
Abstract
Agar, a major polysaccharide of red algal cells, is degraded by β-agarases into neoagarobiose, which is further hydrolyzed into the monomers, d-galactose and 3,6-anhydro-l-galactose, by 1,3-α-3,6-anhydro-l-galactosidases including α-1,3-l-neoagarooligasaccharide hydrolase (α-NAOSH). A novel cold-adapted alkaline α-NAOSH, Ahg558, consisting of 359 amino acids (40.8 kDa) was identified from Gayadomonas joobiniege G7. It was annotated as a glycosyl hydrolase family 43 based on genomic sequence analysis, showing 84% and 74% identities with the characterized α-NAOSHs from Agarivorans gilvus WH0801 and Saccharophagus degradans 2–40, respectively. The recombinant Ahg558 (rAhg558) purified from Escherichia coli formed dimers and cleaved α-1,3 glycosidic bonds at the non-reducing ends of the neoagarobiose, neoagarotetraose, and neoagarohexaose, which was confirmed by thin-layer chromatography and mass spectrometry. The optimum pH and temperature for rAhg558 activity were 9.0 and 30 °C, respectively. Unusually, it retained over 93% activity in a broad range of temperatures between 0 and 40 °C and over 73% in a broad range of pH between pH 6.0 and pH 9.0, indicating it is a unique cold-adapted alkaline exo-acting α-NAOSH. Its enzymatic activity was dependent on Mn2+ ions. Km and Vmax values toward neoagarobiose were 2.6 mg/mL (8.01 mM) and 133.33 U/mg, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biomass is a sustainable resource in a sense that it can be massively obtained by reducing atmospheric CO2 using solar energy. The reduced forms of CO2 that constitute the biomass will eventually be oxidized to CO2 in nature, which contributes to carbon recycling on the earth. Therefore, biomass has been regarded as one of the most important resources for obtaining sustainable and renewable bioenergy. Among them, marine biomass has several advantages compared to starch- or lignocellulose-based biomasses. For instance, it does not compete with agricultural food production and can be efficiently cultivated under the simple growth requirements of light and CO2, without the utilization of pesticides and fertilizers. In addition, it does not contain lignin, which is the main obstacle for practical application of lignocellulosic biomass. In these respects, algae-based production of biofuels has been highlighted by many researchers as third-generation biofuels [1].
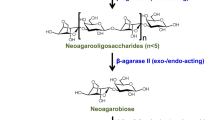
The red algae such as Gelidium amansii have high carbon content (60% of galactan and 20% of cellulose), and thus are expected to be an economically valuable carbon source for mono-sugar and biofuel production [2]. The main component of red algal galactan is agar, a linear polysaccharide consisting of alternately arranged 3-O-linked β-d-galactopyranose (G) and 4-O-linked α-l-galactopyranose (L). Agarose is a major constituent of agar (Fig. 1), where the component L is replaced by 3,6-anhydro-α-l-galactose (AHG) [3]. Therefore, development of an efficient process for agar hydrolysis into AHG and G is a prerequisite for utilizing red algal biomass.

Schematic representation of the β-agarolytic pathway. Agarose, a polymer galactan, is first degraded into oligosaccharides by endo-type β-agarase I, which are further degraded to neoagarobiose by exo-/endo-type β-agarase II. Finally, neoagarobiose is hydrolyzed into monomeric d-galactose (G) and 3,6-anhydro-l-galactose (AHG) by the action of 1,3-α-3,6-anhydro-l-galactosidase. The cleavage sites of β-agarase I and β-agarase II are indicated by filled inverted triangles and arrows, respectively
Several attempts have been reported regarding the complete hydrolysis of agar, such as acid treatment [2], enzymatic treatment [4], or a combination of acid and enzymatic treatments [5]. Although acid-catalyzed hydrolysis of agar seems to be a simple and convenient process, it has many problems such as high-temperature processing, neutralization, and salt removal, as well as the accumulation of toxic compounds including 5-hydroxymethylfurfural [6]. All these together imply that the development of an efficient enzymatic process for agar hydrolysis is preferred to a chemical process for the production of biofuels, by fermenting the hydrolysate.
On the basis of the cleavage pattern on agarose polysaccharide, they are grouped as α-agarase (EC 3.2.1.158) acting on the α-1,3 linkages and β-agarase (EC 3.2.1.81) acting on the β-1,4 linkages of agarose. In comparison to only a few reports on α-agarase [7,8,9], plenty of β-agarases have been reported from different microorganisms [10]. Therefore, the β-agarolytic pathway seems to be a main pathway for agar hydrolysis adopted by microorganisms. In the β-agarolytic pathway (Fig. 1), agarose is rapidly fragmented by endo-type β-agarase I into neoagarooligosaccharides (NAOSs) with G residues at their reducing ends [10, 11]. Then, the NAOSs are further hydrolyzed by exo-/endo-acting (mainly exo-) β-agarase II, resulting in neoagarobiose (NA2). Finally, the NA2 (AHG-G) is hydrolyzed into its monomers, AHG and G, by the exo-acting 1,3-α-3,6-anhydro-l-galactosidases (EC 3.2.1.-) including the α-1,3-l-neoagarobiase/neoagarobiose hydrolase (α-NABH) and the α-1,3-l-neoagarooligosaccharide hydrolase (α-NAOSH). Strictly, α-NABHs act only on NA2 as substrate and release AHG and G by cleaving the α-1,3 linkage of NA2. On the other hand, α-NAOSH acts on NA2 as well as neoagarotetraose (NA4) and neoagarohexaose (NA6) as substrates and releases AHG by cleaving the α-1,3 linkage of NAOS from the non-reducing end. Therefore, the complete hydrolysis of agar into AHG and G requires a consortium of several hydrolytic enzymes such as β-agarase I, β-agarase II, and α-NABH/NAOSH.
