
Abstract
Background
Agarose is hydrolyzed easily to yield oligosaccharides, designated as agaro-oligosaccharides (AGOs). Recently, it has been demonstrated that AGOs induce heme oxygenase-1 (HO-1) expression in macrophages and that they might lead to anti-inflammatory property. Nevertheless, the molecular mechanism of AGO-mediated HO-1 induction remains unknown, as does AGOs’ ability to elicit anti-inflammatory activity in vivo. This study was undertaken to uncover the mechanism of AGO-mediated HO-1 induction and to investigate the therapeutic effect of AGOs on intestinal inflammation.
Methods
Mice were treated with 2,4,6-trinitrobenzene sulfonic acid (TNBS) to induce colitis. The respective degrees of mucosal injury of mice that had received AGO and control mice were compared. We investigated HO-1 expression using Western blotting, quantitative real-time PCR (qRT-PCR), and immunohistochemistry. The expression of tumor necrosis factor-α (TNF-α) was measured using qRT-PCR and enzyme-linked immunosorbent assay.
Results
AGO administration induced HO-1 expression in colonic mucosa. The induction was observed mainly in F4/80 positive macrophages. Increased colonic damage and myeloperoxidase activity after TNBS treatment were inhibited by AGO administration. TNBS treatment induced TNF-α expression, and AGO administration suppressed induction. However, HO inhibitor canceled AGO-mediated amelioration of colitis. In RAW264 cells, AGOs enhanced HO-1 expression time-dependently and concentration-dependently and suppressed lipopolysaccharide-induced TNF-α expression. Furthermore, agarotetraose-mediated HO-1 induction required NF-E2-related factor 2 function and phosphorylation of c-jun N-terminal kinase.
Conclusions
We infer that AGO administration inhibits TNBS-induced colitis in mice through HO-1 induction in macrophages. Consequently, oral administration of AGOs might be an important therapeutic strategy for inflammatory bowel disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel diseases (IBD), including ulcerative colitis and Crohn’s disease, are chronically relapsing disorders of the gastrointestinal tract. Genetic and environmental factors, infection agents, and abnormal activation of mucosal immune system have been linked to onset of IBD, the precise pathogenesis of which remains poorly understood [1]. Consequently, radical therapies for IBD are unavailable. The pharmacologic agents used in IBD therapies including corticosteroids and immunosuppressive agents are only moderately effective. They have been associated with severe side effects and long-term toxicity [2]. Therefore, it is important to investigate safe and effective agents for IBD therapies.
Heme oxygenase (HO) catalyzes the first and rate-limiting enzymatic step of heme degradation and produces carbon monoxide, free iron, and biliverdin [3]. In general, three distinct HO isozymes are known: HO-1, HO-2 and HO-3. One of them, HO-1, is a stress-inducible protein. HO-1 expression is induced by various stressors such as oxidative stress, ultraviolet irradiation, inflammatory cytokine, and heavy metals [4–6]. Because of the strong adaptive response of HO-1 to various stressors, HO-1 expression has been regarded as an adaptive cellular response against inflammatory response and oxidative injury [7]. The stress-mediated induction of HO-1 is regulated by several transcriptional factors (NF-E2-related factor 2 (Nrf2), BTB and CNC homolog 1 (Bach1), hypoxia-inducible factor-1) and signaling cascades [mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)] [8–11]. These transcriptional factors and signaling cascades that are putatively involved in HO-1 induction mutually differ depending on stressor and cell species [11].
Previously, we demonstrated that HO activity and HO-1 expression increased drastically in experimental models of intestinal inflammation [12]. The HO-1 expression was localized in inflammatory cells, mainly mononuclear cells in the intestinal submucosal layer, but not in epithelial cells [13]. Furthermore, transferring HO-1 highly expressing macrophages ameliorates colonic inflammation in mice [14]. Conversely, the intestinal inflammation was potentiated by administration of HO inhibitor [15]. Consequently, HO-1 expression is regarded as a therapeutic target in patients with IBD [16]. However, although several reports describe the therapeutic effect of HO-1 inducers, such as hemin and cobalt-protoporphyrin, in intestinal inflammation, safe and useful inducers for IBD therapies are not known [17, 18].
Agar, which is extracted easily from red algae, is widely used as a food and gelling agent in Asian countries. Agarose, the main component of polysaccharides in agar, is hydrolyzed easily to yield oligosaccharides. The resultant oligosaccharides termed agaro-oligosaccharides (AGOs) have been investigated widely in terms of structure and bioactivity [19–21]. AGOs have repeating agarobiose (AB) units composed of D-galactose at the non-reducing end and 3,6-anhydro-L-galactose at the reducing end. In recent years, previous reports show that AGOs induced HO-1 expression in macrophages and that they might engender an anti-inflammatory effect [22]. Nevertheless, the molecular mechanism of AGO-mediated HO-1 induction remains unknown. Moreover, AGOs' capability to elicit anti-inflammatory activity in vivo through the induction of HO-1 remains unknown. This study was undertaken to investigate the therapeutic effect of AGOs in murine 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis, which shares both immunological and pathological features with human IBD. Furthermore, we clarify the molecular mechanism of AGO-mediated induction of HO-1 using a mouse macrophage-like cell line.
Materials and methods
Animals
Seven-week-old or eight-week-old male C57BL/6 wild type mice used for this study were obtained from Shimizu Laboratory Supplies Co., Ltd. (Kyoto, Japan). Mice were kept at 18–24 °C and 40–70 % relative humidity, with a 12-h light/dark cycle, and were allowed free access to water and a diet for one week during their acclimatization period. Mice were fed a rodent diet (CE-2; Nihon Clea, Tokyo, Japan). Mice were maintained and the experimental procedures conducted in accordance with the NIH guidelines for the use of experiments animals. All experimental protocols were approved by the Animal Care Committee of Kyoto Prefectural University of Medicine (the permission number M23-113).
Administration of agaro-oligosaccharides
The commercial AGOs used for this study were gifts from Takara Bio Inc. (Shiga, Japan). AGOs contain four kinds of oligosaccharides which have different degrees of polymerization, termed agarobiose (AB), agarotetraose (AT), agarohexaose (AH), and agarooctaose (AO), respectively [22]. Mice were treated with 4 g/kg/day of AGOs administered orally via a catheter for 3 or 6 days. The sham group was treated with the vehicle used to administer the AGOs for 6 days. The mice were killed 24 h after final administration of AGOs, and the colon of each was harvested. Colonic mucosa was scraped off using two glass slides.
