
Abstract
Purpose of review Alzheimer’s disease (AD) and frontotemporal dementia can often be diagnosed accurately with careful clinical history, cognitive testing, neurological examination, and structural brain MRI. However, there are certain circumstances wherein detection of specific biomarkers of neurodegeneration or underlying AD pathology will impact the clinical diagnosis or treatment plan. We will review the currently available biomarkers for AD and frontotemporal dementia (FTD) and discuss their clinical importance.
Recent findings With the advent of 18F-labeled tracers that bind amyloid plaques, amyloid PET is now clinically available for the detection of amyloid pathology and to aid in a biomarker-supported diagnosis of AD or mild cognitive impairment (MCI) due to AD. It is not yet possible to test for the specific FTD pathologies (tau or TDP-43); however, a diagnosis of FTD may be “imaging supported” based upon specific MRI or FDG-PET findings. Cerebrospinal fluid measures of amyloid-beta, total-tau, and phospho-tau are clinically available and allow detection of both of the cardinal pathologies of AD: amyloid and tau pathology.
Summary It is appropriate to pursue biomarker testing in cases of MCI and dementia when there remains diagnostic uncertainty and the result will impact diagnosis or treatment. Practically speaking, due to the rising prevalence of amyloid positivity with advancing age, measurement of biomarkers in cases of MCI and dementia is most helpful in early-onset patients, patients with atypical clinical presentations, or when considering referral for AD clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia and typically presents with the gradual onset of short-term memory impairment. AD then progresses to involve other domains of cognition including language, visuospatial abilities, and executive function. Advancing age is the greatest risk factor for AD. However, AD is also a common cause of dementia among patients younger than 65 years (early-onset AD). Frontotemporal dementia (FTD) is one of the most common forms of dementia in patients younger than 65 years and may present with behavioral symptoms or language impairment.
Both conditions are neurodegenerative diseases, characterized by unique pathologies on brain autopsy and by progressive neuronal cell death in specific cortical regions. Although often clinically very distinct, an expert clinical diagnosis of dementia due to AD or FTD may be only 70–80% sensitive to match with autopsy findings [1, 2]. In particular, dementia with predominant aphasia, apraxia, dysexecutive function, or personality change may be caused by either AD or FTD pathology.
In this review, we will discuss the background, evidence, and clinical scenarios for the use of the main categories of clinically available biomarkers for the diagnosis of AD and FTD. These include structural imaging (MRI), functional imaging (FDG-PET, SPECT), and markers of AD pathology, namely, amyloid PET and cerebrospinal fluid (CSF) biomarkers including amyloid beta 1–42 (Aβ1–42), total tau (t-tau), and phosphorylated tau (p-tau). Specifically, the measurement of biomarkers is helpful in the clinical diagnosis and treatment of dementia to detect (1) the presence and spatial pattern of neurodegeneration in an early clinical stage, and (2) a specific pathology, such as the cardinal pathologies of AD, amyloid-beta, and phospho-tau.
We will also briefly mention those biomarkers that have shown promise in recent research and are likely to enter the clinical arena in the not-distant future. Moreover, challenges to the use of biomarkers in clinical practice will be discussed, including the high prevalence of AD biomarkers in cognitively intact elderly, the lack of identified biomarkers for the specific FTD pathologies of TDP-43 and tau, the lack of coverage for biomarker testing by insurance or national health plans, and the importance of seeking patient consent for biomarker testing and disclosure.
Illustrative cases
Case 1
A 70-year-old right-handed man presented with 1 year of gradually progressive personality change with irritability, apathy, lack of hygiene, and susceptibility to financial scams. His wife reported he was less interested in and caring toward his family and he had isolated himself to play Solitaire on his computer and read political conspiracy websites. On neuropsychological testing, he had moderate impairment in executive function, low-normal verbal memory, and otherwise intact cognition. His neurologic examination was intact. An MRI brain showed mild diffuse cortical atrophy without a specific pattern. Initial diagnosis was behavioral variant FTD. His CSF testing showed a reduced level of Aβ1–42 and elevated T-tau/Aβ1–42 and P-tau/Aβ1–42 ratios. His diagnosis was revised to “frontal variant” Alzheimer’s disease and he was started on donepezil. He was also enrolled in an AD clinical trial testing an anti-amyloid antibody.
Case 2
A 64-year-old right-handed man presented with a 2-year history of word-finding difficulty. Clinical and neuropsychological assessments confirmed a profound anomia and relatively preserved function in other cognitive domains. His brain MRI showed evidence of medial and anterior temporal lobe atrophy with greater left-sided emphasis. With a clinical diagnosis of Alzheimer’s disease, he was commenced on memantine that did not lead to a tangible benefit. He underwent CSF analysis for amyloid and tau measurement as he was being considered for enrollment in an AD clinical trial, but the CSF profile was not compatible with AD. A second opinion was sought. Clinical assessment confirmed the presence of profound anomia, but it was also noted that there was a striking preservation of non-verbal episodic memory and visuospatial function. An FDG-PET demonstrated strikingly asymmetrical temporal hypometabolism (mainly left sided). On longitudinal follow-up, the patient developed behavioral features including mental rigidity, apathy, and lack of empathy. He also developed a sweet tooth which led to significant weight gain. He underwent amyloid PET scan that was reported as normal. His diagnosis was revised to semantic variant of frontotemporal dementia with behavioral features.
Alzheimer’s disease
Alzheimer’s disease most often presents with the gradual onset of short-term memory impairment, and patients may present to clinicians while in an amnestic mild cognitive impairment (MCI) stage, wherein memory impairment is evident on testing, but activities of daily living remain generally independent. Over time, patients will develop impairment in other domains of cognition including language, visuospatial function, executive function, and attention, and patients will develop impairments in their activities of daily living. Behavioral symptoms are common in Alzheimer’s disease, particularly depression, anxiety, and irritability in early stages, and paranoid delusions and agitation in moderate-to-severe stages. However, personality and social behavior often remain relatively preserved.