Although hundreds of β-agarase have been cited in the literature, only 12 α-NABHs/α-NAOSHs were reported, mostly in marine bacteria as listed in Table 1. Gayadomonas joobiniege G7, belonging to the family Alteromonadaceae, was isolated as a marine agarolytic microorganism [24]. The genomic studies predicted that the G7 strain had many genes encoding hydrolytic enzymes capable of completely degrading the sulfated polysaccharides [25], and thus might be useful for industrial processing of agar. In order to utilize the algal biomass efficiently, it is necessary to construct a complete agar decomposition system, but research on the α-NABH/α-NAOSH for the final stage of the digestion process has been insufficient. To meet this need, we recently reported a cold-adapted α-NAOSH, Ahg786 from G. joobiniege G7 [23]. We then identified a new α-NAOSH that is distinct from Ahg786 from the same strain, and its unique biochemical characteristics are described in this article.
Materials and Methods
Bacterial Strains, Culture Conditions, and Plasmid
As a source of genomic DNA, the agarolytic eubacterium Gayadomonas joobiniege G7 (ATCC BAA-2321 = DSM25250T = KCTC23721T) was used [24]. Escherichia coli ER2566 and pET-28a(+) were used as host and vector for gene cloning and expression, respectively. Cultivation of G. joobiniege G7 was performed using artificial seawater (ASW) solid medium or ASW-YP liquid medium supplemented with 0.1% agar (w/v) [24]. E. coli was cultured at 37 °C in LB medium and kanamycin was added if necessary.
Enzymes and Chemicals
DNA modification enzymes were purchased from New England Bio Labs (Ipswich, MA, USA), and all other chemicals were from Sigma-Aldrich Corporation (St. Louis, MI, USA) except NAOSs from DyneBio Inc. (Seongnam, Korea). Thin-layer chromatography (TLC) silica gel plates (60G F254) were purchased from Merck KGaA (Darmstadt, Germany). Primers for polymerase chain reaction (PCR) were synthesized from Genotech (Daejeon, Korea).
Gene Cloning of ahg558
To clone the predicted glycosyl hydrolase gene, ahg558 (NCBI reference sequence: WP_017446558), the genomic DNA from G. joobiniege G7 was isolated in the same manner as described by Asghar et al. [23]. The 1080-bp DNA fragment containing the entire coding region of Ahg558 was amplified by PCR under the same conditions as described [23] using a TaKaRa PCR Thermal Cycler Dice® Gradient (Takara Bio, Japan). The forward primer (5′-ATCACTCATATGTCTGAAAAAAAATTAAGT-3′; NdeI site is underlined) and the reverse primer (5′-CTCCACAGGATCCTTTATTTTTAGTTGGAG-3′; BamHI site is underlined) were used. The PCR product was digested by NdeI and BamHI restriction enzymes, and then ligated with pET28a(+) digested with the same enzymes. The resultant recombinant plasmid (pET28a-Ahg558) was transformed into E. coli ER2566.
Expression, Purification, and Molecular Mass Determination of rAhg558
The recombinant His-Ahg558 (rAhg558) protein was expressed and purified by culturing E. coli ER2566/pET28a-Ahg558. Cell culture, induction of gene expression, and protein purification with a TALON metal affinity resin (Takara Bio, Japan) were carried out in the same manner as described [23]. Molecular weight and purity of rAhg558 were confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) [26]. The protein concentrations were determined according to the method of Bradford [27]. Molecular mass of purified rAhg558 was determined by molecular sieve chromatography using an ÄKTA-FPLC system (GE Healthcare Life Sciences, Chicago, IL, USA) at a flow rate of 0.5 mL/min under the same conditions as described [23].
Estimation of α-NABH/NAOSH Activity by 3,5-Dinitrosalicylic Acid (DNS) Method
The α-NABH/NAOSH activity was determined by measuring the concentration of reducing sugar liberated from the substrate by hydrolysis of the enzyme. The concentration of reducing sugar was measured by the DNS method [28] with a slight modification as previously described [23]. Precisely, the reaction was performed using NA2 as substrate and 50 mM glycine-NaOH buffer (pH 9.0) at 30 °C for 15 min. After reacting with the DNS reagent solution, each tube was heat-treated at 100 °C for 10 min, and after 2 min in ice water, the absorbance at 540 nm (A540) was measured using a Spectronic Unicam Genesys 8 Spectrophotometer (Thermo Scientific™, MA, USA). The blank was prepared by adding the substrate without enzyme and reacting under the same conditions. The standard curve for quantification was prepared using G. The enzyme activity (U) was defined as the amount of enzyme producing 1 μmol of G per minute.
Enzymatic Properties
The biochemical properties of the enzyme were investigated using NA2 as a substrate. To investigate the optimum pH for rAhg558, the enzyme activity was determined over a range of pH 4–11 at 30 °C for 10 min. The buffer solutions used were 50 mM citrate buffer for pH 4–6, 50 mM sodium phosphate buffer for pH 6–9, and 50 mM glycine-NaOH buffer for pH 9–11. The optimal temperature for rAhg558 activity was tested in the range from 0 to 70 °C in 50 mM glycine-NaOH buffer (pH 9.0) for 10 min. In this time, the reaction temperature was maintained at 0 °C in ice water. The thermal stability of rAhg558 was determined by preincubating the enzyme at the indicated temperature for 1 h and then reacting in 50 mM glycine-NaOH buffer (pH 9.0) at 30 °C for 10 min to measure the remaining enzyme activity. Comparisons of activity were calculated as a percentage relative to the maximal enzyme activity. The effect of various metal ions or chelators on enzyme activity was measured by adding each chemical to 50 mM glycine-NaOH buffer (pH 9.0) at the indicated concentration and reacting at 30 °C for 10 min.
