
Abstract
Insulin resistance (IR) is present in pathologies such as diabetes, obesity, metabolic syndrome, impaired glucose tolerance, hypertension, inflammation, cardiac disease, and dyslipidemias. Population studies show that IR is multifactorial and has genetic components, such as defects in the insulin-signaling pathway (as serine phosphorylation on insulin substrate or decreased activation of signaling molecules) and RAS/MAPK-dependent pathways. IR is connected to mitochondrial dysfunction, overproduction of oxidants, accumulation of fat, and an over-activation of the renin-angiotensin system linked to the NADPH oxidase activity. In addition, nitric oxide (NO), synthesized by nitric oxide synthases (endothelial and inducible), is also associated with IR when both impaired release and reduced bioavailability of all which lead to inflammation and hypertension. However, increased NO may promote vasculoprotection. Moreover, reduced NO release induces heat shock protein 70 kDa (HSP70) expression in IR and diabetes, mediating beneficial effects against oxidative stress injury, inflammation and apoptosis. HSP70 may be used as biomarker of the chronicity of diabetes. Hsp72 (inducible protein) is linked to vascular complications with a high-fat diet by blocking inflammation signaling (cytoprotective and anti-cytotoxicity intracellular role). Elucidating the IR signaling pathways and the roles of NO and HSPs is relevant to the application of new treatments, such as heat shock and thermal therapy, nitrosylated drugs, chemical chaperones or exercise training.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Insulin resistance (IR) and type-2 diabetes mellitus (t-2DM)—together with other related metabolic disorders, metabolic syndrome (MS) and obesity—are growing dramatically all over the world. The WHO estimates that, by the year 2030, approximately 366 million people will be affected with diabetes [1]. The prevalence of a high-fat diet and low physical activity are very important factors for those diseases to appear, contributing to the current pandemic of obesity and its associated complications such as ischemic heart and vascular disease, neuropathy, retinopathy, and nephropathy [2, 3, 4•].
Insulin exhibits diverse biologic functions in mammalian cells and organs. Its classic physiological functions are maintaining normal blood glucose levels through glucose uptake and promoting glucose disposal in skeletal muscle and adipose tissue as well as suppressing both the production and storage of glucose in the liver [5].
IR, or the resistance to the metabolic action of insulin, is a key event necessary to better understand the pathophysiologic of both t-2DM and obesity [6].
However, IR is not restricted to individuals with abnormal glucose tolerance (GT) and IR-stimulated glucose uptake, and it also occurs in non-obese individuals with normal oral GT. Documented results from a population study indicate that many individuals with associated metabolic disorders (e.g., dyslipemia and hyperuricemia) are insulin-resistant and, in the general population, IR can be found even in the absence of any mayor metabolic disorders [7]. About 25 % of non-diabetic subjects exhibits IR within the same ranges found in t2-DM patients. IR in patients with t-2DM occurs many years before the onset of diabetes and with the consequence of declined insulin secretion. IR was observed in two-thirds of subjects with impaired glucose tolerance (IGT) [7, 8].
In addition, other biologic actions for insulin have been described. As recently recognized, one of its functions is the modulation of nitric oxide (NO) bioavailability which, by increasing NO production, promotes vasodilatation in the endothelium [6, 9]. The effects of NO on cellular functions are complex and appear to be contradictory: NO may be cytotoxic but can also protect cells from toxic injury acting as an antioxidant, and it may activate or inhibit signal transduction pathways and gene transcription [10•]. Of special interest to this review, many research works, both in vivo and in vitro, have explained the pathophysiology role of NO in IR as well as in development complications [9, 11].
Moreover, cells have been observed to exhibit different mechanisms that protect them from diverse physiological and environmental stressors. The induction of stress response proteins (SRPs) is a conserved protection mechanism that may slow the damaging effects of oxidative stress (OS) and inflammation [12•]. These proteins are represented by a group of chaperones in protein folding as well as by heat shock proteins (HSP) [13]. HSPs solved the problem of protein misfolding and aggregation by preventing the irreversible aggregation of nonnative conformations [14].
Notably, diabetes and IR have been showed to induce HSP [15]. Interestingly for our studies, HSP 70 kDa (HSP70) participates in inflammation by inducing different inflammation-related responses according to its location (intra- or extracellular), which makes this protein a master regulator for controlling the immune system, inflammation and associated IR. It was suggested that the extracellular HSP70/intracellular HSP70 ratio may represent a better marker not only for the immune-inflammatory status of many types of diseases such as obesity-induced insulin insensitivity and diabetes, but also for other inflammation-related states, e.g., atherosclerosis, heart disease and obesity-related nonalcoholic fatty liver disease [16••].
The relationship between NO, HSP70 and IR has been recently discussed [17••] and, for this reason, we deem appropriate to review their comments.
Insulin Resistance
IR has been described by Reaven (1988) as a common phenomenon appearing after glucose uptake, and which is characterized by a chronic hyperinsulinemia state with subsequent impaired release of glucose. Into insulin secretion compensatory responses, occurs deterioration on ITG lead to IR continuous, increases or decreases, for have to maintain euglycemia [18•]. If IR occurs before chronic hyperglycemia development, the difference from IR in a pre-diabetic state, results from OS activation by increased glucose levels (pathway-selective IR) [19]. In IR, the impairment of insulin ability to exert its effects on glucose, protein and lipid metabolism in target tissues produced a lower biologic response at physiological concentrations because there is a decreased sensitivity such as insulin-mediated glucose disposal [20, 21].
At present, evidence from large population studies shows that IR is multifactorial [19, 22, 23, 24•], and has genetic components [25–27] whose understanding exceeds the purpose of this document.
The molecular and cellular mechanisms of IR are relevant to understanding its pathogenesis as well as various associated diseases such as diabetes, obesity, MS, IGT, hypertension, inflammation, coronary artery disease and dyslipidemias resulting from defects in both insulin secretion and action. Insulin-resistant patients may develop overt t-2DM when pancreatic cells cannot produce enough insulin to maintain euglycemia due to deficient glucose sensing [28].
Firstly, it should be noted the importance of signaling pathways in the biological actions of insulin mediated by intracellular signaling transduction of protein kinase cascades [29].
In various tissues, such as vascular endothelium, skeletal muscle, and adipose tissue, phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-dependent insulin-signaling pathways regulate vasodilator and metabolic actions of insulin. However, mitogen-activated protein kinase (MAPK)-dependent insulin-signaling pathways tend to promote pro-hypertensive and pro-atherogenic actions of insulin [30••].
The PI3K-dependent insulin-signaling pathway is developed through insulin binding to the extracellular α-subunits of its receptor and increasing the β-subunit tyrosine kinase activity which can phosphorylate insulin receptor substrates (IRS). The IRS proteins activate PI3K (a lipid kinase) by interacting with the Src homology 2 (SH2) domains of the p85 regulatory subunit. PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate, generating phosphatidylinositol 3,4,5-triphosphate (activating the p110 catalytic subunit). The phosphoinositide produced then will subsequently phosphorylate protein kinase B, Akt, (a serine/threonine kinase), into phosphoinositide-dependent kinase-1, atypical protein kinase C (PKC) and other serine kinases [29, 31].
Thus, IR can be caused by defects at multiple sites in the insulin-signaling pathways, such as increased serine phosphorylation of IRS proteins, increased degradation of IRS and activity of various phosphatases or decreased activation of insulin receptor downstream-signaling molecules, including Akt and atypical PKC [30••].
Other intracellular signaling system is the MAPK related to cell growth, differentiation and survival [32]. MAPK is activated through the renin-angiotensin system (RAS) which not only activates MAPK but also RAF and MAPK/extracellular signal-regulated kinase (MEK). The small GTP protein RAS is activated by GTP exchange factor Sos, which in turn is activated by binding of the SH2 domain of Grb-2 to Shc [30••].