Induction of colitis
For induction of colitis, 100 mg/kg TNBS (Sigma-Aldrich Corp., St. Louis, MO, USA) dissolved in 30 % ethanol was administered intrarectally to anesthetized (ketamine/xylazine) mice via a catheter [23]. The sham group received 30 % ethanol. Three days after TNBS administration, the mice were killed humanely. Then the colon damage was graded in terms of the presence of visible damage, serosal adhesions, diarrhea, strictures, and bowel wall thickening, as described previously [24]. Subsequently, colonic mucosa was harvested. All scoring was performed by the same individual under blind conditions to prevent observer bias. In some experiments, 10 mg/kg tin protoporphyrin (SnPP; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), which is an HO inhibitor, that had been dissolved in PBS was administered intraperitoneally. The sham group received PBS.
For histologic evaluation, specimens of the distal colon were fixed in 10 % neutral buffered formalin. After fixation, specimens were embedded in paraffin, cut to 3-μm thickness using a microtome cryostat, and mounted on MAS-coated slides. Sections were stained with hematoxylin and eosin.
Measurement of myeloperoxidase activity
Tissue-associated myeloperoxidase (MPO) activity was determined using a modification of the method of Grisham et al. [25] as an index of neutrophil accumulation. Colonic mucosa was homogenized with 1.0 ml of PBS using a Teflon Potter–Elvehjem homogenizer with subsequent centrifugation at 20,000×g for 15 min. The resultant precipitation was resuspended in 0.3 ml of 50 mM potassium phosphate buffer (pH 5.4) containing 0.5 % hexadecyl trimethylammonium bromide and sonicated, with subsequent centrifugation at 20,000×g for 15 min. The supernatant was stored at −80 °C. MPO activity was assessed by measuring the H2O2-dependent oxidation of 3,3′,5,5′-tetramethylbenzidine. One unit of enzyme activity was defined as the amount of MPO that caused a change in the 1.0/min absorbance at 655 nm and 25 °C.
Preparation of each oligosaccharide
The commercial AGOs contain AB, AT, AH, and AO. For preparation of each oligosaccharide, AGOs were subjected to gel filtration chromatography in a Toyopearl HW-40C column (26 mm dia. 100 cm; Tosoh Corp. Tokyo, Japan) at the rate of 0.2 ml/min with pure water as the carrier to separate the AGOs of various sizes. For preparation of neoagarotetraose (NAT), agar was suspended in 100 mM sodium phosphate buffer (pH 7.0) to 10 % (w/v) and maintained at 37 °C for 7 days in the presence of β-agarase (Sigma-Aldrich Corp.) with subsequent removal of the insoluble agars. The resultant reaction mixture was subjected to gel filtration chromatography as described above. The fractions containing individual oligosaccharide were identified using Thin Layer Chromatography with silica gel 60 F254 (Merck and Co. Inc., Darmstadt, Germany) with butanol:ethanol:water (5:5:1) as the solvent system.
Cell culture
RAW264 (a mouse macrophage-like cell line) cells were cultured in Dulbecco’s modified Eagle’s medium–high glucose (4.5 g/l glucose) supplemented with 10 % fetal bovine serum and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin). The RAW264 cells were obtained from the RIKEN BioResource center. Cells were maintained at 37 °C in a 5 % CO2/95 % air atmosphere at 100 % humidity unless otherwise specified.
Immunohistochemical and immunofluorescence staining
Immunohistochemical and immunofluorescence staining of HO-1 and F4/80 in mouse colon were performed as described previously [26]. In brief, for immunohistochemical staining of HO-1, sections of the distal colon were incubated with rabbit polyclonal anti-HO-1 antibody (SPA-895; Enzo Life Sciences Inc., Farmingdale, NY, USA) with subsequent incubation with Histofine Simple Stain mouse MAX PO (Nichirei Biosciences Inc., Tokyo, Japan) as a secondary antibody. The bound antibodies were visualized using diaminobenzidine as a chromogen substrate reagent. Negative controls for non-specific binding incubated with secondary antibodies were confirmed to produce no signal. All sections were counterstained with hematoxylin. Sections were finally dehydrated, cleared, and coverslipped. For immunofluorescence staining of HO-1 and F4/80, sections of the distal colon were incubated with rabbit polyclonal anti-HO-1 (SPA-895) and rat monoclonal anti-F4/80 (ab6640; Abcam, Cambridge, MA, UK) antibodies. The sections were incubated further with Alexa Fluor 594-conjugated secondary anti-rabbit IgG and Alexa Fluor 488-conjugated secondary anti-rat IgG (Invitrogen Corp., Carlsbad, CA, USA) with subsequent inspection using a confocal laser scanning microscope (FV10i; Olympus Corp., Tokyo, Japan).
For immunofluorescence staining of HO-1 in RAW264 cells, RAW264 cells seeded on μ-Dish 35 mm (ibidi GmbH, Munich, Germany) were cultured in the presence of AGOs. Cells were fixed in 4 % paraformaldehyde in PBS, followed by incubation with PBS containing 0.1 % Triton X-100. Cells were incubated with Protein Block Serum-Free (Dako Corp., Carpenteria, CA, USA) and then incubated with rabbit polyclonal anti-HO-1 antibody (SPA-895). Cells were further incubated with Alexa Fluor 488-conjugated secondary anti-rabbit IgG (Invitrogen Corp.) and labeled with Hoechst 33342 to stain nuclear chromatin, with subsequent inspection using a confocal laser scanning microscope.
Immunosorbent assay of tumor necrosis factor-α
Colonic mucosal homogenates were centrifuged at 20,000×g for 15 min with subsequent harvesting the resultant supernatants. RAW264 cells were cultured with 100 μg/ml AGOs for 12 h. Then the cells were stimulated by 1 ng/ml lipopolysaccharide (LPS) for 6 h with subsequent collection of the supernatants. The tumor necrosis factor-α (TNF-α) concentrations in these supernatants were measured using an enzyme-linked immunosorbent assay (ELISA) kit (eBioscience, San Diego, CA, USA) specific for mouse TNF-α. Assays were performed according to the manufacturer’s instructions. After color development, optimal densities were measured at 450 nm using a plate reader (Spectramax M2; Molecular Devices Corp. Sunnyvale, CA, USA).