Determination of Kinetic Parameters
The kinetic parameters Km and Vmax of rAhg558 toward NA2 were determined in 50 mM glycine-NaOH buffer (pH 9.0) at 30 °C. Km and Vmax were calculated from the Lineweaver-Burk plot [29]. The reaction time was limited to 2 min to restrict the substrate utilization to below 5%. Values of catalytic efficiency (Kcat/Km) and turnover number (Kcat) were calculated based on the Km, Vmax, and (E) values.
Thin-Layer Chromatography and Mass Analysis of the Hydrolyzed Products
For TLC analysis, 5 μg of rAhg558 enzyme and 50 μg of substrate (NA2, NA4, or NA6) were used. The reaction volume was 15 μL, and the enzyme reaction was carried out at 30 °C for 16 h in 50 mM glycine-NaOH buffer (pH 9.0). Ten microliters of the reaction solution was spotted on a TLC silica gel plate (60G F254). After developing the plate with a solvent (n-butanol:ethanol:water = 3:1:1, v/v), the hydrolysis products were observed by spraying 20% (v/v) sulfuric acid in methanol and heating at 90 °C [20].
For mass spectrometry, 15 μg of rAhg558 and 100 μg of substrate were used, and the reaction was performed under the same conditions as the TLC sample at a reaction volume of 30 μL. The reaction mixture was dried in a centrifugal evaporator (Eyela CVE-2000, EYELA, Japan). Then, the methanol extract of dried sample was analyzed using an Agilent 1200 series high-performance liquid chromatography system coupled to a 4000 QTRAP MS/MS detector (AB SCIEX, Foster City, CA, USA) with an electrospray ionization Turbo V ion source. The MS spectrum of the analyzed mass range was 150–1000 m/z with the resolution of 80,000 (200 m/z).
In Silico Analysis of Ahg558
Sequence comparisons of amino acid of Ahg558 with homologous proteins were analyzed using the Clustal program (https://www.ebi.ac.uk/Tools/msa/clustalo/). The phylogenetic tree was generated by the neighbor-joining (NJ) method [30] of the Mega 6 program [31]. The topology of the completed tree was evaluated by carrying out 1000 resampling and displaying the branching points through bootstrap analysis. Evolutionary distances between homologous proteins were calculated using the Poisson correction method as described [23]. The three-dimensional (3-D) structure modeling of Ahg558 was performed using SdNABH (SMTL ID 3r4z.1) [17] as a template on the Swiss-Model (http://swissmodel.expasy.org/) web page [13].
Results
In Silico Analysis of Ahg558
Ahg558 (WP_017446558.1) was annotated as a putative glycosyl hydrolase from the genomic analysis of G. joobiniege G7 [25]. The Ahg558 consisted of 359 amino acids with an expected molecular weight of 40.8 kDa. NCBI BLAST search revealed that Ahg558 had the highest identity (84%) with the α-NAOSH AgaWH117 characterized from Agarivorans gilvus WH0801 [14] and 74%, 60%, and 59% identities with the three α-NAOSHs, namely SdNABH from Saccharophagus degradans 2–40 [17], BpGH117 from Bacteroides plebeius [16], and AhgA from Zobellia galactanivorans [18], respectively, whose three-dimensional structures have been characterized. Moreover, a well-conserved protein domain (cd08992) of the glycosyl hydrolase family 43 (GH43) was found in a broad region spanning Ser-7 and Lys-346 with an e value of 0 (Fig. 2). Although all the biochemically characterized α-NAOSHs have the GH43 domain, they were proposed to be classified into a new family GH117 based on their unique catalytic activity [18]. Based on in silico analysis, Ahg558 was proposed to be an α-NABH/α-NAOSH candidate, which led us to characterize its biochemical property. Gene organization analysis revealed that several proteins putatively related to agar or G/AHG catabolism, such as two β-agarases, sugar kinase, sugar phosphate isomerase, and phosphogluconate dehydrogenase, are encoded in the vicinity of ahg558 in the chromosome, implying the importance of Ahg558 in agar metabolism by G. joobiniege G7 (Fig. 2). Contrastingly, except for the galactose oxidase gene, there was no gene expected to be involved in agar metabolism near the ahg786 gene.

Distribution of peripheral genes including 1,3-α-3,6-anhydro-l-galactosidase gene and conserved domain of Ahg558. (Upper) The two gene clusters including 1,3-α-3,6-anhydro-l-galactosidase gene from [18], [16] Gayadomonas joobiniege G7 [23] were aligned. Arrows indicate individual ORFs and arrowheads indicate the stop codon of each ORF. The number of each ORF is indicated in the arrow. The expected function of each ORF is shown in the row below. The ORFs corresponding to 1,3-α-3,6-anhydro-l-galactosidase are indicated by arrows filled with black, and the other ORFs related to agar metabolism are indicated by shaded arrows. DH, dehydrogenase; TR, transcriptional regulator. (Lower) The conserved domain (cd08992) of GH family 43 in Ahg558 (WP_017446558.1). The domain occupies a broad region spanning Ser-7 and Lys-346 with an e value of 0
Purification and Molecular Weight Determination of rAhg558
SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP/) analysis predicted that Ahg558 did not have any signal peptide, implying that it is an intracellular protein. Therefore, the whole Ahg558 protein with a 6x His-tag at the N-terminal was expressed in E. coli and purified to homogeneity. The purified rAhg558 protein was estimated to have a molecular weight of 43 kDa, coinciding with the expected size (43.14 kDa), with 20 vector-derived additional amino acids including 6x His-tag at the N-terminus, on SDS-PAGE analysis (Fig. 3a). Analysis of molecular sieve chromatography using a Superose 12 column resulted in an apparent molecular weight of 85 kDa, indicating Ahg558 is present as a dimer (Fig. 3b).

Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and molecular sieve chromatogram of the purified rAhg558. a SDS-PAGE analysis. rAhg558 with N-terminus His-tag was purified by TALON metal affinity chromatography from E. coli ER2566/pET28a-Ahg558. Lanes: M, molecular mass marker; 1, total cell protein before IPTG induction; 2, total cell protein after IPTG induction; 3, purified rAhg558. Proteins corresponding to rAhg558 were indicated by arrows. b Determination of molecular mass of rAhg558 by molecular sieve chromatography. (a) A sample (600 μg of rAhg558) was loaded onto a Superose 12 column. The chromatography was performed at a flow rate of 0.5 mL/min and monitored by measuring the absorbance at 280 nm. (b) Molecular mass of rAhg558 was calculated by using the size marker proteins on a Superose 12 column. Position a, β-amylase (200 kDa); position b, yeast alcohol dehydrogenase (150 kDa); position c, bovine serum albumin (66 kDa); position d, bovine carbonic anhydrase (29 kDa). The position corresponding to the elution peak of rAhg558 was indicated by an arrow
Enzymatic Properties of rAhg558
Although rAhg558 did not show any detectable hydrolyzing activity toward 0.2% agarose (data not shown), it was able to hydrolyze NA2 into AHG and G as shown by TLC analysis. Furthermore, it also hydrolyzed NA4 and NA6, producing the common products corresponding to AHG and the remainders (Fig. 4a).


Analysis of neoagarooligosaccharide hydrolysates generated by rAhg558. a Thin-layer chromatography (TLC). The neoagarooligosaccharide hydrolysates by rAhg558 were analyzed on a silica gel (60G F254) TLC plate. The composition of each reaction is shown at the bottom of the figure. b–d Ion-trap mass spectrometry. The hydrolysates of neoagarooligosaccharide in a were dried in vacuo and extracted with methanol. The molecular mass distribution was then determined using a high-performance liquid chromatography system coupled to a 4000 QTRAP MS/MS detector with an electrospray ionization Turbo V Ion Source. b Hydrolysate of neoagarobiose. Because the peak of 3,6-anhydro-l-galactose was too small to be visible in the case of mass range of 150–1000 m/z, the MS spectra were extended to the mass range of 150–280 m/z. The peaks for the molecular ion at m/z 203 (M + Na)+ and m/z 185 (M + Na)+, corresponding to d-galactose and 3,6-anhydro-l-galactose, are indicated by arrows, respectively. c The hydrolysate of neoagarotetraose. The peaks for the molecular ion at m/z 509 (M + Na)+ and m/z 185 (M + Na)+, corresponding to neoagarotriose and 3,6-anhydro-l-galactose, are indicated by arrows, respectively. d The hydrolysate of neoagarohexaose. The peaks for the molecular ion at m/z 815 (M + Na)+ and m/z 185 (M + Na)+, corresponding to neoagaropentaose and 3,6-anhydro-l-galactose, respectively, are indicated by arrows
Mass spectrometry analysis confirmed that rAhg558 indeed hydrolyzed NA2 into the molecular masses of AHG (m/z 185 [M + Na]+) and G (m/z 203 [M + Na]+; Fig. 4b). NA4 was hydrolyzed into the molecular masses of AHG (m/z 185 [M + Na]+) and neoagarotriose (m/z 509 [M + Na]+; Fig. 4c), and NA6 was hydrolyzed into AHG (m/z 185 [M + Na]+) and neoagaropentaose (m/z 815 [M + Na]+; Fig. 4d). These results indicate that Ahg558 is an exo-type 1,3-α-3,6-anhydro-l-galactosidase (α-NAOSH) cleaving α-1,3-glycosidic linkages and thus releasing AHG from the non-reducing end of various NAOSs.
Biochemical Characteristics of rAhg558
The biochemical property of rAhg558 was studied using NA2 as a substrate. The temperature profile showed that rAhg558 had maximum activity at 30 °C and retained its activity in a broad range between 0 °C (93% activity) and 40 °C (93% activity), indicating Ahg558 is a cold-adapted NAOSH (Fig. 5a). However, a drastic decrease in enzyme activity was observed above 50 °C. Similarly, it was quite stable to heat treatment for 1 h in a broad range between 0 and 40 °C, but lost most of the enzyme activity above 50 °C (Fig. 5a).


Biochemical characteristics of rAhg558 toward neoagarobiose. a Effect of temperature. The optimum temperature for rAhg558 activity was determined in 50 mM glycine-NaOH buffer (pH 9.0) ranging from 0 to 70 °C. The thermo-stability of the enzyme was determined after pre-incubation for 1 h at temperatures ranging from 0 to 70 °C. Circles, optimum temperature; squares, thermo-stability. b Effect of pH. NAOSH activity was determined at 30 °C in various pH conditions: circles, 4.0–6.0 (50 mM citrate buffer); squares, 6.0–9.0 (50 mM sodium phosphate buffer); diamonds, 9.0–11.0 (50 mM glycine-NaOH buffer). In a and b, the highest enzyme activity was considered 100% and the relative activity of the remaining measurements was calculated. c Effect of metal ions and chelator. Effect of metal ions and EDTA on NAOSH activity was determined at a final concentration of 0.5 mM or as indicated. The enzyme activity without the addition of the chemicals was regarded as 100%, and the relative activity of the remaining measurements was calculated. d Determination of kinetic parameters. A Lineweaver-Burke plot was used to determine the kinetic parameters of Ahg558 acting on neoagarobiose. In a–d, all data shown are mean values from at least three replicate experiments
rAhg558 showed maximum activity at alkaline pH 9.0 in 50 mM glycine-NaOH buffer (Fig. 5b). It also showed good activity between pH 6.0 (82% activity) and pH 8.0 (79% activity), and even retained 58% of enzyme activity at pH 11.0, indicating its unique alkaliphilic nature (Fig. 5b).
