
Abstract
People vary genetically in their susceptibility to the effects of environmental risk factors for many diseases. Genetic variation also underlies the extent to which people respond appropriately to clinical therapies. Defining the basis to the interactions between the genome and the environment may help elucidate the biologic basis to diseases such as type 2 diabetes, as well as help target preventive therapies and treatments. This review examines 1) some of the most current evidence on gene × environment interactions in relation to type 2 diabetes; 2) outlines how the availability of information on gene × environment interactions might help improve the prevention and treatment of type 2 diabetes; and 3) discusses existing and emerging strategies that might enhance our ability to detect and exploit gene × environment interactions in complex disease traits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The global prevalence of type 2 diabetes is expected to reach around 438 million people in 2030 [1]. When diabetes takes a grip, glycemic control often continues to decline and micro and macrovascular complications occur; even in diabetic patients treated with the best available therapies, most eventually die of cardiovascular disease [2, 3].
Obesity is a major risk factor for diabetes. Although preventive strategies involving weight loss have a considerable favorable impact on the short-term incidence of diabetes [4, 5], most at-risk persons eventually develop the disease, irrespective of treatment [6]. A pressing need exists therefore, to better understand the causes, processes, and consequences of type 2 diabetes.
The genome is the conduit through which the environment conveys many of its effects on the phenotypes involved in health and disease. The disruption of this signaling circuit in the form of germline DNA variation impacts the extent to which healthful environmental exposures protect against the development of disease and may enhance the pathophysiologic effects of unhealthful environmental exposures. This concept is broadly referred to as “gene × environment interaction” (GEI), in which the “gene” is usually one or more DNA variants and the “environment” can be any nongenetic factor that impacts risk.
In animal, plant, and bacterial genetics, numerous, concrete examples of GEI exist. In drosophila, for example, there is a critical period in which environmental temperature impacts the development of eye facets in larvae; in general, the warmer the environment, the fewer facets develop [7]. Interestingly, the extent to which temperature affects eye facet development is modified by the genetic background of the fly [7], representing one of the first documented examples of a GEI. Mechanistic studies of GEI were first documented in 1961 by Jacob and Monod [8] for their Nobel Prize winning work a few years later on transcriptional regulation in Escherichia coli [9], in which they showed that bacterial enzyme synthesis is controlled at “structural” and “functional” genetic levels through interactions with cytoplasmic components that can be induced or repressed by specific metabolites.
There are also numerous tangible examples of GEI in humans that are exploited to improve human health. Perhaps the most frequently cited example of a gene × diet interaction relates to a rare autosomal-recessive mutation in the gene encoding the hepatic enzyme phenylalanine hydroxylase (PAH, also often referred to as PKU), an enzyme that influences the metabolism of phenylalanine to the amino acid tyrosine. Under normal dietary conditions, carriers of the PAH mutation are susceptible to severe impairments in cognitive development, but phenylalanine-free diets substantially improve prognosis.
Many speculate that GEIs also impact susceptibility to type 2 diabetes and that elucidating these interactions may help improve prevention and management of the disease [10••]. As discussed later in this review, the discovery and elucidation of GEIs will also greatly expand our knowledge of the genetic loci within which diabetes-associated variants reside.
Why GEIs exist has been the topic of intense debate, with some postulating that the loci involved conveyed a survival advantage throughout much of human evolution and have consequently been under positive selective pressure [11], whereas others argue that these variants occurred randomly and have remained in the gene pool either because their harmful effects on reproductive fitness have only recently been triggered by a population-wide shift in lifestyle and the development of obesity [12•] or because the mutation has evaded natural selection, owing to the manifestation of disease-related deleterious consequences after the reproductive phase in the life of the affected individual.
Although understanding the origins of GEI in diabetes and related traits is interesting and important, it is perhaps more important at this stage to determine which of the many studies that have reported on this topic are likely to be reliable, as few have been adequately replicated. Thus, firmly establishing if, as well as why, genes and environmental factors might interact in type 2 diabetes should be prioritized.