At the skeletal muscle level, the RAS/MAPK-dependent pathway is caused by hyperglycemia which increases insulin to levels similar to those of non-resistant pathways of insulin-signaling (pathway-selective IR) [33].
IR in skeletal muscle is a critical feature, because this is the major site of induction of peripheral IR, and its early defect leads to the initial development of IGT (reduced ability to dispose of an oral glucose load) in prediabetes and subsequently to overt t-2DM [34].
Interestingly, relevant evidences consider that IR, SM, diabetes and obesity are linked with mitochondrial dysfunction [30••, 35], which is associated with decreased mitochondrial number, abnormal morphology, lower levels of oxidative enzymes and lower ATP synthesis [24•, 36•, 37••].
In addition, the mutations of mitochondrial DNA (mtDNA) and decreased gene expression are possible causes for the variability of baseline mitochondrial function seen in insulin’s primary target tissues: skeletal muscle cells, adipocytes, and hepatocytes [38].
Skeletal muscle IR has been proposed in various studies, both in humans and animal models, that show possible alterations in oxidative mitochondrial function (decreased oxidative phosphorylation) and mitochondrial morphology (diminished mitochondrial biogenesis), as well as a decrease in the activity of the respiratory chain [39••, 40].
Moreover, the reactive oxidative species (ROS) formed in mitochondria hydroxyl radical (OH−), singlet oxygen (O), anion superoxide (O2 −), NO and peroxynitrite (NOOO−), all of which can damage cells and macromolecules (proteins, DNA, and lipids) by different pathways resulting in mitochondrial dysfunction [41].
Numerous systemic and cellular dysfunctions can contribute to ROS overproduction, such as hyperglycemia, dyslipidemia, endoplasmic reticulum (ER) stress, advanced glycation end-products (AGE), nitric oxide synthases (NOS), lipid peroxides, and activate reduced insulin actions [42, 43]. This overabundance of oxidants is associated with the multifactorial etiology of IR, primarily in skeletal muscle tissue [44]. OS may also be linked to lipid-induced ER stress contributing to IR through activation of serine kinases such as c-Jun N-terminal kinase (JNK), increasing serine phosphorylation of IRS proteins [30••, 45]. ROS stimulates pro-inflammatory signaling by activation of IκB kinase (IKK-β) that phosphorylates IRS into serine residues [46].
Several studies suggest that defects in lipid metabolism leading to an impairment mechanism for IR, both in human subjects and rodents, reduces insulin-stimulated glucose disposal [47, 48].
Moreover, free fatty acids (FFAs) stimulate Toll-like receptor (TLR)-mediated inflammatory signaling, which activates IκB kinase and JNK. It also stimulates the production of other cytokines, including tumor necrosis factor-alpha, interleukin-1beta, and interleukin-6 [49].
More recently, it has been suggested that lipid-induced mitochondrial dysfunction and consequent increases in ROS, in turn, activate various serine kinases that phosphorylate IRS proteins, promote intracellular accumulation of diacylglycerol (DG) and activate PKC, which increase serine phosphorylation of IRS proteins leading to IR [50].
Other mechanisms have been implied, such as lipid activation of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase activity through the downstream production of ceramides, and signaling by pathways that converge on necrosis factor-beta (NFkβ) [51, 52].
Several investigations have demonstrated that excess of angiotensin II (ANG II) action both in vivo and in vitro induces a state of whole-body and skeletal muscle IR. These investigations present the concept that over-activation of the RAS, in particular ANG II acting via angiotensinogen 1 (AT1) receptor linked to NADPH oxidase activity, can mediate skeletal muscle IR, at least in part via a mechanism involving the production of superoxide ions [53, 54].
Several recent reports support the concept that exposure of mammalian skeletal muscle to an OS results in stimulation of the serine kinase p38MAPK, and that the occurrence of stress-activated p38MAPK signaling is associated with diminished stimulation of insulin signaling and glucose transport activity [55]. Finally, diverse findings have shown that IR associated with endothelial dysfunction is accompanied by decreased PI3K-NO pathway [56].
Nitric Oxide Linked to Insulin Resistance
NO appears as regulator of cell and tissue function throughout body. NO is a molecule synthesized by NOS enzymes from L-arginine, NADPH and O2 as substrates that consequently produced L-citrulline, nicotinamide adenine dinucleotide (NAD) and H2O, in the cytosol [57•]. NO also shows intramitochondrial production regulated by mitochondrial NOS (mtNOS) and non-enzymatic reactions with O2 and ubiquinol (UQH2) [58]. Compared with other reactive species, NO has a rather high diffusion distance in biological systems given its lipophilic nature, neutral charge, and relative low reactivity [59].
In the mitochondrial matrix, NO reacts with the superoxide ion (O2 −) giving peroxynitrite (ONOO−) which is a powerful cytotoxin that is easily diffusible from the intramitochondrial space, unlike what occurs with NO [60]. NO such as ONOO− is a pro-oxidant potentially leading to oxidative stress and cellular damage by oxidizing and nitrating lipids, proteins and DNA, and it impairs mitochondrial function at high levels [61].
However, the effects of NO on cellular functions are complex and appear contradictory. Originally, it was described as a cytotoxic agent [62]. This molecule may be cytotoxic but it may also protect cells from toxic injury by acting as an antioxidant, and it may activate or inhibit signal transduction pathways and gene transcription [10•, 63]. Another key function of NO is the regulation of mitochondrial respiration inhibiting O2 consumption and modulating O2 gradients in cells and tissues by regulating hemoglobin action [64•]. Moreover, NO is most well-known as a potent regulator of blood flow, similar to the endothelial-derived relaxing factor (EDRF) [65].
NO is generated in many locations by the different NOS isoforms, and it is the local production which determines the physiological actions [66]. The NOS-3 or eNOS, is most abundant in vascular endothelium but it is also found in cardiomyocytes, neurons, epithelial cells, adipocytes, and hepatocytes [67, 68•]. The iNOS or NOS-2, which has the highest capacity to generate NO, is inducible and expressed in multiple cells, such as macrophages, in response to inflammatory stimuli by cytokines, lipopolysaccharides and other immunologic agents. Expression of iNOS is regulated at the transcriptional and posttranscriptional level by signaling pathways that involve agents such as factor NFkβ or MAPK [66]. Last, the NOS-1 or nNOS is expressed mostly in neurons, skeletal muscle and epithelial cells. nNOS is a Ca2+/calmodulin-dependent isoform that can be activated by agonists of the N-methyl-D-aspartate receptor [69]. In contrast, the nNOS and eNOS are constitutively expressed, but their activity is regulated by the intracellular calcium concentration. Thus, iNOS as well as NO are involved in a variety of acute or chronic disease states such as inflammation, ischemia-reperfusion, diabetes, cancer, neurological diseases, and aging [59, 70].
In renal disease, there is important evidence from animal investigations showing that experimentally induced chronic iNOS inhibition causes glomerular ischemia, glomerulosclerosis, tubulointerstitial injury, proteinuria, and systemic and glomerular hypertension [71]. Total NO production decrease in renal disease likely evidences the endothelial dysfunction and NO renal production, contributing to the progression of chronic kidney disease itself [72•]. Recent studies from our group suggest an even more critical cytoprotective role for NO in the developing kidney. More specifically, it has been implied in the regulation of renal and glomerular hemodynamics, natriuresis, blunting of tubuloglomerular feedback (TGF), inhibition of tubular sodium reabsorption, and modulation of renal sympathetic nerve activity [73].
Another investigation revealed that NO induces apoptosis through mechanisms associated with decreased activity of mitochondrial electron transport chain, and release of mitochondrial cytochrome-C into cytosol [74].