EDTA treatment showed severe inhibition of the enzyme activity at final concentrations of 0.5 mM (46% inhibition) to 5 mM (83% inhibition), implying that it requires some metal ions as a cofactor (Fig. 5c). Enzyme activities were inhibited to different extents depending on the type of metal ion, except MnCl2 at 0.5 mM. The inhibitory effect conferred by EDTA on the enzyme activity was completely circumvented by the addition of MnCl2. Furthermore, the enzyme activity was remarkably enhanced by the addition of MnCl2 in a concentration-dependent manner, strongly indicating that rAhg558 is dependent on manganese ion for its enzymatic activity.
Enzyme Kinetics
Km and Vmax values toward NA2 were 2.6 mg/mL and 133.33 U/mg, respectively (Fig. 5d). Kcat and Kcat/Km were 90.82 s−1 and 0.01135 μM−1 s−1, respectively.
Analysis of the Phylogenetic Tree and 3-D Structural Model of Ahg558
According to a protein BLAST in NCBI (http://blast.ncbi.nlm.nih.gov), there were numerous sequences with high homology to Ahg558 from various microorganisms. However, only 12 proteins have been validated as α-NABHs/NAOSHs among the homologues, and the gene information for eight proteins of them is available (Table 1). In the phylogenetic tree analysis of Ahg558 with the eight α-NAOSs (Fig. 5a), Ahg558 formed a clade with AgaWH117 from Agarivorans gilvus WH0801 and showed a closer evolutionary distance to SdNABH from S. degradans 2–40 [17], than AhgA from Z. galactanivorans [18] and BpGH117 from B. plebeius [16], consistent with the identity in their amino acid sequences (Fig. 6a). Ahg558 formed a distinct clade with Ahg786, a novel cod-adapted NAOSH, which was recently identified from G. joobiniege G7 [23].

Phylogenetic tree and three-dimensional structure of Ahg558. a Phylogenetic tree of Ahg558 with other α-NAOSHs. The evolutionary history was constructed using the neighbor-joining (NJ) method of the MEGA 6 program. The subjects used to construct the phylogenetic tree included all of the α-NAOSH listed in Table 1, whose genetic information was fully known. Two GH96 family α-agarases from Thalassotalea agarivorans and from Alteromonas agarilytica are also presented in the tree. The accession number of each protein is indicated in parentheses. The evolutionary distance was calculated using the Poisson correction method by removing gaps and missing data. b Three-dimensional structures of Ahg558 and sdNABH. (Upper) The sdNABH (SMTL ID 3r4z.1) was used to model the three-dimensional structure of the Ahg558 in Swiss-Model (http://swissmodel.expasy.org/). The predicted narrow substrate-binding clefts and metal-binding pockets are shown in arrows and gray circles, respectively. (Lower) Amino acid sequence alignment of Ahg558 and sdNABH. The regions responsible for the β-sheet structure are marked with arrows.
The 3-D structure of Ahg558 was constructed using SdNABH [17] as the template in Swiss-Model [32], as recommended with the first priority by the program. The 3-D structures of the two proteins were very similar. Moreover, the presence of a narrow substrate-binding cleft and the metal-binding pocket present in the five-bladed β-propeller catalytic domain of Ahg558 indicated the exo-mode of catalytic action (Fig. 6b). The three catalytic amino acids in the active site commonly found in the GH43 domain were well conserved as Asp-48, Asp-203, and Glu-261 in Ahg558 (Supplementary Fig. S1). Moreover, the N-terminal helices and the conserved signature proposed for domain swapping for dimerization in SdNABH were also well conserved in Ahg558 (7-SxAxxR), supporting our data for dimerization of rAhg558.
Discussion
In this study, we identified a unique cold-adapted alkaline exo-type α-NAOSH, Ahg558. BlastP search in the NCBI revealed more than 100 proteins with 95% or more identity with Ahg558; however, their enzymatic properties have not been characterized. All of them have the well-conserved GH family 43 protein domain (cd08992), which forms a five-bladed β-propeller domain with the catalytic residues. GH43 proteins are inverting enzymes that directly invert the stereochemistry of the anomeric carbon atom of the substrate, and three catalytic amino acids, Asp (catalytic general base), Glu (catalytic general acid), and Asp (orienting the catalytic acid), are involved in the hydrolytic reaction [33].
Despite the presence of GH43 domains, α-NAOSH and α-NABH were proposed to be classified into a new GH family 117 because of their unique enzymatic properties [18]. To date, 221 proteins are listed in the GH117 group, but only 8 proteins have been biochemically characterized (Table 1). Therefore, additional information on GH117 hydrolases is required to cater to their scientific and industrial interests.