Although an amnestic presentation is most typical in Alzheimer’s disease, variant presentations are possible, including primary progressive aphasia due to underlying AD pathology, posterior cortical atrophy presentation, or a frontal variant of AD, characterized by personality change and a dysexecutive syndrome.
The cardinal pathologies of Alzheimer’s disease are amyloid plaques and neurofibrillary tangles, and longitudinal studies indicate that these pathologies may accumulate years prior to any cognitive decline [3•, 4]. In the clinical setting, biomarker testing may be pursued for prognosis, future planning, and to guide treatment. The National Institute on Aging–Alzheimer’s Association guidelines for the diagnosis of Alzheimer’s disease dementia recommend biomarkers for research purposes and as “optional clinical tools for use where available and when deemed appropriate by the clinician.” Biomarker testing allows a diagnosis of AD to be made to a greater level of certainty “with evidence of AD pathophysiological process” (Table 1) [5]. The majority of clinical trials for AD now require biomarker confirmation as inclusion criteria.
Frontotemporal dementias
Frontotemporal dementia is the umbrella term used to denote degenerative brain diseases that, as the name implies, predominantly affect frontal and temporal lobes of the brain [6]. The frontotemporal lobar degeneration (FTLD) spectrum also includes corticobasal degeneration and progressive supranuclear palsy, in addition to FTD; however, this review will focus on imaging and CSF biomarkers for FTD.
FTD syndromes are the second most common type of dementia in the younger-than-65 age group [7] (AD is the most common) and the third most common type overall [8]. Clinically, FTD syndromes are broadly categorized to behavioral variant FTD (bvFTD) [9] and primary progressive aphasias (PPAs) [10], and with the caveat that evolution from one clinical syndrome to another is a common occurrence [11]. bvFTD is characterized by a variable combination of behavioral features. According to the latest consensus recommendations [9] (Table 1), presence of at least three out of six clinical features is required for the diagnosis of possible bvFTD. Significant functional decline and consistent neuroimaging is required for moving to the next level of certainty (probable bvFTD). The presence of one of the FTD-causing mutations [chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), or progranulin (GRN)] or histopathological evidence of FTLD is necessary for diagnosis of bvFTD with definite FTLD pathology. Language variants of FTD are divided to semantic variant PPA (svPPA) and non-fluent/agrammatic variant (nfvPPA). According to the latest consensus criteria [12], svPPA is characterized by impairment in confrontation naming and single-word comprehension whereas nfvPPA patients suffer from a variable combination of grammatical violations and effortful halting speech. Neuroimaging is helpful in identifying different patterns of atrophy and hypometabolism and presence of atrophy and/or hypometabolism in the relevant areas (Table 1) will improve the degree of confidence in clinical diagnosis [12, 13]. Similar to bvFTD, presence of either histopathological evidence of degenerative pathology or presence of known pathogenic mutation is required for the diagnosis of PPA with definite pathology.
Abnormal phosphorylation of MAPT without concomitant amyloid pathology (primary tau pathology) and cytoplasmic aggregation of trans-activation response DNA binding protein of 43 kDa (TDP-43) are the two main pathological hallmarks of FTD with almost equal contribution to FTD pathology [14]. Also, a minority of patients (~ 10%) harbor fused-in-sarcoma pathology [15].
Imaging biomarkers
Imaging biomarkers are mainly divided into two main categories: structural imaging such as MRI or high-resolution CT and functional imaging which includes PET and advanced MR-based techniques such as functional MRI or diffusion tensor imaging. For the purpose of this review, we focus mainly on imaging techniques available in clinical setting. The recommendations from all current guidelines on imaging biomarkers including European Federation of Neurological Societies, Fourth Canadian Consensus Conference on Diagnosis and Treatment of Dementia, and UK National Institute of Health and Care Excellence have been summarized in Table 2.
Structural imaging
There is a consensus between all current guidelines [16,17,18] that cases with clinical dementia syndrome should be investigated by structural imaging at least once during their work-up. Structural imaging is not only used to identify the pattern of atrophy specific for different dementia syndromes but also to exclude the presence of non-degenerative pathologies manifesting with cognitive impairment. Examples of such conditions include tumors, vascular disease, inflammatory or infective processes (e.g., encephalitis), subdural collections, or normal-pressure hydrocephalus.
MRI is considered the modality of choice because of its superiority in depicting vascular and inflammatory changes. High-resolution CT, however, can be used as an alternative when MRI is not available or contraindicated. The minimum MRI sequences required to investigate dementia are high-resolution 3D or volumetric T1-weighted images in coronal and one additional plane mainly to identify the pattern and degree of atrophy, and T2-weighted and fluid attenuation inversion recovery (FLAIR) images to determine the degree of vascular changes. T2* or gradient echo or susceptibility-weighted imaging is used to assess the presence of cerebral microbleeds or vascular amyloid deposition in Alzheimer’s disease. Diffusion-weighted imaging, on the other hand, shows foci of restricted diffusion in cases with acute cerebrovascular event and is very helpful to exclude Creutzfeldt-Jacob disease presenting with restricted diffusion in posterior thalamus or cortical ribboning.
Although specific patterns of atrophy have been described for each dementia syndrome, these patterns are commonly observed in group level compared with healthy controls. Given the considerable anatomical overlap between different degenerative diseases, making accurate diagnosis could be challenging for an individual case.