However, cell types such as endothelial cells from the microvasculature are resistant to induction of apoptosis by NO [75]. Low concentrations of NO provide protection in various cells by inhibiting certain caspases [76]. A potent antiapoptotic activity of NO has been proposed as alternative mechanism of inducing HSP70, by means of NO mediated modification in intracellular antioxidants levels [77, 78•].
On the other hand, primary NO production is also stimulated in response to insulin. Many evidences show that mitochondrial NOS activity may change from NO to O2 − generation in response to hyperglycemia [79••, 80•]. Insulin induces NO production by increasing expression and/or activation of eNOS [70, 81].
The insulin-signaling pathway in vascular endothelium-regulating production of NO has been elucidated. Insulin-stimulated activation of eNOS is mediated via the PI3K branch of the signaling cascade, which later phosphorylates and activates Akt into PKC. Akt directly phosphorylates eNOS into Ser1177 [64•, 82•]. Interestingly, insulin stimulates the protein complex formation consisting of eNOS binding to calmodulin and to HSP90. This requires HSP90 binding to eNOS which facilitates insulin-stimulated activation of eNOS by phosphorylation into Ser1177. The insulin-induced eNOS activation is calcium-independent [83]. In addition, RAS/MAP-kinase branch of insulin-signaling pathways does not affected significantly to activation of eNOS in response to insulin [84].
Furthermore, ceramide (which is increased in obesity) promotes the disruption of the eNOS–Akt complex from HSP90, which normally increases eNOS activity by promoting the displacement of caveolin-1 from eNOS [85].
eNOS activity is also regulated by other posttranslational modifications including acylation and S-nitrosylation [86]. Also, insulin stimulates NO production in vascular smooth muscle cells (VSMC) in a PI3K-dependent manner where it activates guanylate cyclase. NO attenuates production of pro-inflammatory cytokines, decreases expression of vascular cell adhesion molecules, inhibits VSMC proliferation, offers resistance to apoptosis, and attenuates platelet aggregation and monocyte adhesion to vascular wall [4•, 6].
Moreover, inactivation of NO during the enhanced ROS production vasculature can significantly reduce NO bioavailability (maladapted endothelial phenotype) with abnormal vasoreactivity associated with elevated expression of pro-inflammatory and pro-thrombotic factors and increased OS [70].
Many research works, both in vivo and in vitro, have explained the patho-physiological role of NO in IR as well as in developing complications. Original studies show that insulin-mediated vasodilation is dependent on NO and that insulin-mediated skeletal muscle vasodilation contributes to insulin sensitivity in humans [79••, 87, 88••].
Decreased NO generation and/or bioavailability may be the pathological link between NO, obesity and diabetes, rather than an excessive NO production. The endothelial dysfunction is associated with hypertension in both conditions [89]. Insulin-resistant subjects with obesity, exhibit attenuated insulin-vasodilation in muscle smooth cells under NO impaired effects [90].
The role of NO in regulating metabolic actions of insulin was evidenced by the presence of IR and hypertension in eNOS knockout mice (eNOS −/−) [91•]. These findings suggest that endothelium-derived NO has additional and direct metabolic effects on mitochondrial function and induces defects in endothelial function characterized by reduced NO bioavailability [92].
This metabolic improvement was at least partially due to eNOS-mediated activation of MAPK in the liver, which suppressed hepatic gluconeogenesis. Hence, the activity of eNOS uncoupling in the liver may be important for regulating systemic glucose metabolism [93•].
This OS and inflammatory cytokines may contribute to IR states affecting different PI3K and MAPK pathways through most independent and interdependent mechanisms in the endothelium. The balance between PI3K/Akt/eNOS/NO and MAPK/Endothelin-1vascular actions of insulin caused by ROS and RAS led to impaired both vascular and metabolic actions of insulin [93•]. Thus, impaired eNOS phosphorylation derived IR was shown to be responsible for diminished glucose uptake in the skeletal muscle of mice subjected to nutrient excess [94•].
Some studies, showing lower NO bioavailability in cultured cells, isolated arteries, animal models, and humans with diminished eNOS phosphorylation under conditions of nutrient excess and obesity, for example, elevated FFAs [95, 96•, 97].
NO and other nitrogen species generated from iNOS could also regulate systemic metabolism [98]. In the liver, iNOS expression is very important for regulating insulin sensitivity. Overexpression of iNOS may cause hepatic IR, hyperglycemia and hyperinsulinemia, and the use of an iNOS-specific inhibitor (L-NIL) reversed hyperglycemia, hyperinsulinemia and IR in ob/ob mice [99•]. Since iNOS is induced by inflammatory signals, it frequently coincides with increased superoxide generation. iNOS induces IR through mechanisms such as nitrosative posttranslational modifications of proteins in the insulin-signaling pathway. Increases in iNOS expression in skeletal muscle of obese mice are associated with increased S-nitrosation of the insulin receptor, IRS and Akt. Another mechanism is by promoting proteasomal degradation which decreases the abundance of IRS in presence of iNOS and NO donors [100].
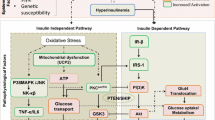
In obesity, pro-inflammatory macrophages of adipose tissue are the main cells responsible for iNOS expression and may propagate the inflammatory signaling involved in IR [101]. Furthermore, lack of iNOS does not evidence induced IR, which suggests that not all insulin-resistant states are alike. The mice lacking the iNOS isoform show IR that appears associated with a sympathetic, α-adrenergic mechanism [102] Fig. 1.

Effect of nitric oxide as a mediator of the insulin resistance
Heat Shock Proteins Linked to Insulin Resistance
HSPs, well-known chaperones, are a family of polypeptides that assist the damaged molecule in regaining its functional conformation. HSPs are synthetized in response to different stressors (heat shock, hypothermia, hypoxia, free radicals, ischemia, ethanol, ultraviolet radiation, viral infection, and others) [14]. HSPs protect proteins, lipids and nucleic acids from damage by reducing oxidation, and are, therefore, cytoprotective. They also modulate cell function and gene expression, and contribute to protein homeostasis [12•].
The major functions of HSPs are protection against apoptotic exchanges, assistance in folding of nascent polypeptides, prevention of misfolding and aggregation of nonnative conformations, and aiding protein folding [13]. Other functions include suppressing pro-inflammatory cytokines and repairing ion channel, preserving mitochondria, membranes, ER and nucleus, and participating in intracellular transport [15, 103].
The most important HSP families are HSP40, HSP60, HSP70, HSP90-kDa, and the small heat shock proteins [103]. HSP70, the most ubiquitous and highly conserved, helps proteins adopt native conformation after misfolding. Also, they protect nascent translating proteins, promote the organellar transport of proteins and reduce toxic aggregates. The inducible HSP 72 and 73 kDa forms are found at the highest level [104, 105]. Protein folding regulated by HSP70 is a complex ATP/ADP-dependent process [13].
HSP expression is regulated in multiple organs during development [106]. In the kidney, for example, HSP72 and HPS90 have individual characteristic distribution with differential responses to challenges [107••]. Particularly relevant to our area of knowledge, HSP70 is involved in the adaptive response of the human kidney to congenital unilateral ureteropelvic junction obstruction (UPJO), a condition involving abnormal nephrogenesis with renal injury, leading to vasoconstriction, macrophage infiltration, OS and tubulointerstitial fibrosis and apoptosis [108]. In another investigation, we have shown that HSP70 may provide cytoprotection against diverse factors, such as protection against tubulointerstitial fibrosis, independent from changes in blood pressure; this includes decreased OS linked to upregulation of HSP70 expression [109]. Furthermore, HSP70 expression is associated with the inhibition of renal tubule epithelial cell apoptosis during recovery from low-protein feeding, as mentioned above [110].
Different types of cells release HSP70, which has an important signaling role in the inflammatory and immune response [111]. Regarding cellular OS and mitochondrial apoptosis, these can be prevented by HSP70 expression [112].
