori T (2013) Neutrophils are essential as a source of IL-17 in the effector phase of arthritis. PLoS One 8(5):e62231. doi:10.1371/journal.pone.0062231
Maicas N, Ferrandiz ML, Brines R, Ibanez L, Cuadrado A, Koenders MI, van den Berg WB, Alcaraz MJ (2011) Deficiency of Nrf2 accelerates the effector phase of arthritis and aggravates joint disease. Antioxid Redox Signal 15(4):889–901. doi:10.1089/ars.2010.3835
Kelkka T, Hultqvist M, Nandakumar KS, Holmdahl R (2012) Enhancement of antibody-induced arthritis via Toll-like receptor 2 stimulation is regulated by granulocyte reactive oxygen species. Am J Pathol 181(1):141–150. doi:10.1016/j.ajpath.2012.03.031
Servettaz A, Goulvestre C, Kavian N, Nicco C, Guilpain P, Chereau C, Vuiblet V, Guillevin L, Mouthon L, Weill B, Batteux F (2009) Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J Immunol 182(9):5855–5864. doi:10.4049/jimmunol.0803705
Marut W, Jamier V, Kavian N, Servettaz A, Winyard PG, Eggleton P, Anwar A, Nicco C, Jacob C, Chereau C, Weill B, Batteux F (2013) The natural organosulfur compound dipropyltetrasulfide prevents HOCl-induced systemic sclerosis in the mouse. Arthritis Res Ther 15(5):R167. doi:10.1186/ar4351
Pereira S, Lowell C (2003) The Lyn tyrosine kinase negatively regulates neutrophil integrin signaling. J Immunol 171(3):1319–1327
Campbell AM, Kashgarian M, Shlomchik MJ (2012) NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med 4 (157):157ra141. doi:10.1126/scitranslmed.3004801
Grimm MJ, Vethanayagam RR, Almyroudis NG, Dennis CG, Khan AN, D’Auria AC, Singel KL, Davidson BA, Knight PR, Blackwell TS, Hohl TM, Mansour MK, Vyas JM, Rohm M, Urban CF, Kelkka T, Holmdahl R, Segal BH (2013) Monocyte- and macrophage-targeted NADPH oxidase mediates antifungal host defense and regulation of acute inflammation in mice. J Immunol 190(8):4175–4184. doi:10.4049/jimmunol.1202800
Dwivedi N, Radic M (2014) Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis 73(3):483–491. doi:10.1136/annrheumdis-2013-203844
Wipke BT, Allen PM (2001) Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol 167(3):1601–1608
Gal I, Bajnok E, Szanto S, Sarraj B, Glant TT, Mikecz K (2005) Visualization and in situ analysis of leukocyte trafficking into the ankle joint in a systemic murine model of rheumatoid arthritis. Arthritis Rheum 52(10):3269–3278. doi:10.1002/art.21532
Griffiths RJ, Pettipher ER, Koch K, Farrell CA, Breslow R, Conklyn MJ, Smith MA, Hackman BC, Wimberly DJ, Milici AJ et al (1995) Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proc Natl Acad Sci U S A 92(2):517–521
Sadik CD, Kim ND, Iwakura Y, Luster AD (2012) Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcgammaR signaling. Proc Natl Acad Sci U S A 109(46):E3177–E3185. doi:10.1073/pnas.1213797109
Hoffmann MH, Bruns H, Backdahl L, Neregard P, Niederreiter B, Herrmann M, Catrina AI, Agerberth B, Holmdahl R (2013) The cathelicidins LL-37 and rCRAMP are associated with pathogenic events of arthritis in humans and rats. Ann Rheum Dis 72(7):1239–1248. doi:10.1136/annrheumdis-2012-202218
Agier J, Efenberger M, Brzezinska-Blaszczyk E (2015) Cathelicidin impact on inflammatory cells. Cent Eur J Immunol 40(2):225–235. doi:10.5114/ceji.2015.51359
Cesaro A, Anceriz N, Plante A, Page N, Tardif MR, Tessier PA (2012) An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis. PLoS One 7(9):e45478. doi:10.1371/journal.pone.0045478
Vossenaar ER, Nijenhuis S, Helsen MM, van der Heijden A, Senshu T, van den Berg WB, van Venrooij WJ, Joosten LA (2003) Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum 48(9):2489–2500. doi:10.1002/art.11229
Rohrbach AS, Hemmers S, Arandjelovic S, Corr M, Mowen KA (2012) PAD4 is not essential for disease in the K/BxN murine autoantibody-mediated model of arthritis. Arthritis Res Ther 14(3):R104. doi:10.1186/ar3829
Askenasy N (2016) Mechanisms of autoimmunity in the non-obese diabetic mouse: effector/regulatory cell equilibrium during peak inflammation. Immunology 147(4):377–388. doi:10.1111/imm.12581