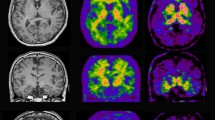
In typical amnestic AD, atrophy mainly involves the medial temporal lobes and hippocampi and extends to parietal lobes (Fig. 1). In atypical AD presentations such as posterior cortical atrophy (PCA), the volume loss is more prominent in posterior–superior parietal lobes involving posterior cingulate cortex (PCC), while atrophy in logopenic variant of PPA (lvPPA) mainly involves the posterolateral and inferior temporal and parietal lobes which can also extend to PCC [19].

Patient with AD (top row) shows atrophy in medial temporal lobe and hippocampi in MRI (a), hypometabolism in temporal lobes in FDG-PET (b), and widespread cortical amyloid deposition in frontal, parietal, and temporal lobes in amyloid PET (c). Patient with nfvPPA (bottom row) demonstrates atrophy in lateral and anterior temporal lobes and frontal operculum on the left side in MRI (d), hypometabolism in left peri-insular region in FDG PET (e) with no evidence of amyloid deposition (f).
The pattern of atrophy in different FTD syndromes are as follows: in bvFTD, the atrophy is mainly in the prefrontal cortex, anterior temporal lobes, and insula with involvement of striatum and thalamus [20]. There is a group of patients who meet the diagnostic criteria for bvFTD but have very slow disease course. They have normal MRI and PET and are classified as FTD phenocopy [8]. In semantic variant of PPA (svPPA), atrophy is asymmetric, usually more severe on the left, and involves the anterior and inferior temporal lobes. The MRI changes in svPPA precede the clinical symptoms. Patients can have remarkable atrophy in the temporal poles while they are still independent in daily activities [21]. In non-fluent variant of PPA (nfvPPA), atrophy is also left predominant and mostly seen in the inferior frontal lobe (mainly pars opercularis), dorsolateral prefrontal region, superior temporal gyrus, and insula. Also, atrophy can extend to caudate head bilaterally and putamen on the left [22] (Fig. 1).
To help the clinicians and radiologists in reporting cases with dementia, different visual scales have been developed to assess the pattern of atrophy and determine the degree of small vessel disease.
-
1-
MTA (Scheltens) visual scale scores the degree of atrophy from 0 to 4 based on the maximum height of choroid fissure (CSF space above the hippocampus), width of the temporal horn (CSF space lateral to the hippocampus), and maximum height of hippocampal formation in T1W coronal image [23]. 0 means no change in the above measures while 4 means severe loss of height in hippocampal formation and widening of both choroid fissure and temporal horn.
-
2-
Global cortical atrophy (GCA) (Pasquier) scale assesses general brain atrophy in 13 regions based on degree of sulcal dilatation with a score range from 0 (no atrophy) to 3 (knife-blade atrophy) [24]. Although GCA has more extensive coverage, it has less reliability because it includes regions susceptible to partial volume effect [25•].
-
3-
White matter changes (Fazekas) scale is based on size, shape, and extension of small vessel changes (hyperintensity in T2/FLAIR or hypodensity in CT) in the white matter of both cerebral hemispheres. The scores are 0, no change; 1, punctuate focal lesion; 2, beginning of confluence of lesions; and 3, diffuse involvement of the entire region with or without U fiber involvement [26].
To optimize the utility of structural imaging in clinical setting, it is crucial to have a specific protocol for acquisition and a standard, structured reporting system. Having a standardized method becomes more important when the images are required to be assessed longitudinally or across different centers. In a recent paper by Imaging Cognitive Impairment Network, they suggested that standard radiological report on cases with dementia should include MTA, GCA, and white matter scores to improve the accuracy of diagnosis [27].
Functional imaging
18F-FDG PET and perfusion hexamethylpropyleneamine oxime (HMPAO) SPECT
18F-FDG PET provides information on the first stages of glucose metabolism and is a proxy of synaptic activity and neurodegeneration in dementia. HMPAO SPECT or perfusion SPECT measures cerebral blood flow, which represents an indirect estimate of metabolism.
In all current guidelines (Table 2) [16,17,18], both 18 F-FDG PET and perfusion SPECT are considered as the second tier of imaging biomarkers when the combination of clinical evaluation and structural imaging fails to secure the diagnosis. Most guidelines recommended perfusion SPECT as an alternative to FDG when PET is not available. It has been shown that HMPAO SPECT has less sensitivity and specificity [28]. In the USA, Medicare and Medicaid services have approved the utility of 18F-FDG PET in differentiating AD from other dementia (particularly FTD).
The main roles of 18F-FDG PET are to
-
1.
Confirm the presence of neurodegeneration and differentiate dementia from normal aging or psychiatric illnesses mimicking degenerative brain disease. Overall sensitivity of FDG for diagnosis of AD from control is around 76% with the median specificity of 82% [29•].
-
2.
Differentiate AD from other types of dementias. The pattern of hypometabolism can help to distinguish AD from FTD with 86% sensitivity and 97% specificity [30] and AD from dementia with Lewy bodies (DLB) with > 90% sensitivity and 70% specificity [31, 32]. These early studies have very small subject numbers and differ between specific regional measures used and are best used with close attention to clinical context of PET acquisition.
Hypometabolism in 18F-FDG PET, as a marker of neurodegeneration, precedes atrophy in structural imaging and cognitive decline in clinical examination [33, 34], although it has strong spatial correlation with MRI changes and its pattern is highly associated with clinical symptoms [34].
Nevertheless, neurodegeneration is a relatively late finding in the course of AD and 18F-FDG PET might still be within normal limits in preclinical or early MCI cases. Therefore, while absence of hypometabolism makes neurodegeneration less likely, it cannot exclude the early stages of dementia. Presence of hypometabolism, on the other hand, has predictive value in identifying individuals progressing from MCI to AD [35].