NADPH oxidases are an important link in the inflammatory mechanism as they catalyze the production of superoxide and other ROS, both recognized as major causes of cellular damage with subsequent disease [113]. In a previous study, we showed that NADPH oxidase activity can be reverted with paricalcitol, a vitamin D3 analogue, on mitochondrial fractions from animal kidney treated with this inducer [114•].
Vitamin D receptor (VDR) produces HSP70/AT1 expression, collaborating with protection at renal structural and functional levels. We have proposed that low AT1 expression through VDR induction could be a consequence of heat shock response through HSP70-mediated cell protection [115•].
Moreover, HSP70 plays a role in controlling VDR concentrations within the cell [116•]. Adams et al. suggested that HSP70-related intracellular vitamin D-binding proteins act as regulators of vitamin D (VD) metabolism [117]. Thus, HSP70 and VD are linked by controlling the expression of protein and VDR in the cell [108].
Here, evidence suggests that VD affects directly and/or indirectly pancreatic beta cell dysfunction, IR, systemic inflammation and t-2DM [118•]. VD and VDR play important roles in the cardiovascular system and in IR given that one of vitamin D’s pleiotropic effects is its interaction with RAS components [119]. Hence, the upregulation of islet RAS genes in combination with VD insufficiency can result in an increase in IR and subsequent tissue inflammatory mechanisms [120].
There is much evidence that VD insufficiency seems to lead to heart failure, left ventricular hypertrophy, hypertension, chronic vascular inflammation and MS. A deficiency or insufficiency of serum VD may be predictive of IR in individuals with prediabetes [121•].
In this sense, diabetes and associated OS increase HSP production in response to various inducers [122, 123]. Thus, levels of HSP70 expression increase under stress conditions. The induction of HSPs leads to metabolic improvement in diabetic rodents, monkeys, and in humans because it helps to enhance lipid accumulation in liver and adipose tissue, inflammatory signaling and IR [124, 125••, 126].
From a correlation of eHSP70 with IR, pancreatic β-cell dysfunction and reduced insulin sensitivity, it was observed cellular death through the activation of NADPH oxidase isoforms leading to OS due to its chronically systemic pro-inflammatory effect [127•, 128].
In diabetes, the HSP levels differ according to the tissue, i.e., levels are higher in some tissues and lower in others [129••, 130••]. The eHSP70 (extracellular) and iHSP70 (intracellular) levels in subjects with diabetes compared with healthy subjects turned out to be different [130••].
iHSPs, e.g., iHSP72 and iHSP73, show low levels in insulin sensitive tissues, such as skeletal muscle, heart, liver and monocytes in both type-1 and type-2 diabetes, and they are protective and anti-inflammatory. iHSP levels are inversely correlated with glucose disposal, IR, inflammatory cytokines, GLUT4 levels and mitochondrial function. eHSP70 is produced in response to low-grade inflammation related to a pro-inflammatory response, decreased expression of iHSP70 and reduced insulin sensitivity [130••].
In addition, iHSP is anti-inflammatory by inactivation of NF-κB while eHSP70 causes adverse effects [131]. The impaired induction of iHSPs emerges as a consequence of the deactivation of heat shock factor 1 (HSF1). iHSF1 is deactivated through inhibiting the phosphorylation of HSF1 by glycogen synthase kinase-3β (GSK-3β) that would promote the activation of inflammatory c-JUNK and NF-κB [132]. Interestingly, HSP90 represses HSF1 and, therefore, selective Hsp90 inhibitors activate HSF1-dependent transcription in insulin signaling [133].
Krause et al. suggest that the eHSP70/iHSP70 ratio may be determinant to trigger a chronic pro-inflammatory state that leads to IR and t-2DM development, and that is a marker for the immune-inflammatory status in many others diseases [134••].
Additionally, increased HSP70 in noninsulin-sensitive diabetic tissues like the endothelium, have been reported to be linked to inflammation from an advanced diabetic state. Here, it was shown that HSP70 is increased in the circulation of diabetic patients and correlates positively with the chronicity of the disease [135•]. This data also suggest that HSP70 may have the potential to be used as a biomarker in diabetes.
A recent study showed that HSP72 (an inducible HSP70 form) is related to vascular complications associated with a high-fat diet inducing IR. HSP72 promotes vasodilation and inhibits cell proliferation thrombosis by augmenting angiotensin-(1–7) via eNOS expression [136••].
However, other data indicated that the expression of HSP72 decreased in t-2MD patients with IR when HSP72 was restored through various methods, such as HSP72 transgenic, HSP72 inducer administration or long-term hyperthermia [137•].
Therefore, an essential role of HSP72 would be blocking inflammation signaling [138] and preventing IR in genetic obesity or high-fat diet, suggesting cytoprotective and anti-autotoxicity roles for intracellular HSP70 [139•]. Similarly, a beneficial effect of HSP70 on IR has been reported in transgenic animals [140].
Other findings in patients with polycystic ovary syndrome revealed that serum HSP70 was positively correlated with high levels of C-reactive protein and TNF-α [141••].
Atalay et al. reported impaired rise in HSP70 in exercising diabetic animals, while there was an increased mRNA expression in the subjects [142]. However, increased levels of HSP70 and HSP60 have been reported, respectively, in the kidney and liver of diabetic animals [132].
It should be noted that increased HSP70 expression would be initiated by NO-induced chronic elevation (NO-HSP70 cycle) [143••]. If elevated levels of HSP70 are continued, as in heat therapy, they would eventually enhance the phosphorylation of AKT, AMPK, and eNOS. Thus, more NO is produced, improving vasomotricity and vasoprotection in IR and diabetes [144•].
The function of HSP70 can be debilitated due to glycation, which blocks its protein refolding ability [145•].
Therefore, a vicious cycle is created in which inflammation-induced IR leads to lower HSPs and further inflammation. Here, Hooper et al. proposes that impaired HSP activity is a key event in the pathogenesis of t-2DM [143••].
Finally, IR and diabetes are associated with impaired NO release from endothelial cells mediated by both eNOS and iNOS induction leading to inflammation and atherosclerosis associated to OS [135•, 146].
Reduced NO release from endothelial cells have an oxidizing effect which increases HSP70 expression during IR, diabetes and MS [17••]. This data suggests that HSP70 may be used as a biomarker, and that the changes in its ratio value may be applied for the management of inflammatory response [135•, 143••]. Also, HSP70 shows beneficial effects on an IR state by protecting cells from damage due to oxidative stress injury, inflammation and apoptosis [147•].
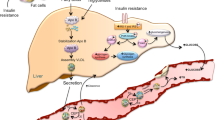
However, increased NO promotes vasculoprotection which is attributed to the actions of VD. As mentioned above, HSP70 and VD are linked by controlling the expression of protein and VDR. These findings suggest relevant pathways of interplay between VD and IR, MS and t-2DM [148•] Fig. 2.

Implication of Hsp70 as a mediator of the insulin resistance
Further studies are required to continue elucidating the events in IR signaling pathways and to provide more clear insights about the roles of NO and HSPs in the development of these diseases.
Also, it is necessary to investigate the actions of chemical chaperones as potential pharmacological applications [149], new nitrosylated drugs [150], rinse solution containing polyethylene glycol 35 for liver graft protection against ischemia-reperfusion injury [151••], exercise training and the potential benefits of heat [144•] and thermal therapy [152••, 153] against metabolic diseases.