RNA interference
As a target sequence for Nrf2 siRNA, we used 5′-GCAUGUUACGUGAUGAGGAUGGAAA-3′ (Invitrogen Corp.). The stealth RNAi negative control Low GC was used as a control siRNA. The duplex (10 nM) was introduced into RAW264 cells using Lipofectamine RNAiMAX reagent and Opti-MEM (Invitrogen Corp.) for 48 h according to the manufacturer’s instructions.
Western blotting
Colonic mucosa of the experimental animal was lysed in CelLytic M Tissue Lysis Reagent (Sigma-Aldrich Corp.). RAW264 cells cultured in the presence of AGOs and NAT were harvested and lysed in CelLytic M Cell Lysis Reagent (Sigma-Aldrich Corp.). These lysates were centrifuged at 20,000×g for 15 min. The supernatants were analyzed using Western blot with a NuPAGE 10 % Bis–Tris Gel and a NuPAGE western transfer system according to the manufacturer’s instructions (Invitrogen Corp.) with rabbit polyclonal anti-HO-1 (SPA-895), anti-JNK (c-jun N-terminal kinase), anti-phospho-JNK, anti-ERK (extracellular signal-regulated kinase), anti-phospho-ERK, anti-p38, anti-phospho-p38, anti-STAT3 (signal transducers and activators of transcription 3), anti-phospho-STAT3 (Y705) (Cell Signaling Technology Inc., Beverly, MA, USA), anti-TLR2 (toll-like receptor 2), anti-TLR9 (IMGENEX, San Diego, CA, USA), and anti-lamin B1 (Ab16048; Abcam) antibodies, goat polyclonal anti-β-actin (Ab8229; Abcam), anti-TLR4 (Ab53629; Abcam) antibodies, and mouse monoclonal anti-p65 (F-6; Santa Cruz Biotechnology Inc.) antibody with subsequent immunoreaction with the horseradish peroxidase-conjugated anti-rabbit, anti-goat, and anti-mouse IgG, respectively. The immunoreactive proteins were visualized with ECL Plus Western Blotting Detection System (GE Healthcare, Piscataway, NJ, USA).
Quantitative real-time PCR
Total RNA was extracted from colonic mucosa and RAW264 cells with Isogen (Nippon Gene Co. Ltd., Tokyo, Japan). It was then reverse transcribed. The resultant cDNA was subjected to quantitative real-time PCR (qRT-PCR) using specific primers for TNF-α (forward primer: 5′-ATCCGCGACGTGGAACTG-3′, reverse primer: 5′-ACCGCCTGGAGTTCTGGAA-3′), β-actin (forward primer: 5′-TATCCACCTTCCAGCAGATGT-3′, reverse primer: 5′-AGCTCAGTAACAGTCCGCCTA-3′), HO-1 (forward primer: 5′-CCTCACTGGCAGGAAATCATC-3′, reverse primer: 5′-CCTCGTGGAGACGCTTTACATA-3′), Nrf2 (forward primer: 5′-CTCCTTAGACTCAAATCCCACCTTAA-3′, reverse primer: 5′-TGGGCTCTGCTATGAAAGCA-3′), IL-1β (forward primer: 5′-TCGCTCAGGGTCACAAGAAA-3′, reverse primer: 5′-CATCAGAGGCAAGGAGGAAAAC-3′), IL-6 (forward primer: 5′-GACCTGCAGCAATACCATTGAC-3′, reverse primer: 5′-CCGTTATTTCCAAACTGCATCA-3′), KC (keratinocyte chemoattractant) (forward primer: 5′-TGTCAGTGCCTGCAGACCAT-3′, reverse primer: 5′-CCTGAGGGCAACACCTTCA-3′), IL-12 (forward primer: 5′-CACCCTTGCCCTCCTAAACC-3′, reverse primer: 5′-CACCTGGCAGGTCCAGAGA-3′), and IL-17A (forward primer: 5′-TCATCTGTGTCTCTGATGCTGTTG-3′, reverse primer: 5′-TCGCTGCTGCCTTCACTGT-3′). PCR was performed with a Power SYBR Green PCR Master Mix and a real-time PCR system (7300; Applied Biosystems, Foster City, CA, USA). Relative quantifications of gene expression with qRT-PCR data were calculated relative to β-actin. PCR conditions were denaturation at 95 °C for 15 s, primer-annealing at 60 °C for 1 min, with subsequent melting curve analysis in which the temperature was increased from 60 to 95 °C.
Statistical analysis
Comparisons among three or more groups were performed using one-way analysis of variance (ANOVA) with Tukey’s post hoc testing. Comparisons with two groups were performed using Mann–Whitney’s U test. Statistical analyses were performed using statistical software (JMP ver. 8.0.1; SAS Institute Inc., Cary, NC, USA). All results are expressed as mean ± SEM, and differences were considered statistically significant at a p value of <0.05.
Results
AGO administration up-regulates the expression of HO-1 in colonic mucosa
To study the effect of AGOs on HO-1 expression in mice colonic mucosa, mice were treated with AGOs administered orally for the designated time periods (Fig. 1a). In response to the administration of AGOs, HO-1 expression was up-regulated significantly compared to the vehicle-administered group (Fig. 1b). Next, to investigate HO-1 localization in colonic mucosa, we performed HO-1 immunohistochemical staining. Induced HO-1 expression was observed mainly in mononuclear cells in colonic submucosa (Fig. 1c, middle and right panels). The HO-1-positive mononuclear cells were subtle in vehicle-administered mice (Fig. 1c, left panel). To investigate the HO-1-positive cells precisely, we also performed immunofluorescence staining of the colonic mucosa derived from AGO-administered mice. As presented in Fig. 1d, HO-1 immunoreactivity was coexpressed with F4/80, which is the marker protein of macrophage. Consequently, AGO administration caused a marked increase in the population of HO-1-positive macrophages.