In typical AD, hypometabolism is seen in medial temporal lobe, parieto-temporal regions, precuneus, and posterior cingulate gyrus [36, 37] (Fig. 1). In atypical AD presentations, PCA is associated with hypometabolism in parieto-occipital lobes. lvPPA, on the other hand, is characterized by left temporoparietal junction metabolic deficit [38].
In bvFTD, maximum abnormality is seen in orbitofrontal, dorsolateral, and medial prefrontal cortex and anterior temporal poles [39, 40]. In svPPA, there is evidence of asymmetrical hypometabolism in temporal poles, more severe on the left, with extension to the medial, inferior, and lateral temporal lobes [7]. The prominent hypometabolic regions in nfvPPA are left inferior frontal and superior temporal regions [41] (Fig. 1).
Like structural imaging, the abovementioned syndrome-specific patterns of hypometabolism are derived from group differences. Therefore, the caveat in using them to make a diagnosis for an individual case is that there are overlaps between areas of abnormality across dementia syndromes and many cases might have mixed pathologies.
The presence or absence of hypometabolism in 18F-FDG PET images can be visually assessed by a highly skilled rater although studies from experienced centers have concluded that qualitative evaluation of FDG images can be ambiguous and difficult [42, 43]. To overcome this challenge and improve the diagnostic accuracy of FDG, PET images can be assessed quantitatively using standard uptake value ratio (SUVr) [44]. SUVr is a ratio of activity in each region of brain relative to a reference area where metabolism is unaffected or mildly affected. Examples of such areas include cerebellar cortex, vermis, whole cerebellum, and pons [45].
Amyloid PET
Deposition of extracellular amyloid β (Aβ) plaques as one of the main pathological hallmarks of AD appears well before any other changes in the course of disease [34]. In vivo imaging of amyloid in the last decade has revolutionized AD research. There are different PET ligands binding selectively with high affinity to fibrillar Aβ aggregates in vivo. 11C-Pittsburgh Compound B (11C-PiB) is the best characterized PET tracer, but its short half-life (20 min) has restricted its use to centers with an on-site cyclotron. Recently, several 18F-labeled amyloid derivatives have emerged to overcome the limitation of 11C-PiB. They include 18F-flutemetamol [46] (GE-067; Vizamyl™, GE Healthcare), the stilbene derivative 18F-florbetapir [47] (AV-45; AMYViD™, Eli Lilly), and the strylpyridine derivative 18F-florbetaben [48] (BAY-94-9172; Neuraceq™, Piramal).
These tracers have been approved by the US FDA “for PET imaging of the brain to estimate β-amyloid neuritic plaque density in adult patients with cognitive impairment being evaluated for AD and other causes of cognitive decline.”
The results of 18F-labeled tracers are comparable with 11C-PiB, although PiB is slightly superior in differentiating AD from control [49, 50]. They all show increased cortical binding in regions known to be affected by Aβ deposition such as frontal, parieto-temporal, and posterior cingulate cortices [51,52,53,54] (Fig. 1), and they have strong correlation with autopsy and postmortem findings [55,56,57,58]. Although non-specific retention in white matter is higher in 18F-labeled tracers, there is no evidence that it might affect the interpretation of the images significantly [49, 50, 59].
In comparison to FDG PET, amyloid imaging has higher accuracy in differentiating AD from healthy controls [42]. The sensitivity and specificity of amyloid PET—with either 11C- or 18F-labeled tracers—are above 85% [60, 61•]. Different studies have also claimed that the inter-rater reliability of visual reading is higher in amyloid imaging compared to FDG and it has higher agreement with quantitative classification. PiB PET can differentiate AD from FTD with more than 90% sensitivity [62].
Based on current imaging guidelines, amyloid PET is not part of routine imaging work-up in dementia. The European Federation of the Neurological Societies [18], the Amyloid Imaging Task Force [63, 64], and the recent Canadian Consensus Conference on the Use of Amyloid Imaging [65] all have similar recommendations on utility of amyloid imaging in clinical setting. Similar to FDG, amyloid PET should only be used in cases presenting with cognitive decline in whom the diagnosis is still uncertain despite comprehensive clinical assessment by a dementia expert and structural imaging. The clinical applications of amyloid PET according to different guidelines have been summarized in Table 2. Amyloid imaging is particularly useful in the following scenarios:
-
1-
In AD cases with atypical or early onset presentation (age < 65) or when there is evidence of psychiatric illnesses or heterogeneous syndromes.
-
2-
To differentiate AD pathology from FTD.
-
3-
In MCI cases with progressive unexplained cognitive decline when confirming diagnosis can alter the management [18, 64, 65].
It should be noted that amyloid PET cannot distinguish clinical variants of AD pathology from each other or AD from DLB. It is also not useful in staging the severity of the disease [65]. As 10–30% of cognitively normal individuals can have positive amyloid PET [66], according to current recommendations, it is not appropriate to use amyloid PET on cognitively normal individuals based on family history or genetic predisposition such as APOE4 [64].
Early data from IDEAS (Imaging Dementia-Evidence for Amyloid Scanning) study of amyloid PET in US Medicare beneficiaries showed that amyloid PET can have a substantial impact on patient management in day-to-day practice [67]. There is also a growing body of evidence showing that the early diagnosis and intervention at preclinical stage of AD is paramount in tackling the disease. Therefore, the clinical utility of amyloid PET, as one of the earliest diagnostic biomarkers in AD, may change in the near future.
Amyloid burden can be assessed both visually and quantitatively using SUVr. Cerebellar gray matter or pons with low Aβ plaque density demonstrates the minimum binding in amyloid imaging, hence being used as reference regions for SUVr calculation [52]. Longitudinal studies on Alzheimer Disease Neuroimaging Initiative data have shown using subcortical white matter as reference might improve the accuracy of detecting cortical changes over time [68].