Conclusions
According to the collected evidence, IR and diabetes are associated with impaired NO release from endothelial cells mediated by both eNOS and iNOS induction and lead to inflammation, atherosclerosis, and oxidative stress. Reduced NO release from endothelial cells has an oxidizing effect which increases HSP70 expression during IR, diabetes, and SM. This data suggest that HSP70 may be used as a biomarker and the changes in its ratio value for the management of inflammatory response. Also, HSP70 has beneficial effects on the IR state, protecting cells from damage due to oxidative stress injury, inflammation, and apoptosis. In addition, increased NO promotes vasculoprotection attributed to actions of vitamin D. HSP70, as well vitamin D, are both linked through controlling expression such as protein and vitamin D receptor. These findings suggest relevant pathways of interplay between vitamin D, IR, SM, and t-2DM. Further studies are required to continue elucidation the events in IR signaling pathways and to provide more clear insights about the roles of NO and HSPs in development of these diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–53.
Yip J, Facchini FS, Reaven GM. Resistance to insulin-mediated glucose disposal as a predictor of cardiovascular disease. J Clin Endocrinol Metab. 1998;83(8):2773–6.
Jeon JY, Ko SH, Kwon HS, Kim NH, Kim JH, Kim CS, et al. Prevalence of diabetes and prediabetes according to fasting plasma glucose and HbA1. Diabetes Metab J. 2013;37(5):349–57.
DeFronzo RA, Ferrannini E. Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14(3):173–94. This review discussed that insulin resistance appears to be a syndrome that is associated with a clustering of metabolic disorders.
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806.
Manrique C, Lastra G, Sowers JR. New insights into insulin action and resistance in the vasculature. Ann N Y Acad Sci. 2014;1311(1):138–50.
Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Targher G, et al. Prevalence of IR in metabolic disorders: the Bruneck study. Diabetes. 1988;47(10):1643–9.
Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin N Am. 2011;95(5):875–92.
Muniyappa R, Yavuz S. Metabolic actions of angiotensin II and insulin: a microvascular endothelial balancing act. Mol Cell Endocrinol. 2012;378(1–2):59–69.
Pfeilschifter J, Eberhardt W, Huwiler A. Nitric oxide and mechanisms of redox signaling. J Am Soc Nephrol. 2003;14(8 Suppl 3):S237–40. Generation and action of free radicals such as nitric oxide, is discussed with a special focus on the renal injury.
Steinberg H, Cressman E, Wu Y, Hook G, Cronin J, Johnson A, et al. Insulin mediated nitric oxide production is impaired in insulin resistance. Diabetes. 1997;46:24A.
Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol. 2011;76:91–9. This review examines the properties of the stress-responsive transcription factor in the regulation of the HSR, our current understanding of the stress-sensing mechanisms that recognize and distinguish between acute stress such as heat shock and chronic proteostasis imbalance.
Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381(6583):571–80.
Macario AJL, Conway de Macario E. Sick chaperones, cellular stress, and disease. N Engl J Med. 2005;353(14):1489–501.
Tytell M, Hooper PL. Heat shock proteins: new keys to the development of cytoprotective therapies. Expert Opin Ther Targets. 2001;5(2):267–87.
Krause M, Bock PM, Takahashi HK, Homem De Bittencourt Jr PI, Newsholme P. The regulatory roles of NADPH oxidase, intra- and extra-cellular HSP70 in pancreatic islet function, dysfunction and diabetes. Clin Sci (Lond). 2015;128(11):789–803. This review describes possible mechanisms by which HSP70 participates in cell function/dysfunction, focusing on the possible role of HSPs in pancreatic islet α- and β-cell physiological function in health and Type 2 diabetes mellitus.
Hooper P. Diabetes, nitric oxide, and heat shock protein. Diabetes Care. 2003;26(3):951–2. Dr. Hooper describes that decreased Hsps linked to nitric oxide imbalance in type 1 and type 2 diabetes may be a primary factor leading to the development of diabetes and its diverse, widespread organ damage.
Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–607. The resistance to insulin-stimulated glucose uptake and hyperinsulinemia are involved in the etiology and clinical course of three major related diseases- non-insulin-dependent diabetes mellitus, hypertension, and coronary artery disease.
Faerch K, Borch-Johnsen K, Holst JJ, Vaag A. Pathophysiology and aetiology of impaired fasting glycaemia and impaired glucose tolerance: does it matter for prevention and treatment of type 2 diabetes? Diabetologia. 2009;52(9):1714–23.
Kahn CR. Insulin resistance, Insulin insensitivity, and insulin unresponsiveness: a necessary distinction. Metab Clin Exp. 1978;27(12 Suppl 2):1893–902.
Reaven G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol Metab Clin N Am. 2004;33(2):283–303.
Groop LC, Saloranta C, Shank M, Bonadonna RC, Ferrannini E, DeFronzo RA. The role of free fatty acid metabolism in the pathogenesis of insulin resistance in obesity and noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1991;72(1):96–107.
Ritz P, Berrut G. Mitochondrial function, energy expenditure, aging and insulin resistance. Diabetes Metab. 2005;31(Spec No2):5S67–73.
Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55 Suppl 2:S9–15. Recent magnetic resonance spectroscopy studies in healthy lean elderly subjects and healthy lean insulin-resistant offspring of parents with type 2 diabetes have demonstrated that reduced mitochondrial function may predispose these individuals to insulin resistance.
Dedoussis GV, Kaliora AC, Panagiotakos DB. Genes, diet and type 2 diabetes mellitus: a review. Rev Diabetes Stud. 2007;4(1):13–24.
Franks PW. Gene & environment interactions in type 2 diabetes. Curr Diabetes Rep. 2011;11(6):552–61.
Ashcroft FM, Rorsman P. Diabetes mellitus and the cell: the last ten years. Cell. 2012;148(6):1160–71.
Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414(6865):807–12.
Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106(2):165–9.
Kim J, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102(4):401–14. The authors propose that mitochondrial dysfunction may be a central cause of insulin resistance and associated complications.
Zierath JR, Krook A, Wallberg-Henriksson H. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 2000;43(7):821–35.
Cohen P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol. 1997;7(9):353–61.
Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104(31):12587–94.
DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in non-insulin-dependent (Type II) diabetes mellitus. J Clin Invest. 1985;76(1):149–55.
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71.
Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–7. This review presents emerging evidence that supports the potentially unifying hypothesis that prominent features of type 2 diabetes are caused by mitochondrial dysfunction.
Jelenik T, Roden M. Mitochondrial plasticity in obesity and diabetes mellitus. Antioxid Redox Signal. 2013;19(3):258–68. The variability of baseline mitochondrial function in the main target tissue of insulin action, skeletal muscle and liver, may be attributed to inherited and acquired changes in either mitochondrial quantity or quality.
Lee HK. Evidence that the mitochondrial genome is the thrifty genome. Diabetes Res Clin Pract. 1999;45(2–3):127–35.
Crescenzo R, Bianco F, Mazzoli A, Giacco A, Liverini G, Iossa S. Mitochondrial efficiency and insulin resistance. Front Physiol. 2015;5:512. The authors present evidence suggesting that an increase in mitochondrial efficiency precede and therefore can contribute to the development of high-fat-induced insulin resistance in skeletal muscle.
Ren J, Pulakat L, Whaley-Connelland A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl). 2010;88(10):993–1001.
Murphy MP. Induction of mitochondrial ROS production by electrophilic lipids: a new pathway of redox signaling? Am J Physiol Heart Circ Physiol. 2006;290:H1754–5.
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40.
Solinas G, Karin M. JNK1 and IKKβ: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24:2596–611.
Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and b-cell dysfunction? Diabetes. 2003;52:1–8.
Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaeffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–37.
Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25.
Kim JK, Wi JK, Youn JH. Metabolic impairment precedes insulin resistance in skeletal muscle during high-fat feeding in rats. Diabetes. 1996;45:651–8.
Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–9.
Kim F, Tysseling KA, Rice J, Pham M, Haji L, Gallis BM, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol. 2005;25:989–94.
Boden G, Lebed B, Schatz M, Homko C, Lemieux S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes. 2001;50:1612–7.
Zhang DX, Zou AP, Li PL. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am J Physiol Heart Circ Physiol. 2003;284:H605–12.