Effect of AGO administration on HO-1 expression in mouse colonic mucosa. a Schematic representations of AGO administration for individual mouse groups are depicted. Black arrowheads indicate the administration of AGOs. b Protein contents in colonic mucosa were analyzed by Western blot with anti-HO-1 (upper panel) and anti-β-actin antibodies (middle panel). Expression levels of HO-1 were quantified by normalization to expression levels of β-actin (lower panel). Data represent the mean ± SE of eight mice. Statistically significant differences (p < 0.05) are denoted by different letters. c Colonic specimens from mice receiving vehicle (left panel) or AGOs (middle and right panels) were analyzed by immunohistochemical staining of HO-1. All sections were counterstained with hematoxylin (×40). d The colonic specimen from mice receiving AGOs (Day 6) was analyzed using coimmunofluorescence staining of HO-1 and F4/80. Arrowheads indicate both HO-1 and F4/80 immunopositive cells (×120)
AGO administration inhibits TNBS-induced colitis
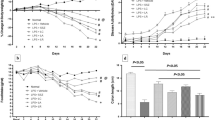
To investigate the effect of AGO administration on colitis symptoms, colitis was induced in vehicle-treated or AGO-treated mice, as indicated in the experimental schedule in Fig. 2a. TNBS treatment caused severe weight loss. The reduction was improved significantly by AGO administration (Fig. 2b). In sham groups, AGO administration had no effect on weight variations. In mice exposed to TNBS, macroscopic findings in the colon showed severe colitis with hyperemia, edema, thickening, and ulceration. The AGO administration inhibited macroscopic lesions without influence in the sham groups (Fig. 2c). Furthermore, the damage score of TNBS-induced colitis was evaluated as described in the “Materials and methods”. The TNBS treatment-induced increase in the colonic damage score was suppressed significantly by AGO administration (Fig. 2d). Consequently, the AGO administration drastically inhibits TNBS-induced colitis both macroscopically and histologically.

Effect of AGO administration on TNBS-induced colitis. a A schematic representation of the experimental schedule for the individual mouse groups: black and gray arrowheads, respectively, indicate the administration of AGOs and TNBS. b Body weight variance of mice treated with or without TNBS, in the presence or absence of AGOs. Data represent the mean ± SE of 13 mice. c Macroscopic findings in the colon from mice treated with or without TNBS were represented. In sham groups, AGO administration had no effect on macroscopic finding in the colon. In TNBS-treated groups, AGO administration inhibited TNBS-induced colitis. d The damage score of TNBS-induced colitis was evaluated as described in the “Materials and methods”. Data are expressed as a scatter plot. Bar indicates the mean of 13 mice. Statistically significant differences (p < 0.05) are denoted by different letters
AGO administration suppresses neutrophil accumulation in colonic mucosa
Representative histological images from TNBS-treated mice are depicted in Fig. 3a. Histological grading of the hematoxylin/eosin-stained colonic sections of mice that received TNBS alone demonstrated severe mucosal and submucosal inflammation, as well as extensive crypt distortion (Fig. 3a, upper panel). The AGO administration drastically reduced these typical colitic features (Fig. 3a, lower panel). Furthermore, tissue-associated MPO activity, which is the index of neutrophil accumulation, was measured in colonic mucosal homogenates. Compared to that in the sham groups, MPO activity increased drastically after TNBS treatment. The AGO administration inhibited the increase in MPO activity significantly (Fig. 3b). In the sham groups, AGO administration had no effect on the MPO activity.

Effect of AGO administration on neutrophil accumulation. a Histological appearance of the colon is shown for TNBS-treated mice receiving vehicle (upper panel) or AGOs (lower panel). All sections were stained with H&E (×40). b The colonic mucosa was scraped off and then homogenized. Tissue-associated MPO activity was determined as an index of neutrophil accumulation in the colonic mucosa. Data represent the mean ± SE of seven mice. Statistically significant differences (p < 0.05) are shown as different letters
AGO administration suppresses expressions of inflammatory cytokine in colonic mucosa
Regarding HO-1 expression, both in vehicle- and AGO-treated mice, HO-1 expression in colonic mucosa was markedly increased after TNBS treatment (Fig. 4a). In sham groups, HO-1 expression tended to increase in AGO-treated mice compared with vehicle-treated mice. Additionally, we performed HO-1 immunohistochemical staining. Both in vehicle- and AGO-treated mice, TNBS treatment resulted in intestinal inflammation accompanied with the accumulation of HO-1-positive staining mononuclear cells (Fig. 4b). To analyze the effect of AGO administration further, we determined the expression levels of inflammatory cytokine in the colonic mucosa. Comparison to the sham groups showed that TNBS treatment markedly increased the expressions of TNF-α, IL-1β, KC (a functional mouse homolog for human IL-8), IL-12, and IL-17A, but not of IL-6. The increase of TNF-α, IL-12, and IL-17A expressions were suppressed significantly by AGO administration (Fig. 4c, Supplementary Fig. 1d and 1e). The increase of KC expression was tended to be lower in AGO-treated mice compared with vehicle-treated mice (Supplementary Fig. 1c). In contrast, the elevation of IL-1β expression by TNBS treatment was not affected in AGO-treated mice (Supplementary Fig. 1a). Similar results were obtained when the protein contents of TNF-α in the colonic mucosa were determined using ELISA (Fig. 4d). These results indicate that the AGO administration significantly suppresses TNBS-induced colitis biochemically.

Analysis of HO-1 and TNF-α expressions in TNBS-induced colitis model. a Protein content of HO-1 in the colonic mucosa was analyzed by Western blot with anti-HO-1 (upper panel) and anti-β-actin antibodies (middle panel). Expression levels of HO-1 were quantified by normalization to expression levels of β-actin (lower panel). b Colonic specimens from TNBS-treated mice receiving vehicle (left panel) or AGOs (right panel) were analyzed by immunohistochemical staining of HO-1. All sections were counterstained with hematoxylin (×40). c Colonic mucosa was scraped off. Then total RNA was extracted. The resultant RNA was subjected to qRT-PCR, and the level TNF-α mRNA was evaluated. Data were normalized to the level of β-actin. d The protein content of TNF-α in the colonic mucosa was determined using an enzyme-linked immunosorbent assay ELISA kit specific for mouse TNF-α. Data represent the mean ± SE of eight mice. Statistically significant differences (p < 0.05) are denoted by different letters
Inhibition of HO-1 cancels the AGO-mediated amelioration of colitis
To confirm whether the AGO-mediated inhibition of colitis results from the protective activity of HO-1, we evaluated the TNBS-induced colitis in the presence of SnPP, which is an HO inhibitor, as indicated in the experimental schedule in Fig. 5a. Macroscopic findings in the colon showed that TNBS-induced severe colitis was inhibited in AGO-treated mice (Fig. 5b). The inhibitory effect of AGOs was canceled by SnPP administration. Similar results were obtained when the damage score of TNBS-induced colitis was evaluated as described in the “Materials and methods” (Fig. 5c). Furthermore, histological grading of hematoxylin/eosin-stained colonic sections of mice showed TNBS-induced severe mucosal and submucosal inflammation and crypt distortion were reduced in AGO-treated mice (Fig. 5d). The AGO-mediated reduction of these typical colitic features was canceled by SnPP administration.