One of the main issues in performing amyloid PET on a wider population and at an early stage of the disease is interpretation of amyloid images. It is important to remember that positive amyloid PET alone does not confirm the diagnosis of AD or MCI. As mentioned above, 10–30% of normal individuals can have positive amyloid PET. The long-term prognosis of this group is still not completely clear. A meta-analysis on prevalence of amyloid PET positivity in dementia syndromes showed amyloid PET can be positive in 51% of cases with dementia with Lewy bodies, 38% in corticobasal syndrome, 30% in vascular dementia, and 12% in cases with FTD. As the authors have concluded, presence of Aβ in non-AD dementia syndromes could be either because of clinical misdiagnosis or secondary to concordance/mixed pathologies in many cases where the clinical manifestation is not driven by Aβ [69]. Moreover, although negative PET excludes the diagnosis of AD, it cannot exclude the diagnosis of non-amyloid-related dementia. Therefore, interpretation of images should only be done by a dementia expert and based on clinical evaluation, risk factors, and cognitive status.
Another sensitive issue with amyloid PET is disclosure of the result. It can be stressful for patients and might have a legal or social impact. The Canadian consensus on the use of amyloid imaging [65] has recommended using the disclosure methods developed by Harkins et al. [70]. Recommendations for communicating amyloid PET results to patients with MCI were recently formulated [71]. Before amyloid PET becomes available for wider clinical use, more studies are required on disclosure issues.
Tau PET
The presence of neurofibrillary tangles, phosphorylated tau protein aggregates, is another pathological hallmark of AD. Tau deposition occurs in a particular spatiotemporal pattern starting from transentorhinal/entorhinal cortex to hippocampus and then extending to the rest of temporal lobe and neocortical regions [72]. Tau deposition has a close association with cognitive decline [73], severity of dementia symptoms [74], and brain atrophy [75]. Recently, different tau selective PET tracers have been developed and used for human studies: 18F-THK523, 18F-THK5117, 18F-THK5105, 18F-THK5351, 18F-AV1451(T807), and 11C-PBB3 [76]. Despite promising results in initial studies, Tau PET is still some way from being qualified to be used in a clinical setting.
CSF
Cerebrospinal fluid (CSF) testing can be important in the diagnosis of dementia, first and foremost to rule out an infectious or inflammatory cause. CSF markers of the cardinal Alzheimer’s disease pathologies can also be measured and are helpful in determining if Alzheimer’s disease pathology is present. The core CSF biomarkers for AD are amyloid-b1–42 (Aβ1–42), total-tau (t-tau), and phospho-tau181 (p-tau) and are felt to correspond, respectively, to Aβ deposition in senile plaques, neuronal death, and hyperphosphorylation of tau in neurofibrillary tangles. Lower levels of Aβ1–42 and higher levels of t-tau and p-tau, and especially a high ratio of t-tau/Aβ1–42 or p-tau/Aβ1–42, are found in patients with AD compared to patients with FTD or normal controls, with a sensitivity and specificity reaching 85–90% [77•]. The AD CSF biomarkers are also helpful in identifying if AD is the underlying pathology in PPA. The Alzheimer’s Biomarkers Standardization Initiative (2014) reached a consensus that “lumbar puncture for AD CSF biomarker analysis be considered as a routine clinical test in patients with early-onset dementia, at the prodromal (MCI) stage or with atypical AD” [78]. However, specifically in MCI patients, the Cochrane Collaboration described “a state of uncertainty” and cautioned about the risk of misdiagnosis and overdiagnosis of AD when measuring CSF t-tau, p-tau, or p-tau/Aβ1–42 ratio in patients with MCI [79•].
An elevated level of CSF t-tau is seen in several conditions associated with rapid neuronal cell death including Creutzfeldt-Jakob disease and brain trauma. Thus, in the diagnosis of AD, it is important to use both the tau and Aβ1–42 markers, and not simply rely on elevated t-tau levels. An important practical note is that standard CSF collection tubes are polyethylene, but collection of CSF for measurement of Aβ1–42 must utilize polypropylene tubes because of the possibility of Aβ1–42 adhering to the walls of a polyethylene tube leading to underestimation of CSF Aβ1–42 [80].
While it is often appropriate to measure the AD CSF biomarkers in patients with clinical FTD, there are currently no FTD-specific CSF biomarkers. Overall, patients with FTD do not show the gross elevations in CSF t-tau or p-tau as seen in patients with AD. Several previous studies did not find a difference in CSF p-tau between patients with FTD with or without underlying tau pathology. However, a recent study comparing CSF p-tau levels to postmortem tau pathology on autopsy found that there was a positive association [81]. Specifically, patients with pathologic FTD with underlying tau pathology had higher CSF p-tau levels than patients with underlying TDP-43 pathology. In both groups, p-tau levels were significantly lower than in AD patients. Further studies will be needed to test this finding in other cohorts and to develop potential diagnostic cut points.
One CSF marker that appears promising for the diagnosis of FTD is neurofilament light chain (NfL), a major component of the axonal cytoskeleton and a putative marker of axonal injury. CSF levels of NfL are greatly elevated (2.5–11-fold) in all forms of FTLD, including bvFTD and PPA subtypes, when compared to normal controls, and correlate with disease severity [82]. NfL are also elevated, to a lesser extent, in AD and other neurodegenerative diseases. There is also a strong correlation between NfL levels in CSF and serum [83].
There are currently no blood or serum biomarkers available in clinical practice for the diagnosis of AD or FTD, but this is an area of great research activity. Potential candidates include more sensitive measures of amyloid-beta and tau, as well as novel biomarkers such as lipids, microRNAs, and immunologic markers [84].