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–45.
Ogihara T, Asano T, Ando K, Chiba Y, Sakoda H, Anai M, et al. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension. 2002;40(6):872–9.
Diamond-Stanic MK, Henriksen EJ. Direct inhibition by angiotensin II of insulin-dependent glucose transport activity in mammalian skeletal muscle involves a ROS-dependent mechanism. Arch Physiol Biochem. 2010;116:88–95.
Henriksen EJ, Diamond-Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2011;51:993–9.
Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord. 2013;14(1):5–12.
Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev. 1991;43:109–42. Original review, which highlights the physiological, patho-physiological and, pharmacological aspects of the nitric oxide.
Poderoso JJ, Lisdero C, Schopfer F, Riobo N, Carreras MC, Cadenas E, et al. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem. 1999;274:37709–16.
Boveris A, Valdez LB, Zaobornyj T, Bustamante J. Mitochondrial metabolic states regulate nitric oxide and hydrogen peroxide diffusion to the cytosol. Biochim Biophys Acta. 2006;1757(5–6):535–42.
Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem Res Toxicol. 1997;10:1285–92.
Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–86.
Albina JE, Reichner JS. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 1998;17:39–53.
Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424.
Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A. Negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev. 2009;89:27–71. The cellular and molecular mechanisms by which ROS and RNS are thought to participate in normal insulin action and in the induction of insulin resistance are mainly described.
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84(24):9265–9.
Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615.
Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75(2):247–60.
Sansbury BE, Cummins TD, Tang Y, Hellmann J, Holden CR, Harbeson MA, et al. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circ Res. 2012;111:1176–89. Increased eNOS activity prevents the obesogenic effects of high-fat diet without affecting systemic insulin resistance, in part, by stimulating metabolic activity in adipose tissue.
Sadler CJ, Wilding JP. Reduced ventromedial hypothalamic neuronal nitric oxide synthase and increased sensitivity to NOS inhibition in dietary obese rats: further evidence of a role for nitric oxide in the regulation of energy balance. Brain Res. 2004;1016:222–8.
Muniyappa R, Iantorno M, Quon MJ. An integrated view of insulin resistance and endothelial dysfunction. Endocrinol Metab Clin N Am. 2008;37(3):685–711.
Baylis C. Nitric oxide deficiency in chronic kidney disease. Am J Physiol Ren Physiol. 2008;294:F1–9.
Vallés P, Manucha W. Nitric oxide in the kidney: physiological roles and regulation. In: Gimenéz MS, Gómez NN, editors. Advances in chemistry and biology of nitric oxide. Kerala: Research Signpost; 2007. p. 53–80. This review is focused on NOS isoforms distribution and regulation and on nitric oxide roles in renal physiology.
Mazzei L, Manucha W. Wt-1 expression linked to nitric oxide availability during neonatal obstructive nephropathy. Adv Urol. 2013;2013:401750.
Ushmorov A, Ratter F, Lehmann V, Dröge W, Schirrmacher V, Umansky V. Nitric oxide- induced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome c release. Blood. 1999;93:2342–52.
Kim YM, Bombeck CA, Billiar TR. Nitric oxide as a bifunctional regulator of apoptosis. Circ Res. 1999;84(3):253–6.
Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–48.
Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol. 1997;17(9):5317–27.
Manucha W, Vallés P. Cytoprotective role of nitric oxide associated whit Hsp70 expression in neonatal obstructive nephropathy. Nitric Oxide Biol Chem. 2008;18(3):204–15. In this original study, the authors postulated that the mechanism of apoptosis inhibition by nitric oxide would include stimulation of heat shock protein 70 expression.
Kaneki M, Shimizu N, Yamada D, Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal. 2007;9:319–29. S-nitrosylation has recently been proposed to play an important role in the pathogenesis of insulin resistance.
Wang H, Wang AX, Aylor K, Barrett EJ. Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes. 2013;62:4030–42. Nitric oxide directly promotes endothelial cell insulin transport by enhancing protein S-nitrosylation.
Scherrer U, Randin D, Vollenweider P, Vollenweider L, Nicod P. Nitric oxide release accounts for insulin’s vascular effects in humans. J Clin Invest. 1994;94:2511–5.
Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, et al. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–45. Receptor kinase activity is necessary to mediate production of nitric oxide through the insulin receptor. Both PI3K and Akt contribute importantly to this process.
Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser1179. J Biol Chem. 2001;276(32):30392–8.
Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28(5):463–91.
Zhang QJ, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes. 2012;61(7):1848–59.
Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284(1):R1–12.
Steinberg H, Cressman E, Wu Y, et al. Insulin mediated nitric oxide production is impaired in insulin resistance. Diabetes. 1997;46:24A.
Sansbury BE, Hill BG. Regulation of obesity and insulin resistance by nitric oxide. Free Radic Biol Med. 2014;73:383–99. This review discusses the role of nitric oxide in regulating adiposity and insulin sensitivity and places its modes of action into context with the known causes and consequences of metabolic disease.
Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Chayama K, Oshima T. Effect of obesity on endothelium-dependent, nitric oxide-mediated vasodilation in normotensive individuals and patients with essential hypertension. Am J Hypertens. 2001;14:1038–45.
Williams IL, Wheatcroft SB, Shah AM, Kearney MT. Obesity, atherosclerosis and the vascular endothelium: mechanisms of reduced nitric oxide bioavailability in obese humans. Int J Obes Relat Metab Disord. 2002;26:754–64.
Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–5. eNOS is important for the control not only of arterial pressure but also of glucose and lipid homeostasis.
Abudukadier A, Fujita Y, Obara A, Ohashi A, Fukushima T, Sato Y, et al. Tetrahydrobiopterin has a glucose-lowering effect by suppressing hepatic gluconeogenesis in an endothelial nitric oxide synthase-dependent manner in diabetic mice. Diabetes. 2013;62:3033–43.
Paneni F, Costantino S, Cosentino F. Role of oxidative stress in endothelial insulin resistance. World J Diabetes. 2015;6(2):326–32. The authors describe emerging theories concerning endothelial insulin resistance, with particular emphasis on the role oxidative stress. Complex molecular circuits including endothelial nitric oxide synthase, mitochondrial adaptor p66(Shc), and nicotinamide adenine dinucleotide phosphate-oxidase are discussed.
Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307. Improving endothelial insulin signaling may serve as a therapeutic strategy for ameliorating skeletal muscle insulin resistance.
Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, et al. Blockade of the nuclear factor-κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012;125:1122–33.
Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1982–8. The authors have demonstrated that during obesity induced by high-fat diet feeding, inflammation and insulin resistance develop in the vasculature well before these responses are detected in muscle, liver, or adipose tissue. This observation suggests that the vasculature is more susceptible than other tissues to the deleterious effects of nutrient overload.
Bender SB, Herrick EK, Lott ND, Klabunde RE. Diet-induced obesity and diabetes reduce coronary responses to nitric oxide due to reduced bioavailability in isolated mouse hearts. Diabetes Obes Metab. 2007;9:688–96.
Noronha BT, Li JM, Wheatcroft SB, Shah AM, Kearney MT. Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes. 2005;54:1082–9.
Fujimoto M, Shimizu N, Kunii K, Martyn JA, Ueki K, Kaneki M. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes. 2005;54:1340–8. iNOS plays a role in fasting hyperglycemia and contributes to hepatic insulin resistance in ob/ob mice.
Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, et al. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem. 2005;280:14203–11.
Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23.
Turini P, Thalmann S, Jayet PY, Cook S, Mathieu C, Burcelin R, et al. Insulin resistance in mice lacking neuronal nitric oxide synthase is related to an alpha-adrenergic mechanism. Swiss Med Wkly. 2007;137:700–4.
Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425–49.
Asea A. Hsp70: a chaperokine. Novartis Found Symp. 2008;291:173–9. discussion 179–183, 221–4.