Involvement of HO-1 in AGO-mediated inhibition of colitis. a Schematic representation of the experimental schedule for the individual mouse groups is depicted. The black, gray, and white arrowheads, respectively, indicate the administration of AGOs, TNBS, and SnPP. b Macroscopic findings in the colon from mice treated with or without SnPP were represented. In AGO-treated groups, the inhibitory effect of AGOs on TNBS-induced colitis was canceled by SnPP administration. c The damage score of TNBS-induced colitis was evaluated as described in the “Materials and methods”. Data are expressed as a scatter plot. Bar indicates the mean of 10 or 12 mice. Statistically significant differences (p < 0.05) are denoted by different letters. d Histological appearance of the colon is shown for TNBS-treated mice receiving AGOs or/and SnPP. All sections were stained with H&E (×40)
AGOs enhance HO-1 expression in RAW264 cells
To characterize the molecular mechanism of HO-1 induction by AGOs further, we examined HO-1 expression in RAW264 cells cultured in the presence of AGOs. RT-PCR analysis showed that AGOs increased the HO-1 mRNA level in a time-dependent manner (Fig. 6a). In association with the increased HO-1 mRNA level, the expression of HO-1 protein was also increased in a time-dependent manner with a peak at 12 h (Fig. 6b). The increase in HO-1 protein expression was dependent on the AGO concentration (Fig. 6c). To investigate the intracellular distribution of HO-1 in RAW264 cells in response to AGOs, we performed immunofluorescence staining using confocal microscopy. AGOs induced the expression of HO-1 remarkably. The expression was detected mainly in cytoplasm, but not in Hoechst-stained nuclei (Fig. 6d).

Effect of AGOs on HO-1 expression in RAW264 cells. a, b RAW264 cells were cultured with 100 μg/ml AGOs for the indicated time period. The RNA was extracted and subjected to qRT-PCR. Then the level of HO-1 mRNA was evaluated. Data were normalized to the level of β-actin. Data represent the mean ± SE (n = 5). Statistically significant differences (p < 0.05) are denoted by different letters (a). The cell lysates were analyzed using Western blot with anti-HO-1 and anti-β-actin antibodies (b). c RAW264 cells were cultured with increasing concentrations of AGOs (10, 25, 50, 100, and 200 μg/ml) for 12 h. The cell lysates were analyzed using Western blot with anti-HO-1 and anti-β-actin antibodies. d RAW264 cells were cultured with 100 μg/ml AGOs for 12 h. The expression and localization of HO-1 were analyzed using immunofluorescence
AGOs suppress LPS-induced expressions of inflammatory cytokine in RAW264 cells
To confirm the anti-inflammatory effect of AGO-induced HO-1 in RAW264 cells, we determined the expressions of inflammatory cytokine. Stimulation of LPS increased the TNF-α and IL-1β expressions significantly, and these increase were suppressed by AGO treatment without influence on TLR expressions (Fig. 7a, Supplementary Fig. 2a and 3). The AGO-mediated inhibition of TNF-α induction was canceled by SnPP treatment. Unstimulated RAW264 cells expressed a small amount of TNF-α during the same time period. In contrast, stimulation of LPS had no effect on IL-6, KC, IL-12, and IL-17A expressions (Supplementary Fig. 2b). With ELISA also, LPS increased the production of TNF-α in RAW264 cells significantly, and this induction was inhibited by AGO treatment (Fig. 7b). The AGO-mediated suppression of TNF-α induction was canceled by SnPP treatment. Results show that up-regulation of HO-1 by AGOs contributes to the inhibition of inflammatory cytokine expressions from LPS-stimulated RAW264 cells.

Effect of AGOs on TNF-α expression in LPS-stimulated RAW264 cells. a RAW264 cells were cultured with 100 μg/ml AGOs for 12 h. Then the cells were stimulated by 1 ng/ml LPS for 2 h in the presence or absence of 1 μM SnPP. The RNA was extracted and subjected to qRT-PCR, and the level of TNF-α mRNA was evaluated. Data were normalized to the level of β-actin. b RAW264 cells were cultured with 100 μg/ml AGOs for 12 h. Then the cells were stimulated by 1 ng/ml LPS for 6 h in the presence or absence of 1 μM SnPP. The concentration of TNF-α in the supernatant was determined using an enzyme-linked immunosorbent assay ELISA kit specific for mouse TNF-α. Data represent the mean ± SE (n = 4). Statistically significant differences (p < 0.05) are signified by different letters
Individual components of AGOs enhance HO-1 expression in RAW264 cells
We examined the effects of individual components of commercial AGOs on HO-1 expression. The AGOs were separated into AB, AT, AH, and AO, as described in “Materials and methods”. Irrespective of the length of each oligosaccharide, AB, AT, AH, and AO showed similar induction of HO-1 expression (Fig. 8a), and AT up-regulated HO-1 expression at concentrations higher than 5 μM (Fig. 8b). Meanwhile, NAT, having D-galactose at the reducing end, had no effect on HO-1 expression even at 100 μM.

Effects of various oligosaccharides on HO-1 expression in RAW264 cells. a, b RAW264 cells were cultured with the designated concentrations of AB, AT, AH, AO, or NAT for 12 h. Cell lysates were analyzed using Western blot with anti-HO-1 and anti-β-actin antibodies
AT increases HO-1 expression Nrf2-dependently and JNK-dependently in RAW264 cells
To determine the effect of Nrf2 on AT-mediated up-regulation of HO-1 expression, RAW264 cells were transfected with Nrf2-specific siRNA. Figure 9a shows that siRNA knocked down endogenous Nrf2 significantly. The siRNA-mediated depression of Nrf2 partially inhibited the AT-induced HO-1 mRNA level (Fig. 9b).