Conclusion
Structural MRI is the most commonly used biomarker to aid in the diagnosis of AD and FTD. MRI often demonstrates atrophy in areas of the cortex evidently impaired based on history and cognitive testing, and in the prototypical areas of the brain affected by these diseases (usually symmetric medial temporal lobes proceeding to biparietal lobes in AD versus often asymmetric frontal or temporal lobes in FTD). However, there are certain circumstances in clinical practice wherein the pursuit of further biomarkers of spatial patterns of neurodegeneration (as in FDG PET or SPECT) or biomarkers of amyloid beta and tau (amyloid PET or CSF) will be helpful. These circumstances include (1) early-onset dementia, (2) atypical dementia not meeting classic clinical criteria, (3) cases with strong confounders by history (vascular dementia, depression, alcohol abuse) but with a suspicion for underlying contributing AD or FTD pathology, and (4) cases of MCI wherein patients are well informed and desiring to know if AD pathology is present. Published recommendations for appropriate use of biomarker testing, particularly amyloid PET and CSF testing for AD biomarkers, do not always agree. Furthermore, in many cases, insurers or national health plans may not cover biomarker testing, and out-of-pocket costs may be prohibitive. Being mindful of those limitations, it is appropriate to pursue biomarker testing if the patient understands the implications and ramifications, and if the clinician deems that performance of the biomarker test will affect diagnostic decisions or change management, whether by way of patient counseling, pursuit (or non-pursuit) of further diagnostic work-up, or guiding treatment (lifestyle modification, pharmacologic treatment, or referral to clinical trials).
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance
Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer disease centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71(4):266–73.
Balasa M, Gelpi E, Martin I, Antonell A, Rey MJ, Grau-Rivera O, et al. Diagnostic accuracy of behavioral variant frontotemporal dementia consortium criteria (FTDC) in a clinicopathological cohort. Neuropathol Appl Neurobiol. 2015;41(7):882–92.
• Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Cedarbaum J, et al. 2014 update of the Alzheimer’s disease neuroimaging initiative: a review of papers published since its inception. Alzheimers Dement. 2015;11(6):e1–120. The Alzheimer’s Disease Neuroimaging Initiative (ADNI) is one of the most important studies of longitudinal changes in cognition and biomarkers in healthy elderly, MCI, and AD patients in the USA, and has developed numerous standard imaging protocols and analytics for the field. Several studies worldwide have been partially modeled on ADNI, in Europe, Japan, and Australia
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804.
McKhann GM, Knopman DS. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2011;7(3):263–9.
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–54.
Diehl-Schmid J, Grimmer T, Drzezga A, Bornschein S, Perneczky R, Forstl H, et al. Longitudinal changes of cerebral glucose metabolism in semantic dementia. Dement Geriatr Cogn Disord. 2006;22(4):346–51.
Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672–82.
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–77.
Mesulam M. Primary progressive aphasia—a language-based dementia. N Engl J Med. 2003;349(16):1535–42.
Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996–2005.
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–14.
Sajjadi SA, Sheikh-Bahaei N, Cross J, Gillard JH, Scoffings D, Nestor PJ. Can MRI visual assessment differentiate the variants of primary-progressive aphasia? AJNR Am J Neuroradiol. 2017;38(5):954–60.
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol. 2007;114(1):5–22.
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(Pt 11):2922–31.
NICE. Dementia: supporting people with dementia and their carers in health and social care | Guidance and guidelines |National Institute for Health and Care Excellence. 2016.
Soucy JP, Bartha R, Bocti C, Borrie M, Burhan AM, Laforce R, et al. Clinical applications of neuroimaging in patients with Alzheimer’s disease: a review from the fourth Canadian consensus conference on the diagnosis and treatment of dementia 2012. Alzheimers Res Ther. 2013;5(Suppl 1):S3.
Filippi M, Agosta F, Barkhof F, Dubois B, Fox NC, Frisoni GB, et al. EFNS task force: the use of neuroimaging in the diagnosis of dementia. Eur J Neurol. 2012;19(12):e131–40. 1487-501
Ossenkoppele R, Cohn-Sheehy BI, La Joie R, Vogel JW, Moller C, Lehmann M, et al. Atrophy patterns in early clinical stages across distinct phenotypes of Alzheimer’s disease. Hum Brain Mapp 2015;36(11):4421–4437.
Pan PL, Song W, Yang J, Huang R, Chen K, Gong QY, et al. Gray matter atrophy in behavioral variant frontotemporal dementia: a meta-analysis of voxel-based morphometry studies. Dement Geriatr Cogn Disord. 2012;33(2–3):141–8.
Rohrer JD, Warren JD, Modat M, Ridgway GR, Douiri A, Rossor MN, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology. 2009;72(18):1562–9.
Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55(3):335–46.
Scheltens P, Leys D, Barkhof F. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol. 1992;8:967–72.
Pasquier F, Leys D, Weerts JG, Mounier-Vehier F, Barkhof F, Scheltens P. Inter- and intraobserver reproducibility of cerebral atrophy assessment on MRI scans with hemispheric infarcts. Eur Neurol. 1996;36(5):268–72.
• Harper L, Barkhof F, Fox NC, Schott JM. Using visual rating to diagnose dementia: a critical evaluation of MRI atrophy scales. J Neurol Neurosurg Psychiatry. 2015;86(11):1225–33. In this systemic review, different cerebral atrophy rating scales for dementia have been examined to highlight the diagnostic potential of these clinically applicable tools
Wahlund LO, Barkhof F, Fazekas F, Bronge L, Augustin M, Sjogren M, et al. A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke. 2001;32(6):1318–22.
Wahlund LO, Westman E, van Westen D, Wallin A, Shams S, Cavallin L, et al. Imaging biomarkers of dementia: recommended visual rating scales with teaching cases. Insights Imaging 2017;8(1):79–90.