Murphy ME. The HSP70 family and cancer. Carcinogenesis. 2013;34(6):1181–8.
Kang SS, Song JH, Lee MY, Kang YH, Lim SS, Ryu SY, et al. Developmental immunolocalization of heat shock protein 70 (HSP70) in epithelial cell of rat kidney. Histol Histopathol. 2011;26(11):1363–73.
Mazzei L, Docherty NG, Manucha W. Mediators and mechanisms of heat shock protein 70 based cytoprotection in obstructive nephropathy. Cell Stress Chaperones. 2015;20(6):893–906. This review summarizes our understanding of how the biological actions of Hsp70 may affect renal cytoprotection in the context of organ injury.
Vallés P, Jorro F, Carrizo L, Manucha W, Oliva J, Cuello-Carrión FD, et al. Heat shock proteins HSP27 and HSP70 in unilateral obstructed kidneys. Pediatr Nephrol. 2003;18(6):527–35.
Manucha W, Carrizo L, Ruete C, Molina H, Vallés P. Angiotensin II type I antagonist on oxidative stress and heat shock protein 70 (HSP 70) expression in obstructive nephropathy. Cell Mol Biol (Noisy-le-grand). 2005;51(6):547–55.
Carrizo LC, Ruete CM, Manucha WA, Ciocca DR, Vallés PG. Heat shock protein 70 expression is associated with inhibition of renal tubule epithelial cell apoptosis during recovery from low-protein feeding. Cell Stress Chaperones. 2006;11(4):309–24.
Borges TJ, Lopes RL, Pinho NG, Machado FD, Souza AP, Bonorino C. Extracellular Hsp70 inhibits pro-inflammatory cytokine production by IL-10 driven down-regulation of C/EBPβ and C/EBPδ. Int J Hyperth. 2013;29(5):455–63.
Rinaldi Tosi ME, Bocanegra V, Manucha W, Gil Lorenzo A, Valles PG. The Nrf2-Keap1 cellular defense pathway and heat shock protein 70 (Hsp70) response. Role in protection against oxidative stress in early neonatal unilateral ureteral obstruction (UUO). Cell Stress Chaperones. 2011;16(1):57–68.
Ferder L, Inserra F, Martinez-Maldonado M. Inflammation and the metabolic syndrome: role of angiotensin II and oxidative stress. Curr Hypertens Rep. 2006;8(3):191–8.
Garcia IM, Altamirano L, Mazzei L, Fornes M, Molina MN, Ferder L, et al. Role of mitochondria in paricalcitol-mediated cytoprotection during obstructive nephropathy. Am J Physiol Ren Physiol. 2012;302(12):F1595–605. This study evaluates the cytoprotective effects of paricalcitol, a VDR activator, at the mitochondrial level using an obstructive nephropathy model.
Garcia IM, Altamirano L, Mazzei L, Fornes M, Cuello-Carrion FD, Ferder L, et al. Vitamin D receptor modulated Hsp70/AT1 expression may protect the kidneys of SHRs at the structural and functional levels. Cell Stress Chaperones. 2014;19(4):479–91. Recent data suggest that Hsp70/AT1 modulated by VDR is involved in the mechanism by which paricalcitol provides renal protection in a hypertension model. In addition, lower AT1 expression through VDR induction could be a consequence of the heat shock response Hsp70-mediated cell protection.
Lutz W, Kohno K, Kumar R. The role of heat shock protein 70 in vitamin D receptor function. Biochem Biophys Res Commun. 2001;282(5):1211–9. Hsp70-like chaperone proteins play a role in controlling concentrations of the VDR within the cell.
Adams JS, Chen H, Chun RF, Nguyen L, Wu S, Ren SY, et al. Novel regulators of vitamin D action and metabolism: lessons learned at the Los Angeles zoo. J Cell Biochem. 2003;88(2):308–14.
Mitri J, Pittas AG. Vitamin D and diabetes. Endocrinol Metab Clin N Am. 2014;43(1):205–32. This article summarizes the current evidence from human studies that suggests but does not prove a relation between vitamin D and type 2 diabetes, and briefly reports on the potential association between vitamin D and type 1 diabetes.
Koroshi A, Idrizi A. Renoprotective effects of Vitamin D and renin-angiotensin system. Hippokratia. 2011;15(4):308–11.
Cheng Q, Boucher BJ, Leung PS. Modulation of hypovitaminosis D-induced islet dysfunction and insulin resistance through direct suppression of the pancreatic islet renin-angiotensin system in mice. Diabetologia. 2013;56(3):553–62.
Ferder M, Inserra F, Manucha W, Ferder L. The world pandemic of vitamin D deficiency could possibly be explained by cellular inflammatory response activity induced by the renin-angiotensin system. Am J Physiol Cell Physiol. 2013;304(11):C1027–39. This review attempts to show that there may be a relationship between inflammatory processes induced by chronic overstimulation of the renin-angiotensin system (RAS) and the worldwide deficiency of vitamin D (VitD) and that both disorders are probably associated with environmental factors.
Soti C, Nagy E, Giricz Z, Vigh L, Csermely P, Ferdinandy P. Heat shock proteins as emerging therapeutic targets. Br J Pharmacol. 2005;146:769–80.
Pandey KB, Mishra N, Rizvi SI. Protein oxidation biomarkers in plasma of type 2 diabetic patients. Clin Biochem. 2010;43(4–5):508–11.
Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes. 2009;58:567–78.
Kondo T, Motoshima H, Igata M, Kawashima J, Matsumura T, Kai H, et al. The role of heat shock response in insulin resistance and diabetes. Diabetes Metab J. 2014;38:100–6. Physical medicine using simultaneous stimulation of heat with mild electric current activates heat shock response, thereby reducing visceral adiposity, insulin resistance, chronic inflammation and improving glucose homeostasis in mice models of T2DM, as well as in humans with MS or T2DM.
Kavanagh K, Flynn DM, Jenkins KA, Zhang L, Wagner JD. Restoring HSP70 deficiencies improves glucose tolerance in diabetic monkeys. Am J Physiol Endocrinol Metab. 2011;300:E894–901.
Wei W, Liu Q, Tan Y, Liu L, Li X, Cai L. Oxidative stress, diabetes, and diabetic complications. Hemoglobin. 2009;33(5):370–7. Protective agents such as metallothionein, zinc and FGFs play an important role in preventing the development of diabetes and diabetic complications.
Marucci A, Miscio G, Padovano L, Boonyasrisawat W, Florez JC, et al. The role of HSP70 on ENPP1 expression and insulin-receptor activation. J Mol Med (Berl). 2009;87:139–44. The authors propose that type 2 diabetes results from a vicious cycle of metabolically induced inflammation, impaired insulin responsiveness, and subsequent loss of homeostatic signaling. A crucial and previously under-recognized event contributing to this loss of homeostasis is a reduction in heat shock proteins (HSPs, or stress proteins).
Hooper PL, Hooper PL. Inflammation, heat shock proteins, and type 2 diabetes. Cell Stress Chaperones. 2009;14(2):113–5.
Krause M, Bock PM, Takahashi HK, Homem De Bittencourt Jr PI, Newsholme P. The regulatory roles of NADPH oxidase, intra- and extra-cellular HSP70 in pancreatic islet function, dysfunction and diabetes. Clin Sci (Lond). 2015;128(11):789–803. In this review, authors describe possible mechanisms by which HSP70 participates in cell function/dysfunction, including the activation of NADPH oxidase isoforms leading to oxidative stress, focusing on the possible role of HSPs and signaling in pancreatic islet α- and β-cell physiological function in health and Type 2 diabetes mellitus.
Kavanagh K, Zhang L, Wagner JD. Tissue-specific regulation and expression of heat shock proteins in type 2 diabetic monkeys. Cell Stress Chaperones. 2009;14(3):291–9.