Effect of Nrf2 depression on AT-mediated induction of HO-1 expression. a The siRNA duplexes were introduced into RAW264 cells for 48 h. The RNA was extracted and subjected to qRT-PCR. Then the level of Nrf2 mRNA was evaluated. Data were normalized to the level of β-actin. Experiments were performed in four separate individuals, and *p < 0.05 compared to control siRNA-treated cells. Data are representative of two independent experiments. b The siRNA duplexes were introduced into RAW264 cells for 48 h. Then the cells were cultured in the presence or absence of 50 μM AT for 6 h. The RNA was extracted and subjected to qRT-PCR, and the level of HO-1 mRNA was evaluated. Data were normalized to the level of β-actin. Data represent the mean ± SE (n = 4). Statistically significant differences (p < 0.05) are denoted by different letters
Various signaling cascades such as MAPK, NF-κB signaling, and STAT3 are reported as involved in HO-1 up-regulation [11]. To identify the kinase pathway involved in the AT-mediated induction of HO-1 expression, RAW264 cells cultured in the presence of AT for the designated time periods. As presented in Fig. 10a, AT treatment caused a time-dependent phosphorylation of JNK. The JNK phosphorylation began at 5 min, peaked at 30 min; then decreased to 60 min after AT treatment. The protein level of JNK was unaffected by AT treatment. However, the phosphorylations of ERK and p38 kinase were unaffected. Meanwhile, the inhibitor of JNK, SP600125, suppressed the AT-mediated up-regulation of HO-1 expression (Fig. 10b). In contrast, AGOs had no effect on nuclear translocation of p65 and phosphorylation of STAT3 at Tyr705 (Supplementary Fig. 4 and 5).

Involvement of MAPK in AT-mediated induction of HO-1 expression. a RAW264 cells were cultured with 50 μM AT for the designated time periods. The cell lysates were analyzed using Western blot with anti-phospho-JNK, anti-JNK, anti-phospho-ERK, anti-ERK, anti-phospho-p38, anti-p38, and anti-β-actin antibodies. b RAW264 cells were cultured with 50 μM AT in the presence or absence of 30 μM SP600125 (JNK inhibitor) for 12 h. The cell lysates were analyzed using Western blot with anti-HO-1 and anti-β-actin antibodies
Discussion
In this study, we demonstrated that AGOs which are hydrolyzed products of agarose attenuated TNBS-induced colitic injury and inflammation in mice through the induction of HO-1 expression in macrophages. Furthermore, we clarified that AGOs induced HO-1 expression Nrf2-dependently and JNK-dependently in mouse macrophage-like cells.
Western blot and immunohistochemical analyses revealed that HO-1 expression was up-regulated in colonic mucosa of mice that received AGOs when compared to that of vehicle-received mice, and showed that the induction of HO-1 was observed mainly in macrophages. Other reports related to the localization of HO-1 in colonic mucosa have described that HO-1 is expressed not only in inflammatory cells but also in epithelial cells [27, 28]. However, in various organs such as alveolar spaces, endometrium, and small intestinal mucosa, HO-1 stained cells are macrophages [26, 29, 30]. Additionally, we reported previously that HO-1 was localized in the macrophages, but not in epithelium, in inflamed colonic mucosa in patients with active ulcerative colitis [13]. This finding is consistent with our obtained data. In this study, we evaluated colonic mucosal injury based on the macroscopic damage score and histology using the murine TNBS model, which is a well-known experimental model of IBD. TNBS treatment induced colonic injury with hyperemia, edema, thickening, and ulceration. Then AGO administration suppressed these typical colitic features, and the suppression was canceled by SnPP treatment. However, in the present study, although Zhong et al. [17] showed that SnPP treatment aggravates DSS-induced colitis, SnPP treatment had no effect on TNBS-induced colitis in vehicle-treated mice. As the reason for this contradiction, it has been assumed that the intestinal inflammation of TNBS-induced colitis is more severe inflammation than that of DSS-induced colitis model. Therefore, because the intensity of inflammation is saturated in TNBS-treated group, the apparent aggravation of colitis by SnPP treatment is not observed. Furthermore, our recent study showed that HO-1-overexpressing macrophages manifested M2 macrophage markers such as Arginase-1, Fizz1, Ym1, and MRC-1 [14]. Consequently, even though the localization of HO-1 in colonic mucosa is controversial, HO-1-positive macrophages might play a role in the suppression of TNBS-induced colonic injuries.
MPO activity, which is an index of tissue-associated neutrophil accumulation, increased significantly in the colonic mucosa after TNBS treatment. This increase was inhibited by AGO administration. Many researchers have hypothesized that neutrophil-mediated inflammation participates in the development of colonic mucosal injury [31]. Although it is unknown whether AGOs directly affect the function of neutrophils, we have two ideas for AGO-mediated suppression of MPO activity. First, the suppression of intestinal inflammation by AGO through the induction of HO-1 expression resulted in the inhibition of neutrophils accumulation in the colonic mucosa. Second, according to our previous report, carbon monoxide, which is one of the byproduct of heme degradation by HO-1, suppresses KC induction in inflammatory site [32], leading to the inhibition of neutrophils accumulation in the colonic mucosa. Additionally, results described herein demonstrate that in the colonic mucosa, AGO administration suppressed the up-regulation of TNF-α expression in response to TNBS treatment. Similar results were obtained for LPS-stimulated RAW264 cells. TNF-α has recently attracted attention because of its key role in the pathogenesis of IBD. In fact, TNF-α blocking agents have been used as therapeutic agents for treating IBD worldwide [33]. Consequently, inhibition of neutrophil accumulation and suppression of TNF-α expression by AGO administration might be important protective factors helping to decrease TNBS-induced colonic mucosal injury.
Western blot analysis using RAW264 (a mouse macrophage-like cell line) cells showed that HO-1 expression was up-regulated in the presence of AGOs. Consequently, it caused anti-inflammatory effects in LPS-stimulated RAW264 cells. Irrespective of the length of each oligosaccharide, AB, AT, AH, and AO, having 3,6-anhydro-L-galactose at the reducing end, showed similar induction of HO-1 expression. In contrast, NAT, having D-galactose at the reducing end, had no effect on HO-1 expression. Furthermore, Holma et al. [34] reported that administration of galacto-oligosaccharides, which are composed of D-galactose alone, is unable to attenuate TNBS-induced colitis in a rat model. These results suggest that 3,6-anhydro-L-galactose at the reducing end of AGOs plays a crucial role in HO-1 up-regulation.