O'Brien JT, Firbank MJ, Davison C, Barnett N, Bamford C, Donaldson C, et al. 18F-FDG PET and perfusion SPECT in the diagnosis of Alzheimer and Lewy body dementias. J Nucl Med. 2014;55(12):1959–65.
Smailagic N, Vacante M, Hyde C, Martin S, Ukoumunne O, Sachpekidis C. (1)(8)F-FDG PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane database Syst rev. 2015;1:Cd010632. In this Cochrane systemic review, authors searched all the major databases and included and analyzed all the studies that evaluated the diagnostic accuracy of FDG PET to detemine the conversion from MCI to AD or other forms of dementia.
Foster NL, Heidebrink JL, Clark CM, Jagust WJ, Arnold SE, Barbas NR, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130(Pt 10):2616–35.
Lim SM, Katsifis A, Villemagne VL, Best R, Jones G, Saling M, et al. The 18F-FDG PET cingulate island sign and comparison to 123I-beta-CIT SPECT for diagnosis of dementia with Lewy bodies. J Nucl Med. 2009;50(10):1638–45.
Kono AK, Ishii K, Sofue K, Miyamoto N, Sakamoto S, Mori E. Fully automatic differential diagnosis system for dementia with Lewy bodies and Alzheimer’s disease using FDG-PET and 3D-SSP. Eur J Nucl Med Mol Imaging. 2007;34(9):1490–7.
Arlt S, Brassen S, Jahn H, Wilke F, Eichenlaub M, Apostolova I, et al. Association between FDG uptake, CSF biomarkers and cognitive performance in patients with probable Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36(7):1090–100.
Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–16.
Drzezga A, Lautenschlager N, Siebner H, Riemenschneider M, Willoch F, Minoshima S, et al. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur J Nucl Med Mol Imaging. 2003;30(8):1104–13.
Silverman DH, Small GW, Phelps ME. Clinical value of neuroimaging in the diagnosis of dementia. Sensitivity and specificity of regional cerebral metabolic and other parameters for early identification of Alzheimer’s disease. Clin Positron Imaging. 1999;2(3):119–30.
Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging. 2005;32(4):486–510.
Lehmann M, Ghosh PM, Madison C, Laforce R Jr, Corbetta-Rastelli C, Weiner MW, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain. 2013;136(Pt 3):844–58.
Verfaillie SC, Adriaanse SM, Binnewijzend MA, Benedictus MR, Ossenkoppele R, Wattjes MP, et al. Cerebral perfusion and glucose metabolism in Alzheimer’s disease and frontotemporal dementia: two sides of the same coin? Eur Radiol. 2015;25(10):3050–9.
Diehl-Schmid J, Grimmer T, Drzezga A, Bornschein S, Riemenschneider M, Forstl H, et al. Decline of cerebral glucose metabolism in frontotemporal dementia: a longitudinal 18F-FDG-PET-study. Neurobiol Aging. 2007;28(1):42–50.
Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, et al. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64(4):388–401.
Ng S, Villemagne VL, Berlangieri S, Lee ST, Cherk M, Gong SJ, et al. Visual assessment versus quantitative assessment of 11C-PIB PET and 18F-FDG PET for detection of Alzheimer’s disease. J Nucl Med. 2007;48(4):547–52.
Womack KB, Diaz-Arrastia R, Aizenstein HJ, Arnold SE, Barbas NR, Boeve BF, et al. Temporoparietal hypometabolism in frontotemporal lobar degeneration and associated imaging diagnostic errors. Arch Neurol. 2011;68(3):329–37.
Rabinovici GD, Rosen HJ, Alkalay A, Kornak J, Furst AJ, Agarwal N, et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology. 2011;77(23):2034–42.
Herholz K. The role of PET quantification in neurological imaging: FDG and amyloid imaging in dementia. Clinical and Translational Imaging. 2014;2(4):321–30.
Vandenberghe R, Van Laere K, Ivanoiu A, Salmon E, Bastin C, Triau E, et al. 18F-Flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol 2010;68(3):319–329.
Wong DF, Rosenberg PB, Zhou Y, Kumar A, Raymont V, Ravert HT, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2010;51(6):913–20.
Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O'Keefe G, et al. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 2008;7(2):129–35.
Wolk DA, Zhang Z, Boudhar S, Clark CM, Pontecorvo MJ, Arnold SE. Amyloid imaging in Alzheimer’s disease: comparison of florbetapir and Pittsburgh compound-B positron emission tomography. J Neurol Neurosurg Psychiatry. 2012;83(9):923–6.
Landau SM, Thomas BA, Thurfjell L, Schmidt M, Margolin R, Mintun M, et al. Amyloid PET imaging in Alzheimer’s disease: a comparison of three radiotracers. Eur J Nucl Med Mol Imaging. 2014;41(7):1398–407.
Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, et al. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23(6):2086–92.
Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh compound-B. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2005;25(11):1528–47.
Zhang W, Kung MP, Oya S, Hou C, Kung HF. 18F-labeled styrylpyridines as PET agents for amyloid plaque imaging. Nucl Med Biol. 2007;34(1):89–97.
Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305(3):275–83.
Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(Pt 6):1630–45.
Choi SR, Schneider JA, Bennett DA, Beach TG, Bedell BJ, Zehntner SP, et al. Correlation of amyloid PET ligand florbetapir F 18 binding with Abeta aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord. 2012;26(1):8–16.
Curtis C, Gamez JE, Singh U, Sadowsky CH, Villena T, Sabbagh MN, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015;72(3):287–94.