Hooper PL. Insulin signaling, GSK-3, heat shock proteins and the natural history of type 2 diabetes mellitus: a hypothesis. Metab Syndr Relat Disord. 2007;5(3):220–30.
Lee JH, Gao J, Kosinski PA, Elliman SJ, Hughes TE, Gromada J, et al. Heat shock protein 90 (HSP90) inhibitors activate the heat shock factor 1 (HSF1) stress response pathway and improve glucose regulation in diabetic mice. Biochem Biophys Res Commun. 2013;430(3):1109–13.
Krause M, Heck TG, Bittencourt A, Scomazzon SP, Newsholme P, Curi R, et al. The chaperone balance hypothesis: the importance of the extracellular to intracellular HSP70 ratio to inflammation-driven type 2 diabetes, the effect of exercise, and the implications for clinical management. Mediat Inflamm. 2015;2015:249205. Imbalances in the HSP70 status, described by the [eHSP70]/[iHSP70] ratio, may be determinant to trigger a chronic pro-inflammatory state that leads to insulin resistance and T2DM development.
Nakhjavani M, Morteza A, Khajeali L, Esteghamati A, Khalilzadeh O, Asgarani F, et al. Increased serum HSP70 levels are associated with the duration of diabetes. Cell Stress Chaperones. 2010;15:959–64. Serum level of HSP70 is significantly higher in patients with diabetes and correlates with the duration of disease. Higher HSP70 in prolonged diabetes versus newly diagnosed diabetes may be an indicator of metabolic derangement in the course of diabetes.
Karpe PA, Tikoo K. Heat shock prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1–7) signaling. Diabetes. 2014;63:1124–39. Induction of HSP72 is a novel approach to prevent insulin resistance-induced vascular complications.
Henstridge DC, Bruce CR, Drew BG, Tory K, Kolonics A, Estevez E, et al. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes. 2014;63(6):1881–94. Increased oxidative metabolism associated with activation of HSP72 has potential clinical implications not only for type 2 diabetes but also for other disorders where mitochondrial function is compromised.
Chow AM, Steel R, Anderson RL. Hsp72 chaperone function is dispensable for protection against stress-induced apoptosis. Cell Stress Chaperones. 2009;14:253–63.
Chung J, Nguyen AK, Henstridge DC, Holmes AG, Stanley Chan MH, Mesa JL, et al. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2008;105:1739–44. HSP72 is essential in blocking inflammation and preventing insulin resistance in the context of genetic obesity or high-fat feeding.
Chae JI, Kim J, Son MY, Jeon YJ, Kim DW, Kim HE, et al. Cardioprotective molecules are enriched in beating cardiomyocytes derived from human embryonic stem cells. Int J Cardiol. 2013;165(2):341–54.
Gao H, Meng J, Xu M, Zhang S, Ghose B, Liu J, et al. Serum heat shock protein 70 concentration in relation to polycystic ovary syndrome in a non-obese Chinese population. PLoS ONE. 2013;8(6):e67727. Increased serum Hsp70 levels are associated with the combination of IR, oxidative stress and low-grade chronic inflammation in Polycystic ovary syndrome individuals, which provides supportive evidence that Hsp70 plays a key role in this pathogenesis.
Atalay M, Oksala NK, Laaksonen DE, Khanna S, Nakao C, Lappalainen J, et al. Exercise training modulates heat shock protein response in diabetic rats. J Appl Physiol (1985). 2004;97(2):605–11. Diabetic therapies that induce the stress response, whether via heat, bioactive compounds, or genetic manipulation, improve or prevent all of the morbidities and comorbidities associated with the disease.
Hooper PL, Balogh G, Rivas E, Kavanagh K, Vigh L. The importance of the cellular stress response in the pathogenesis and treatment of type 2 diabetes. Cell Stress Chaperones. 2014;19(4):447–64.
Krause M, Ludwig MS, Heck TG, Takahashi HK. Heat shock proteins and heat therapy for type 2 diabetes: pros and cons. Curr Opin Clin Nutr Metab Care. 2015;18(4):374–80. Transient increments in nitric oxide and heat shock protein 70 levels may explain the benefits of heat therapy in obesity and diabetes.
Bathaie SZ, Jafarnejad A, Hosseinkhani S, Nakhjavani M. The effect of hot-tub therapy on serum Hsp70 level and its benefit on diabetic rats: a preliminary report. Int J Hyperthermia. 2010;26(6):577–85. A decrease in complications in diabetic rats after hot-tub therapy is shown here.
Bhatia S, Shukla R, Venkata Madhu S, Kaur Gambhir J, Madhava Prabhu K. Antioxidant status, lipid peroxidation and nitric oxide end products in patients of type 2 diabetes mellitus with nephropathy. Clin Biochem. 2003;36:557–62.
Henstridge DC, Whitham M, Febbraio MA. Chaperoning to the metabolic party: the emerging therapeutic role of heat-shock proteins in obesity and type 2 diabetes. Mol Metab. 2014;3:781–93. Targeted manipulation of Hsp72 or use of chemical chaperiones may have clinical utility in treating metabolic disorders such as insulin resistance and T2DM.
Prasad P, Kochhar A. Interplay of vitamin D and metabolic syndrome: a review. Diabetes Metab Syndr. 2015. March 6. The paper provides an insight into the physiology of vitamin D and relationship of vitamin D deficiency with risk factors of metabolic syndrome through observational and supplementation studies.
Rajan RS, Tsumoto K, Tokunaga M, Tokunaga H, Kita Y, Arakawa T. Chemical and pharmacological chaperones: application for recombinant protein production and protein folding diseases. Curr Med Chem. 2011;18(1):1–15.
Kashfi K. Nitric oxide-releasing hybrid drugs target cellular processes through S-nitrosylation. For Immunopathol Dis Ther. 2012;3(2):97–108.
Zaouali MA, Bejaoui M, Calvo M, Folch-Puy E, Pantazi E, Pasut G, et al. Polyethylene glycol rinse solution: an effective way to prevent ischemia-reperfusion injury. World J Gastroenterol. 2014;20(43):16203–14. The PEG-35 rinse solution prevented oxidative stress, mitochondrial damage, and liver autophagy. Further, it increased the expression of cytoprotective heat shock proteins such as HO-1 and HSP70, activated AMPK, and contributed to the restoration of cytoskeleton integrity after IRI.
McCarty MF, Barroso-Aranda J, Contreras F. Regular thermal therapy may promote insulin sensitivity while boosting expression of endothelial nitric oxide synthase-effects comparable to those of exercise training. Med Hypotheses. 2009;73(1):103–5. Regular thermal therapy has the potential to improve impaired insulin sensitivity and boost endothelial expression of the “constitutive” isoform of nitric oxide synthase-effects.
Atalay M, Oksala N, Lappalainen J, Laaksonen DE, Sen CK, Roy S. Heat shock proteins in diabetes and wound healing. Curr Protein Pept Sci. 2009;10(1):85–95.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Human and Animal Rights and Informed Consent
The animal experiments described herein were not performed by any of the authors, signifying that no animals were harmed in the course of preparing this manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Research and Technology Council of Cuyo University (SECyT), Mendoza, Argentina, and from the National Council of Scientific and Technical Research (CONICET) PIP 2010–2012, both of which were awarded to Walter Manucha. Grant no. PICT 2012–0234, BID 2777 OC/AR.
Additional information
This article is part of the Topical Collection on Hypertension and Obesity
Rights and permissions
About this article

Cite this article
Molina, M.N., Ferder, L. & Manucha, W. Emerging Role of Nitric Oxide and Heat Shock Proteins in Insulin Resistance. Curr Hypertens Rep 18, 1 (2016). https://doi.org/10.1007/s11906-015-0615-4
Published:
DOI: https://doi.org/10.1007/s11906-015-0615-4