Our results clearly demonstrated that rAhg558 is an α-NAOSH but not α-NABH. According to the available literature, 12 enzymes have been reported to hydrolyze NA2 into monomers. Most of them are α-NAOSH, and only those from Cytophaga flevensis [21] and Pseudomonas atlantica [22] were reported as α-NABH. However, Van der Meulen and Harder reported two types of enzyme activity, neoagarobiase and neoagarotera-ase, in the partially purified proteins from Cytophaga flevensis [21]. Therefore, whether Cytophaga flevensis produces α-NABH or α-NAOSH is uncertain. Judging from this fact, it seems that α-NABH is very rare in nature. Unfortunately, the two α-NABHs reported have no further information on their amino acid or nucleotide sequences, and thus, it is impossible to compare their enzymatic activities with that of rAhg558.
rAhg558 is an intracellular dimeric enzyme and its activity is dependent on Mn2+. Most α-NAOSHs are multimeric (mostly dimer) and showed severe inhibitory effects in the presence of metal ions such as Mg2+, Hg2+, Ni2+, Cu2+, Zn2+, and Co2+ (Table 1). Some NAOSHs have been reported to show increased activity in the presence of Mn2+ [34] or Mg2+ [19] ion, but there is no evidence to show that the activity is Mn2+/Mg2+-dependent.
For the members of the GH117 family, a Zn2+-dependent catalytic mechanism was proposed because a crystallographic study on AhgA revealed that Zn2+ ion existed in the metal-binding pocket; but there was no further evidence for the effect of Zn2+ on the enzyme activity [16, 18]. In contrast, the 3-D structure analysis of SdNABH showed no such metal ion bound in the pocket. On the contrary, the activity of SdNABH enzyme was even inhibited by the addition of Zn2+ [17], which was common to many NAOSHs as well as rAhg558 as summarized in Table 1. Therefore, to date, the results on the metal dependence of these enzymes are very uncertain, and further research is necessary. In this context, it may be necessary to elucidate the mechanism of Mn2+ dependence of rAhg558 in relation to the binding of Mn2+ ion in the metal-binding pocket of the enzyme.
rAhg558 showed optimum activity at pH 9.0 and maintained its activity over a broad range of pH from 6.0 to 10.0, which is very unique. All the α-NAOSHs reported had optimum pH in the neutral region (pH 6.0–8.0), and their activity was very sensitive to change in pH (Table 1). Ahg558 showed optimum activity at 30 °C, which is similar to other reported α-NAOSHs [14, 19, 20] and thermal stability up to 30 °C. However, it could retain its enzyme activity at more than 93% of the maximum, in a broad temperature range of 0–40 °C, which is quite distinct from other α-NAOSHs. Recently, we reported a cold-adapted α-NAOSH, Ahg786 from G. joobiniege G7, which had an optimum temperature and pH of 15 °C and 7.0, respectively [23]. Ahg786 retained enzyme activity up to 61% of the maximum at 0 °C, but lost more than 50% (at 30 °C) and 59% (at pH 8.0) of the maximum. Therefore, Ahg558 has an advantage over Ahg786 as well as other α-NAOSHs in that it can act in a wider range of temperatures and pH and has a faster reaction rate in terms of kinetic parameters (Table 1).
Previously, we reported two cold-adapted β-agarases, GH39 family AgaJ9 [35] and GH86 family AgaJ5 [36], from the same strain. The optimum temperature for AgaJ9 activity was 25 °C, and it retained more than 80% of its activity even at 5 °C [35]. Interestingly, genes for both the cold-adapted enzymes, AgaJ9 and Ahg558, are located in close proximity to the chromosome (Fig. 2). AgaJ5 had its optimum enzyme activity at 30 °C and retained 40% of enzymatic activity at 10 °C [36]. The cold-adapted AgaJ9 was able to hydrolyze agarose in the gel state and, hence, has been successfully applied for extraction of DNA from agarose gel [35]. All these results together suggest that G. joobiniege G7 possesses various cold-adapted hydrolytic enzymes. Genomic sequencing analysis revealed that G. joobiniege G7 had several genes encoding hydrolytic enzymes, such as 50 sulfatases, 17 glycoside hydrolases, 13 agarases, 8 β-galactosidases, 3 altronate hydrolases, and 1 cellulase [25]. Some of them may be active at low temperatures of the seawater, such as AgaJ9, AgaJ5, Ahg558, and Ahg786, and may contribute to the growth of the strain by completely hydrolyzing the insoluble sulfated polysaccharides, including agar, for obtaining the carbon source.
Agarolytic enzymes, including α-NAOSHs, are industrially important enzymes; they can be used for preparing agarooligosaccharides (AOs) and NAOSs with various degrees of polymerization. Moreover, the complete digestion of agar yields G and AHG monomers, which are also useful chemical feedstocks. Recently, studies on various biological activities of AOs and NAOs have been accumulated, e.g., anti-inflammation [37], anti-obesity and anti-diabetes [38], cholesterol homeostasis [39], and immunostimulating and anti-tumor [40]. Anti-inflammatory and anti-cariogenic activities, and whitening effects of AHG were also reported [11]. AHG as well as G can be used for bioconversion into biofuels such as ethanol [41], n-butanol [42], and isoprene [42], and sweeteners such as d-tagatose [43]. Moreover, recent studies also showed that G played a very important role in human metabolism, such as energy transfer and galactosylation of complex molecules, and was very useful for a variety of diseases, particularly those affecting the brain [44].
Therefore, the hydrolytic enzymes of agar including α-NAOSHs will be of great industrial importance and their market and demand are expected to increase in the near future. In particular, the Ahg558 enzyme, which can maintain its activity at a wide range of pH and temperatures, is expected to have many advantages in its application, because it can be applied to reaction conditions in which various changes are expected.
References
Nigam, P. S., & Singh, A. (2011). Production of liquid biofuels from renewable resources. Progress in Energy and Combustion Science, 37(1), 52–68.
Park, J. H., Hong, J. Y., Jang, H. C., Oh, S. G., Kim, S. H., Yoon, J. J., & Kim, Y. J. (2012). Use of Gelidium amansii as a promising resource for bioethanol: a practical approach for continuous dilute-acid hydrolysis and fermentation. Bioresource Technology, 108, 83–88.