Depression of Nrf2 using the siRNA approach partially inhibited AT-mediated up-regulation of HO-1 in RAW264 cells. However, AT induced HO-1 expression even in Nrf2-depressed cells. Multiple protein kinase pathways such as ERK, p38 kinase, and PI3K have been proposed to play a role in HO-1 induction [11]. We show here that AT promoted the phosphorylation of JNK, but not of ERK and p38, and AT-mediated induction of HO-1 was suppressed by SP600125, a specific inhibitor for JNK. The phosphorylation of JNK has been found to regulate HO-1 expression in some reports, but not in others [35–37]. This discrepancy might reflect cell-type-specific and stressor-specific differences in the regulation of HO-1 expression. These results suggest that although the role of JNK pathway in the regulation of HO-1 expression is controversial, AGO-mediated HO-1 induction requires both functions of Nrf2 and JNK pathway.
In conclusion, results presented here indicate that AGO administration remarkably attenuated the intestinal inflammation in mice through the induction of HO-1 expression, which is associated with significant suppression of neutrophil accumulation and TNF-α expression. These data suggest that oral administration of AGOs might be an additional therapeutic strategy for IBD.
Abbreviations
- AB:
-
Agarobiose
- AGOs:
-
Agaro-oligosaccharides
- AH:
-
Agarohexaose
- AO:
-
Agarooctaose
- AT:
-
Agarotetraose
- ELISA:
-
Enzyme-linked immunosorbent assay
- ERK:
-
Extracellular signal-regulated kinase
- HO:
-
Heme oxygenase
- IBD:
-
Inflammatory bowel diseases
- JNK:
-
c-jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MPO:
-
Myeloperoxidase
- NAT:
-
Neoagarotetraose
- Nrf2:
-
NF-E2-related factor 2
- PI3K:
-
Phosphatidylinositol 3-kinase
- qRT-PCR:
-
Quantitative real-time PCR
- SnPP:
-
Tin protoporphyrin
- TNBS:
-
2,4,6-Trinitrobenzene sulfonic acid
- TNF-α:
-
Tumor necrosis factor-α
References
Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34.
Atreya R, Neurath MF. New therapeutic strategies for treatment of inflammatory bowel disease. Mucosal Immunol. 2008;1:175–82.
Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–54.
Ponka P. Cell biology of heme. Am J Med Sci. 1999;318:241–56.
Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem. 1969;244:6388–94.
Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol. 1996;15:9–19.
Ryter SW, Choi AM. Heme oxygenase-1/carbon monoxide: from metabolism to molecular therapy. Am J Respir Cell Mol Biol. 2009;41:251–60.
Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–8.
Igarashi K, Sun J. The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal. 2006;8:107–18.
Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. 1997;272:5375–81.
Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–903.
Naito Y, Takagi T, Yoshikawa T. Heme oxygenase-1: a new therapeutic target for inflammatory bowel disease. Aliment Pharmacol Ther. 2004;20(Suppl 1):177–84.
Takagi T, Naito Y, Mizushima K, Nukigi Y, Okada H, Suzuki T, et al. Increased intestinal expression of heme oxygenase-1 and its localization in patients with ulcerative colitis. J Gastroenterol Hepatol. 2008;23(Suppl 2):S229–33.
Harusato A, Naito Y, Takagi T, Uchiyama K, Mizushima K, Hirai Y, et al. BTB and CNC homolog 1 (Bach1) deficiency ameliorates TNBS colitis in mice: Role of M2 macrophages and heme oxygenase-1. Inflamm Bowel Dis (in press).
Wang WP, Guo X, Koo MW, Wong BC, Lam SK, Ye YN, et al. Protective role of heme oxygenase-1 on trinitrobenzene sulfonic acid-induced colitis in rats. Am J Physiol Gastrointest Liver Physiol. 2001;281:G586–94.
Naito Y, Takagi T, Uchiyama K, Yoshikawa T. Heme oxygenase-1: a novel therapeutic target for gastrointestinal diseases. J Clin Biochem Nutr. 2011;48:126–33.
Zhong W, Xia Z, Hinrichs D, Rosenbaum JT, Wegmann KW, Meyrowitz J, et al. Hemin exerts multiple protective mechanisms and attenuates dextran sulfate sodium-induced colitis. J Pediatr Gastroenterol Nutr. 2010;50:132–9.
Berberat PO, A-Rahim YI, Yamashita K, Warny MM, Csizmadia E, Robson SC, et al. Heme oxygenase-1-generated biliverdin ameliorates experimental murine colitis. Inflamm Bowel Dis. 2005;11:350–9.
Chen HM, Yan XJ. Antioxidant activities of agaro-oligosaccharides with different degrees of polymerization in cell-based system. Biochim Biophys Acta. 2005;1722:103–11.
Chen H, Yan X, Zhu P, Lin J. Antioxidant activity and hepatoprotective potential of agaro-oligosaccharides in vitro and in vivo. Nutr J. 2006;5:31.
Kazlowski B, Pan CL, Ko YT. Separation and quantification of neoagaro- and agaro-oligosaccharide products generated from agarose digestion by beta-agarase and HCl in liquid chromatography systems. Carbohydr Res. 2008;343:2443–50.
Enoki T, Okuda S, Kudo Y, Takashima F, Sagawa H, Kato I. Oligosaccharides from agar inhibit pro-inflammatory mediator release by inducing heme oxygenase 1. Biosci Biotechnol Biochem. 2010;74:766–70.
Sugimoto N, Rui T, Yang M, Bharwani S, Handa O, Yoshida N, et al. Points of control exerted along the macrophage-endothelial cell-polymorphonuclear neutrophil axis by PECAM-1 in the innate immune response of acute colonic inflammation. J Immunol. 2008;181:2145–54.
Takagi T, Naito Y, Mizushima K, Akagiri S, Suzuki T, Hirata I, et al. Inhalation of carbon monoxide ameliorates TNBS-induced colitis in mice through the inhibition of TNF-alpha expression. Dig Dis Sci. 2010;55:2797–804.