Tsuboi N, Ernandez T, Li X, Nishi H, Cullere X, Mekala D, Hazen M, Kohl J, Lee DM, Mayadas TN (2011) Regulation of human neutrophil Fcgamma receptor IIa by C5a receptor promotes inflammatory arthritis in mice. Arthritis Rheum 63(2):467–478. doi:10.1002/art.30141
Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, Weissleder R, Mathis D, Benoist C (2006) Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol 7(3):284–292. doi:10.1038/ni1306
Keeling DM, Isenberg DA (1993) Haematological manifestations of systemic lupus erythematosus. Blood Rev 7(4):199–207
Carli L, Tani C, Vagnani S, Signorini V, Mosca M (2015) Leukopenia, lymphopenia, and neutropenia in systemic lupus erythematosus: prevalence and clinical impact—a systematic literature review. Semin Arthritis Rheum 45(2):190–194. doi:10.1016/j.semarthrit.2015.05.009
Cairns AP, Crockard AD, McConnell JR, Courtney PA, Bell AL (2001) Reduced expression of CD44 on monocytes and neutrophils in systemic lupus erythematosus: relations with apoptotic neutrophils and disease activity. Ann Rheum Dis 60(10):950–955
Donnelly S, Roake W, Brown S, Young P, Naik H, Wordsworth P, Isenberg DA, Reid KB, Eggleton P (2006) Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum 54(5):1543–1556. doi:10.1002/art.21783
Bengtsson AA, Pettersson A, Wichert S, Gullstrand B, Hansson M, Hellmark T, Johansson AC (2014) Low production of reactive oxygen species in granulocytes is associated with organ damage in systemic lupus erythematosus. Arthritis Res Ther 16(3):R120. doi:10.1186/ar4575
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ (2010) A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol 184(6):3284–3297. doi:10.4049/jimmunol.0902199
Kaplan MJ (2013) Role of neutrophils in systemic autoimmune diseases. Arthritis Res Ther 15(5):219. doi:10.1186/ar4325
Midgley A, Beresford MW (2016) Increased expression of low density granulocytes in juvenile-onset systemic lupus erythematosus patients correlates with disease activity. Lupus 25(4):407–411. doi:10.1177/0961203315608959
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3(73):73ra20. doi:10.1126/scitranslmed.3001201
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ (2011) Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187(1):538–552. doi:10.4049/jimmunol.1100450
Bosch X (2011) Systemic lupus erythematosus and the neutrophil. N Engl J Med 365(8):758–760. doi:10.1056/NEJMcibr1107085
Mayadas TN, Rosetti F, Ernandez T, Sethi S (2010) Neutrophils: game changers in glomerulonephritis?Trends. Mol Med 16(8):368–378. doi:10.1016/j.molmed.2010.06.002
Camussi G, Cappio FC, Messina M, Coppo R, Stratta P, Vercellone A (1980) The polymorphonuclear neutrophil (PMN) immunohistological technique: detection of immune complexes bound to the PMN membrane in acute poststreptococcal and lupus nephritis. Clin Nephrol 14(6):280–287
Thieblemont N, Wright HL, Edwards SW, Witko-Sarsat V (2016) Human neutrophils in auto-immunity. Semin Immunol 28(2):159–173. doi:10.1016/j.smim.2016.03.004
Cross A, Barnes T, Bucknall RC, Edwards SW, Moots RJ (2006) Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J Leukoc Biol 80(3):521–528. doi:10.1189/jlb.0306178
Cross A, Bucknall RC, Cassatella MA, Edwards SW, Moots RJ (2003) Synovial fluid neutrophils transcribe and express class II major histocompatibility complex molecules in rheumatoid arthritis. Arthritis Rheum 48(10):2796–2806. doi:10.1002/art.11253
Wright HL, Chikura B, Bucknall RC, Moots RJ, Edwards SW (2011) Changes in expression of membrane TNF, NF-{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann Rheum Dis 70(3):537–543. doi:10.1136/ard.2010.138065
Talbot J, Bianchini FJ, Nascimento DC, Oliveira RD, Souto FO, Pinto LG, Peres RS, Silva JR, Almeida SC, Louzada-Junior P, Cunha TM, Cunha FQ, Alves-Filho JC (2015) CCR2 expression in neutrophils plays a critical role in their migration into the joints in rheumatoid arthritis. Arthritis Rheumatol 67(7):1751–1759. doi:10.1002/art.39117