Knutsen, S. H., Myslabodski, D. E., Larsen, B., & Usov, A. I. (1994). A modified system of nomenclature for red algal galactans. Botanica Marina, 37, 163–169.
Yun, E. J., Shin, M. H., Yoon, J. J., Kim, Y. J., Choi, I. G., & Kim, K. H. (2011). Production of 3,6-anhydro-l-galactose from agarose by agarolytic enzymes of Saccharophagus degradans 2-40. Process Biochemistry, 46(1), 88–93.
Kim, H. T., Yun, E. J., Wang, D., Chung, J. H., Choi, I. G., & Kim, K. H. (2013). High temperature and low acid pretreatment and agarases treatment of agarose for the production of sugar and ethanol from red seaweed biomass. Bioresource Technology, 136, 582–587.
Lee, C. H., Yun, E. J., Kim, H. T., Choi, I. G., & Kim, K. H. (2015). Saccharification of agar using hydrothermal pretreatment and enzymes supplemented with agarolytic β-galactosidase. Process Biochemistry, 50(10), 1629–1633.
Hassairi, I., Ben Amar, R., Nonus, M., & Gupta, B. B. (2001). Production and separation of α-agarase from Altermonas agarlyticus strain GJ1B. Bioresource Technology, 79(1), 47–51.
Zhang, W., Xu, J., Liu, D., Liu, H., Lu, X., & Yu, W. (2018). Characterization of an α-agarase from Thalassomonas sp. LD5 and its hydrolysate. Appllied Microbiology and Biotechnology, 102(5), 2203–2212.
Ohta, Y., Hatada, Y., Miyazaki, M., Nogi, Y., Ito, S., & Horikoshi, K. (2005). Purification and characterization of a novel alpha-agarase from a Thalassomonas sp. Current Microbiology, 50(4), 212–216.
Chi, W. J., Chang, Y. K., & Hong, S. K. (2012). Agar degradation by microorganisms and agar-degrading enzymes. Applied Microbiology and Biotechnology, 94(4), 917–930.
Yun, E. J., Yu, S., & Kim, K. H. (2017). Current knowledge on agarolytic enzymes and the industrial potential of agar-derived sugars. Appllied Microbiology and Biotechnology, 101(14), 5581–5589.
Ramos, K. R. M., Valdehuesa, K. N. G., Maza, P. A. M. M., Nisola, G. M., Lee, W. K., & Chung, W. J. (2017). Overexpression and characterization of a novel α-neoagarobiose hydrolase and its application in the production of D-galactonate from Gelidium amansii. Process Biochemistry, 63, 105–112.
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T. A. P., Rempfer, C., Bordoli, L., Lepore, R., & Schwede, T. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303.
Liu, N., Yang, M., Mao, X., Mu, B., & Wei, D. (2016). Molecular cloning and expression of a new α-neoagarobiose hydrolase from Agarivorans gilvus WH0801 and enzymatic production of 3,6-anhydro-L-galactose. Biotechnology and Applied Biochemistry, 63(2), 230–237.
Ariga, O., Okamoto, N., Harimoto, N., & Nakasaki, K. (2014). Purification and characterization of α-neoagarooligosaccharide hydrolase from Cellvibrio sp. OA-2007. Journal of Microbiology and Biotechnology, 24(1), 48–51.
Hehemann, J. H., Smyth, L., Yadav, A., Vocadlo, D. J., & Boraston, A. B. (2012). Analysis of keystone enzyme in agar hydrolysis provides insight into the degradation of a polysaccharide from red seaweeds. Journal of Biological Chemistry, 287(17), 13985–13995.
Ha, S. C., Lee, S., Lee, J., Kim, H. T., Ko, H. J., Kim, K. H., & Choi, I. G. (2011). Crystal structure of a key enzyme in the agarolytic pathway, α-neoagarobiose hydrolase from Saccharophagus degradans 2–40. Biochemical and Biophysical Research Communications, 412(2), 238–244.
Rebuffet, E., Groisillier, A., Thompson, A., Jeudy, A., Barbeyron, T., Czjzek, M., & Michel, G. (2011). Discovery and structural characterization of a novel glycosidase family of marine origin. Environmental Microbiology, 13(5), 1253–1270.
Suzuki, H., Sawai, Y., Suzuki, T., & Kawai, K. (2002). Purification and characterization of an extracellular alpha-neoagarooligosaccharide hydrolase from Bacillus sp. MK03. Journal of Bioscience and Bioengineering, 93(5), 456–463.
Sugano, Y., Kodama, H., Terada, I., Yamazaki, Y., & Noma, M. (1994). Purification and characterization of a novel enzyme, alpha-neoagarooligosaccharide hydrolase (alpha-NAOS hydrolase), from a marine bacterium, Vibrio sp. strain JT0107. Journal of Bacteriology, 176(22), 6812–6818.
Van der Meulen, H. J., & Harder, W. (1976). Characterization of the neoagarotetra-ase and neoagarobiase of Cytophaga flevensis. Antonie Van Leeuwenhoek, 42(1-2), 81–94.
Day, D. F., & Yaphe, W. (1975). Enzymatic hydrolysis of agar: purification and characterization of neoagarobiose hydrolase and p-nitrophenyl alpha-galactoside hydrolase. Canadian Journal of Microbiology, 21(10), 1512–1518.
Asghar, S., Lee, C. R., Park, J. S., Chi, W. J., Kang, D. K., & Hong, S. K. (2018). Identification and biochemical characterization of a novel cold-adapted 1,3-α-3,6-anhydro-L-galactosidase, Ahg786, from Gayadomonas joobiniege G7. Applied Microbiology and Biotechnology, 102(20), 8855–8866.