Grisham MB, Hernandez LA, Granger DN. Xanthine oxidase and neutrophil infiltration in intestinal ischemia. Am J Physiol. 1986;251:G567–74.
Harusato A, Naito Y, Takagi T, Uchiyama K, Mizushima K, Hirai Y, et al. Suppression of indomethacin-induced apoptosis in the small intestine due to Bach1 deficiency. Free Radic Res. 2011;45:717–27.
Barton SG, Rampton DS, Winrow VR, Domizio P, Feakins RM. Expression of heat shock protein 32 (hemoxygenase-1) in the normal and inflamed human stomach and colon: an immunohistochemical study. Cell Stress Chaperones. 2003;8:329–34.
Paul G, Bataille F, Obermeier F, Bock J, Klebl F, Strauch U, et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin Exp Immunol. 2005;140:547–55.
Maestrelli P, El Messlemani AH, De Fina O, Nowicki Y, Saetta M, Mapp C, et al. Increased expression of heme oxygenase (HO)-1 in alveolar spaces and HO-2 in alveolar walls of smokers. Am J Respir Crit Care Med. 2001;164:1508–13.
Yoshiki N, Kubota T, Aso T. Identification of heme oxygenase in human endometrium. J Clin Endocrinol Metab. 2001;86:5033–8.
Naito Y, Takagi T, Yoshikawa T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J Gastroenterol. 2007;42:787–98.
Takagi T, Naito Y, Uchiyama K, Suzuki T, Hirata I, Mizushima K, et al. Carbon monoxide liberated from carbon monoxide-releasing molecule exerts an anti-inflammatory effect on dextran sulfate sodium-induced colitis in mice. Dig Dis Sci. 2011;56:1663–71.
Rutgeerts P, Vermeire S, Van Assche G. Biological therapies for inflammatory bowel diseases. Gastroenterology. 2009;136:1182–97.
Holma R, Juvonen P, Asmawi MZ, Vapaatalo H, Korpela R. Galacto-oligosaccharides stimulate the growth of bifidobacteria but fail to attenuate inflammation in experimental colitis in rats. Scand J Gastroenterol. 2002;37:1042–7.
Goven D, Boutten A, Lecon-Malas V, Boczkowski J, Bonay M. Prolonged cigarette smoke exposure decreases heme oxygenase-1 and alters Nrf2 and Bach1 expression in human macrophages: roles of the MAP kinases ERK(1/2) and JNK. FEBS Lett. 2009;583:3508–18.
Hou CC, Huang CC, Shyur LF. Echinacea alkamides prevent lipopolysaccharide/D-galactosamine-induced acute hepatic injury through JNK pathway-mediated HO-1 expression. J Agric Food Chem. 2011;59:11966–74.
Takagi T, Naito Y, Okada H, Ishii T, Mizushima K, Akagiri S, et al. Lansoprazole, a proton pump inhibitor, mediates anti-inflammatory effect in gastric mucosal cells through the induction of heme oxygenase-1 via activation of NF-E2-related factor 2 and oxidation of kelch-like ECH-associating protein 1. J Pharmacol Exp Ther. 2009;331:255–64.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (B) to Toshikazu Yoshikawa (Grant No. 21390184), (C) to Yuji Naito (No. 22590705), and (C) to Tomohisa Takagi (No. 22590706) from the Japan Society for the Promotion of Science; An Adaptable and Seamless Technology Transfer Program through target-driven R&D to Yuji Naito from Japan Science and Technology Agency.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
535_2012_719_MOESM1_ESM.tif
Supplementary Fig. 1 Analysis of cytokine expressions in TNBS-induced colitis model. a-e Colonic mucosa was scraped off. Then total RNA was extracted. The resultant RNA was subjected to qRT-PCR, and the levels IL-1β, IL-6, KC, IL-12, and IL-17A mRNA were evaluated. Data were normalized to the level of β-actin. Data represent the mean ± SE of eight mice. Statistically significant differences (p < 0.05) are denoted by different letters (TIFF 467 kb)
535_2012_719_MOESM2_ESM.tif
Supplementary Fig. 2 Analysis of cytokine expressions in LPS-stimulated RAW264 cells. a, b RAW264 cells were cultured with 100 μg/ml AGOs for 12 h. Then the cells were stimulated by 1 ng/ml LPS for 2 h. The RNA was extracted and subjected to qRT-PCR, and the levels IL-1β, IL-6, KC, IL-12, and IL-17A mRNA were evaluated. Data were normalized to the level of β-actin. Data represent the mean ± SE (n = 4). Statistically significant differences (p < 0.05) are signified by different letters. n.s. (not significant) compared to unstimulated RAW264 cells (TIFF 299 kb)
535_2012_719_MOESM3_ESM.tif
Supplementary Fig. 3 Effect of AGOs on TLRs expressions in RAW264 cells. RAW264 cells were cultured with 100 μg/ml AGOs for the indicated time period. The cell lysates were analyzed using Western blot with anti-TLR2, anti-TLR4, anti-TLR9, and anti-β-actin antibodies (TIFF 316 kb)
535_2012_719_MOESM4_ESM.tif
Supplementary Fig. 4 Effect of AGOs on nuclear translocation of p65 in RAW264 cells. RAW264 cells were cultured with 100 μg/ml AGOs or 10 ng/ml LPS for the indicated time period. Nuclear protein fractions were extracted using Nuclear Extract kit (Active Motif, Carlsbad, CA, USA) and analyzed using Western blot with anti-p65 antibody and anti-lamin B1 antibodies (TIFF 121 kb)
535_2012_719_MOESM5_ESM.tif
Supplementary Fig. 5 Effect of AGOs on STAT3 signaling in RAW264 cells. RAW264 cells were cultured with 100 μg/ml AGOs or 50 ng/ml IL-6 for the indicated time period. The cell lysates were analyzed using Western blot with anti-phospho-STAT3 (Y705), anti-STAT3, and anti-β-actin antibodies (TIFF 286 kb)
Rights and permissions
About this article
Cite this article
Higashimura, Y., Naito, Y., Takagi, T. et al. Oligosaccharides from agar inhibit murine intestinal inflammation through the induction of heme oxygenase-1 expression. J Gastroenterol 48, 897–909 (2013). https://doi.org/10.1007/s00535-012-0719-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-012-0719-4