denBroeder AA, Wanten GJ, Oyen WJ, Naber T, van Riel PL, Barrera P (2003) Neutrophil migration and production of reactive oxygen species during treatment with a fully human anti-tumor necrosis factor-alpha monoclonal antibody in patients with rheumatoid arthritis. J Rheumatol 30(2):232–237
Brennan FM, Zachariae CO, Chantry D, Larsen CG, Turner M, Maini RN, Matsushima K, Feldmann M (1990) Detection of interleukin 8 biological activity in synovial fluids from patients with rheumatoid arthritis and production of interleukin 8 mRNA by isolated synovial cells. Eur J Immunol 20(9):2141–2144. doi:10.1002/eji.1830200938
deSiqueira MB, da Mota LM, Couto SC, Muniz-Junqueira MI (2015) Enhanced neutrophil phagocytic capacity in rheumatoid arthritis related to the autoantibodies rheumatoid factor and anti-cyclic citrullinated peptides. BMC Musculoskelet Disord 16:159. doi:10.1186/s12891-015-0616-0
Eggleton P, Wang L, Penhallow J, Crawford N, Brown KA (1995) Differences in oxidative response of subpopulations of neutrophils from healthy subjects and patients with rheumatoid arthritis. Ann Rheum Dis 54(11):916–923
Robinson JJ, Watson F, Bucknall RC, Edwards SW (1992) Stimulation of neutrophils by insoluble immunoglobulin aggregates from synovial fluid of patients with rheumatoid arthritis. Eur J Clin Investig 22(5):314–318
Van denSteen PE, Proost P, Grillet B, Brand DD, Kang AH, Van Damme J, Opdenakker G (2002) Cleavage of denatured natural collagen type II by neutrophil gelatinase B reveals enzyme specificity, post-translational modifications in the substrate, and the formation of remnant epitopes in rheumatoid arthritis. FASEB J 16(3):379–389. doi:10.1096/fj.01-0688com
Pillinger MH, Abramson SB (1995) The neutrophil in rheumatoid arthritis. Rheum Dis Clin N Am 21(3):691–714
Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P (2014) Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther 16(3):R122. doi:10.1186/ar4579
Turesson C, Mathsson L, Jacobsson LT, Sturfelt G, Ronnelid J (2013) Antibodies to modified citrullinated vimentin are associated with severe extra-articular manifestations in rheumatoid arthritis. Ann Rheum Dis 72(12):2047–2048. doi:10.1136/annrheumdis-2013-203510
Bayley R, Kite KA, McGettrick HM, Smith JP, Kitas GD, Buckley CD, Young SP (2015) The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis 74(8):1588–1595. doi:10.1136/annrheumdis-2013-204796
Leffler J, Stojanovich L, Shoenfeld Y, Bogdanovic G, Hesselstrand R, Blom AM (2014) Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol 32(1):66–70
Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez C, Hernandez-Ramirez D, Bockenstedt PL, Liaw PC, Cabral AR, Knight JS (2015) Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol 67(11):2990–3003. doi:10.1002/art.39247
van denHoogen LL, Fritsch-Stork RD, van Roon JA, Radstake TR (2016) Low-density granulocytes are increased in antiphospholipid syndrome and are associated with anti-beta2-glycoprotein I antibodies: comment on the article by Yalavarthi et al. Arthritis Rheumatol 68(5):1320–1321. doi:10.1002/art.39576
Barnes TC, Anderson ME, Edwards SW, Moots RJ (2012) Neutrophil-derived reactive oxygen species in SSc. Rheumatology (Oxford) 51(7):1166–1169. doi:10.1093/rheumatology/ker520
Sambo P, Jannino L, Candela M, Salvi A, Donini M, Dusi S, Luchetti MM, Gabrielli A (1999) Monocytes of patients wiht systemic sclerosis (scleroderma) spontaneously release in vitro increased amounts of superoxide anion. J Invest Dermatol 112(1):78–84. doi:10.1046/j.1523-1747.1999.00476.x
Acknowledgements
Illustrated figures were performed by SciToons (www.scitoons.com).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Funding
There is no funding source.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Morell, M., Varela, N. & Marañón, C. Myeloid Populations in Systemic Autoimmune Diseases. Clinic Rev Allerg Immunol 53, 198–218 (2017). https://doi.org/10.1007/s12016-017-8606-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-017-8606-7