This review examines some of the most current evidence on GEI in relation to type 2 diabetes, outlines how the availability of information on GEI might help improve the prevention and management of type 2 diabetes, and discusses existing and emerging strategies that might enhance our ability to detect and exploit GEIs for the benefit of human health.
Why Search for Evidence of GEI in Type 2 Diabetes?
Disease prevention is generally better than treatment or cure. The optimal prevention of any disease requires knowledge of its modifiable risk factors so that they can be intervened upon. Knowledge of salient risk factors, such as genetic variation, can be useful for disease prevention, especially if this knowledge helps target interventions, such as exercise or pharmacotherapy, at the people most likely to respond well to them or at those for whom the avoidance of specific risk factors, such as tobacco or ultraviolet exposure, has an especially marked impact on disease risk.
Type 2 diabetes is highly heterogeneous in its etiology, clinical presentation, and pathogenesis, albeit with the common denominators of elevated blood glucose concentrations and relative insulin deficiency. The syndromic nature of the disease makes it difficult to optimize strategies for its prevention or treatment. Ideally, both would be achieved using uniform strategies that are effective and can be implemented population-wide. However, the response to primary and secondary preventive interventions differs greatly from one person to the next. Although some of this response variability is owing to varying levels of adherence to therapy and methodologic factors, much is attributable to interindividual biological differences in the way treatments work. Exercise intervention studies in groups of families indicate that the phenotypic response to interventions tends to be more similar between biologically related persons than between those who are unrelated [13], supporting the thesis that inherited factors, such as genotypes, modify treatment effectiveness.
Because genotypes are salient biomarkers, remaining unchanged throughout a person’s lifespan, it may be possible to derive genotype panels that reliably predict a person’s level of risk given specific diabetogenic exposures or the extent to which risk is likely to diminish with specific preventive interventions. Clearly, the usefulness of this strategy demands that the predictive ability of the genetic screening tool exceeds that of conventional, low-cost strategies and that the cost-benefit ratio associated with the genetic screening tool is substantially better than for conventional approaches.
The sensitivity and specificity of conventional diabetes prediction algorithms is generally fairly good (area under the receiver operator characteristic curve is ~ 0.85) when risk is ascertained in the months or years prior to diagnosis [14], but their predictive ability diminishes as time to event increases [15–17], which may be because the strongest predictors of diabetes, such as elevated blood glucose concentrations, are largely a consequence of other causal factors, or because the primary causal factors change with time and/or are imprecisely quantified (eg, adiposity and family history of diabetes). Accordingly, risk markers that do not change with time, such as genotypes, tend to improve in their predictive ability relative to conventional risk markers as time to event increases [15–17], a point that has received limited attention to date, possibly because most risk prediction studies are of fairly short duration.
Identifying and validating GEIs may extend our understanding of how specific environmental exposures cause disease by pinpointing genes through which the environmental effects are conveyed. It is also exciting to consider that evidence of GEI might aid in the prediction of disease [18].
Approaches Used to Study GEI in Type 2 Diabetes
Biologic Candidate Gene Studies
The most widely published method for discovering GEI to date is the biologic candidate gene approach, in which functional evidence, often from animal or in vitro human models, is used to identify loci that play a role in disease etiology and that are up- or downregulated with exposure to specific environmental factors such as dietary fats, aerobic exercise, alcohol, or cognitive stress. Variants within these loci are then identified and studied for interaction with related exposures and outcomes in epidemiologic cohorts or intervention studies.
PPARGC1A is an extensively researched biologic candidate gene for type 2 diabetes; the gene transcriptionally coactivates multiple pathways involved in insulin sensitivity, adipogenesis, hepatic glucose production, mitochondrial biogenesis, and many other metabolic processes [19]. Ppargc1a transgenic mice have a higher density of oxidative muscle fiber and enzymes, as well as increased time to exhaustion following electrical stimulation of the hind limbs [20]. Aerobic exercise in humans also increases PPARGC1A mRNA levels in skeletal muscle [21] and studies comparing diabetic and nondiabetic persons have reported dow-regulation of PPARGC1A and its target genes in diabetic skeletal muscle [22]. This evidence has prompted speculation that the PPARGC1A–exercise axis may be an attractive target for the prevention of diabetes and other chronic diseases [19, 23]. Common DNA variants at PPARGC1A have also been associated with type 2 diabetes [24], although no robust association signals in this region were found in subsequent genome-wide association study (GWAS) meta-analyses [25•]. Nevertheless, variants at this locus have been widely examined in the context of GEI [26–31].