Chi, W. J., Park, J. S., Kwak, M. J., Kim, J. F., Chang, Y. K., & Hong, S. K. (2013). Isolation and characterization of a novel agar-degrading marine bacterium, Gayadomonas joobiniege gen, nov, sp. nov., from the Southern Sea, Korea. Korean Journal of Microbiology and Biotechnology, 23(11), 1509–1518.
Kwak, M. J., Song, J. Y., Kim, B. K., Chi, W. J., Kwon, S. K., Choi, S., Chang, Y. K., Hong, S. K., & Kim, J. F. (2012). Genome sequence of agar degrading marine bacterium Alteromonadaceae sp. strain G7. Journal of Bacteriology, 194(24), 6961–6962.
Laemmli, U,K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227(5259), 680–685.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72(1-2), 248–254.
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry, 31(3), 426–428.
Lineweaver, H., & Burk, D. (1934). The determination of enzyme dissociation constants. Journal of the American Chemical Society, 56(3), 658–666.
Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4(4), 406–425.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., & Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution, 30(12), 2725–2729.
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Gallo Cassarino, T., Bertoni, M., Bordoli, L., & Schwede, T. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research, 42(W1), W252–W258.
Nurizzo, D., Turkenburg, J. P., Charnock, S. J., Roberts, S. M., Dodson, E. J., McKie, V. A., Taylor, E. J., Gilbert, H. J., & Davies, G. J. (2002). Cellvibrio japonicus alpha-L-arabinanase 43A has a novel five-blade beta-propeller fold. Nature Structructural Biology, 9(9), 665–668.
Watanabe, T., Kashimura, K., & Kirimura, K. (2016). Purification, characterization and gene identification of a α-neoagarooligosaccharide hydrolase from an alkaliphilic bacterium Cellvibrio sp. WU-0601. Journal of Molecular Catalysis B: Enzymatic, 133, S328–S336.
Jung, S., Lee, C. R., Chi, W. J., Bae, C. H., & Hong, S. K. (2017). Biochemical characterization of a novel cold-adapted GH39 β-agarase, AgaJ9, from an agar-degrading marine bacterium Gayadomonas joobiniege G7. Applied Microbiology and Biotechnology, 101(5), 1965–1974.
Lee, Y. R., Jung, S., Chi, W. J., Bae, C. H., Jeong, B. C., Hong, S. K., & Lee, C. R. (2018). Biochemical characterization of a novel GH86 β-agarase producing neoagarohexaose from Gayadomonas joobiniege G7. Journal of Microbiology and Biotechnology, 28(2), 284–292.
Higashimura, Y., Naito, Y., Takagi, T., Mizushima, K., Hirai, Y., Harusato, A., Ohnogi, H., Yamaji, R., Inui, H., Nakano, Y., & Yoshikawa, T. (2013). Oligosaccharides from agar inhibit murine intestinal inflammation through the induction of heme oxygenase-1 expression. Journal of Gastroenterology, 48(8), 897–909.
Hong, S. J., Lee, J. H., Kim, E. J., Yang, H. J., Park, J. S., & Hong, S. K. (2017). Anti-obesity and anti-diabetic effect of neoagarooligosaccharides on high-fat diet-induced obesity in mice. Marne Drugs, 15(4). https://doi.org/10.3390/md15040090.
Yang, J. H., Cho, S. S., Kim, K. M., Kim, J. Y., Kim, E. J., Park, E. Y., Lee, J. H., & Ki, S. H. (2017). Neoagarooligosaccharides enhance the level and efficiency of LDL receptor and improve cholesterol homeostasis. Journal of Functional Foods, 38, 529–539.
Lee, M. H., Jang, J. H., Yoon, G. Y., Lee, S. J., Lee, M. G., Kang, T. H., Han, H. D., Kim, H. S., Choi, W. S., Park, W. S., Park, Y. M., & Jung, I. D. (2017). Neoagarohexaose-mediated activation of dendritic cells via Toll-like receptor 4 leads to stimulation of natural killer cells and enhancement of antitumor immunity. BMB Reports, 50(5), 263–268.
Ra, C. H., Kim, Y. J., Lee, S. Y., Jeong, G. T., & Kim, S. K. (2015). Effects of galactose adaptation in yeast for ethanol fermentation from red seaweed, Gracilaria verrucosa. Bioprocess and Biosystems Engineering, 38(9), 1715–1722.
Lim, H. G., Lim, J. H., & Jung, G. Y. (2015). Modular design of metabolic network for robust production of n-butanol from galactose-glucose mixtures. Biotechnology for Biofuels, 8(1), 137. https://doi.org/10.1186/s13068-015-0327-7.
Patel, M. J., Patel, A. T., Akhani, R., Dedania, S., & Patel, D. H. (2016). Bioproduction of D-tagatose from D-galactose using phosphoglucose isomerase from Pseudomonas aeruginosa PAO1. Appllied Biochemistry and Biotechnology, 179(5), 715–727.
Coelho, A. I., Berry, G. T., & Rubio-Gozalbo, M. E. (2015). Galactose metabolism and health. Current Opinion in Clinical Nutrition and Metabolic Care, 18(4), 422–427.
Funding
This work was carried out with the support of “Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01328801)” Rural Development Administration, Republic of Korea, and 2017 Research Fund of Myongji University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Ethics Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PPTX 1.46 mb)
Rights and permissions
About this article

Cite this article
Asghar, S., Lee, CR., Chi, WJ. et al. Molecular Cloning and Characterization of a Novel Cold-Adapted Alkaline 1,3-α-3,6-Anhydro-l-galactosidase, Ahg558, from Gayadomonas joobiniege G7. Appl Biochem Biotechnol 188, 1077–1095 (2019). https://doi.org/10.1007/s12010-019-02963-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-019-02963-w