Ong KT, Villemagne VL, Bahar-Fuchs A, Lamb F, Langdon N, Catafau AM, et al. Abeta imaging with 18F-florbetaben in prodromal Alzheimer’s disease: a prospective outcome study. J Neurol Neurosurg Psychiatry. 2015;86(4):431–6.
Villemagne VL, Mulligan RS, Pejoska S, Ong K, Jones G, O'Keefe G, et al. Comparison of 11C-PiB and 18F-florbetaben for Abeta imaging in ageing and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2012;39(6):983–9.
Archer HA, Edison P, Brooks DJ, Barnes J, Frost C, Yeatman T, et al. Amyloid load and cerebral atrophy in Alzheimer’s disease: an 11C-PIB positron emission tomography study. Ann Neurol. 2006;60(1):145–7.
Yeo JM, Waddell B, Khan Z, Pal S. A systematic review and meta-analysis of (18)F-labeled amyloid imaging in Alzheimer’s disease. Alzheimers Dement (Amst). 2015;1(1):5–13. In this systemic review and meta-analysis, the authors have included 19 studies investigating 682 AD patients. They calculated pooled weighted sensitivity, specificity, and odds ratios for each of 18F-labeled amyloid tracer.
Laforce R Jr, Rabinovici GD. Amyloid imaging in the differential diagnosis of dementia: review and potential clinical applications. Alzheimers Res Ther. 2011;3(6):31.
Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. Amyloid imaging task force of the Alzheimer’s Association and Society for Nuclear Medicine and Molecular Imaging. Alzheimers Dement. 2013;9(4):e106–9.
Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the amyloid imaging task force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement. 2013;(1):9, e-1–e16.
Laforce R, Rosa-Neto P, Soucy JP, Rabinovici GD, Dubois B, Gauthier S. Canadian consensus guidelines on use of amyloid imaging in Canada: update and future directions from the specialized task force on amyloid imaging in Canada. Can J Neurol Sci. 2016;43(4):503–12.
Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, et al. The Alzheimer’s disease neuroimaging initiative positron emission tomography core. Alzheimers Dement. 2010;6(3):221–9.
Rabinovici GD. Impact of amyloid PET on patient management: early results from the IDEAS study. AAIC; London2017.
Landau SM, Fero A, Baker SL, Koeppe R, Mintun M, Chen K, et al. Measurement of longitudinal beta-amyloid change with 18F-florbetapir PET and standardized uptake value ratios. J Nucl Med. 2015;56(4):567–74.
Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BN, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA. 2015;313(19):1939–49.
Harkins K, Sankar P, Sperling R, Grill JD, Green RC, Johnson KA, et al. Development of a process to disclose amyloid imaging results to cognitively normal older adult research participants. Alzheimers Res Ther. 2015;7(1):26.
Grill JD, Apostolova LG, Bullain S, Burns JM, Cox CG, Dick M, et al. Communicating mild cognitive impairment diagnoses with and without amyloid imaging. Alzheimers Res Ther. 2017;9(1):35.
Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18(4):351–7.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81.
Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52(1):81–8.
Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71(10):743–9.
Okamura N, Harada R, Furukawa K, Furumoto S, Tago T, Yanai K, et al. Advances in the development of tau PET radiotracers and their clinical applications. Ageing Res Rev. 2016;30:107–13.
Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–84. This tour de force review and meta-analysis of 231 articles containing 15,699 patients with AD and 13,018 controls identified the most consistent AD biomarkers as CSF T-tau, P-tau, Aβ42, and neurofilament
Molinuevo JL, Blennow K, Dubois B, Engelborghs S, Lewczuk P, Perret-Liaudet A, et al. The clinical use of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: a consensus paper from the Alzheimer’s biomarkers standardization initiative. Alzheimers Dement. 2014;10(6):808–17.
Ritchie C, Smailagic N, Noel-Storr AH, Ukoumunne O, Ladds EC, Martin S. CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev. 2017;3:Cd010803. This Cochrane Systemic Review urged caution in the use of CSF tau and CSF tau/ABeta ratio in patients with MCI due to risk of misdiagnosis or overdiagnosis
Perret-Liaudet A, Pelpel M, Tholance Y, Dumont B, Vanderstichele H, Zorzi W, et al. Risk of Alzheimer’s disease biological misdiagnosis linked to cerebrospinal collection tubes. J Alzheimers Dis. 2012;31(1):13–20.
Irwin DJ, Lleo A, Xie SX, McMillan CT, Wolk DA, Lee EB, et al. Ante mortem cerebrospinal fluid tau levels correlate with postmortem tau pathology in frontotemporal lobar degeneration. Ann Neurol. 2017;82(2):247–58.
Scherling CS, Hall T, Berisha F, Klepac K, Karydas A, Coppola G, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75(1):116–26.
Meeter LH, Kaat LD, Rohrer JD, van Swieten JC. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol 2017;13(7):406–419.
O'Bryant SE, Mielke MM, Rissman RA, Lista S, Vanderstichele H, Zetterberg H, et al. Blood-based biomarkers in Alzheimer disease: current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 2017;13(1):45–58.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
N.S.-B. and S.A.S. each declare no potential conflicts of interest.
A.L.P. reports contracts from Avid Radiopharmaceuticals, Eli Lilly, Transition Therapeutics (previously Elan), Stemedica, Biogen, Janssen, Axovant, and Roche/Genentech, as well as personal fees from Lundbeck, outside the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Dementia
Rights and permissions
About this article

Cite this article
Sheikh-Bahaei, N., Sajjadi, S.A. & Pierce, A.L. Current Role for Biomarkers in Clinical Diagnosis of Alzheimer Disease and Frontotemporal Dementia. Curr Treat Options Neurol 19, 46 (2017). https://doi.org/10.1007/s11940-017-0484-z
Published:
DOI: https://doi.org/10.1007/s11940-017-0484-z