Unfortunately, despite many hundreds of publications using the biologic candidate gene approach, few have been followed up with independent studies replicating or refuting the initial findings. Although replication of interactions is often implied [32], it is important to consider whether the replication study tested the same hypothesis to the original report: did both studies focus on the same genetic locus, environmental exposure, and outcome, were the effects comparable in direction and magnitude, and were the analytical approaches similar?; and if the study reported negative results, was it statistically powered to be confident these are not false-negative findings? In other words, we need to carefully weigh up how strong the evidence for or against replication is before accepting or rejecting the validity of a reported interaction effect. The same level of rigor that is applied to genetic association studies per se should be applied when appraising studies of interaction.
Overall, the biologic candidate gene approach has not been successful in identifying robust examples of GEI in type 2 diabetes. As such, continued investment in this approach is probably unjustified unless combined with other more promising methods, such as those discussed below.
Following Up on GWAS Association Signals
An increasingly favored approach for studying GEI is one in which loci discovered in main-effect GWAS meta-analyses are examined as putative effect modifiers of environmental risk factors for diabetes and other traits. Providing the relevant environmental data are also available, these tests can be easily performed, as investigators can often use the same cohort collections that were included in the initial main-effects meta-analyses, thus allowing a fairly quick in silico analysis of interaction effects. Moreover, because these loci are known to be reliably associated with relevant disease traits, this might strengthen causal inference for an interaction and some believe that this may be a useful screening step that improves power to detect interactions [33].
We recently reported evidence of an interaction between an HNF1B variant (rs4430796) and physical activity in type 2 diabetes incidence [34•]. Our study suggests that in people carrying neither copy of the diabetes risk allele, the rate of progression to diabetes is significantly higher in persons who reported low versus high levels of baseline physical activity, an association that is consistent with data from other epidemiologic studies and clinical trials that disregarded the effect modifying roles of genotypes. By contrast, in persons carrying both copies of the diabetes risk allele, there was no apparent protective effect of physical activity on diabetes incidence. Although this specific example awaits replication and should be interpreted with some caution, data such as these highlight that some risk factors are substantially better predictors of diabetes in certain population subgroups than in others, and that it might be possible to identify these subgroups using genotypes. Moreover, the integration of data on GEI into risk prediction algorithms might substantially improve their effectiveness in specific subgroups of the population.
The DPP (Diabetes Prevention Program) is a randomized clinical trial of intensive lifestyle modification, metformin treatment, and placebo control undertaken in more than 3000 adults at high-risk of type 2 diabetes from the five major ethnic groups in the United States. Moore et al. [35] reported tentative evidence that the minor allele at the CDKN2A/B locus modified the effect of the DPP lifestyle intervention on improved β-cell function (P interaction = 0.05) and on type 2 diabetes incidence (P interaction = 0.01). Our group conducted a follow-up study in approximately 9,000 adults from southern Sweden and reported comparable gene × physical activity interaction effects for the same CDKN2A/B variant on 2-hour glucose concentrations [28]. However, a third small North American trial (N ~ 400) called HERITAGE found no evidence that this variant is associated with changes in glucose disposal, insulin sensitivity, or β-cell function following exercise training [36•].
Probably the most widely studied example of GEI relevant to type 2 diabetes is for the PPARG Pro12Ala variant and dietary fat intake. The seminal report on this hypothesis came from a UK-based study [37], quickly followed by a confirmatory paper from the United States [38]; the authors observed that the relationship of dietary fats with plasma insulin levels and body mass index (BMI) differed by Pro12Ala genotype, such that no relationship was observed in Pro12Pro homozygotes and an inverse association was seen in carriers of the Ala12 allele. Many replication attempts have since been reported (summarized in [39]). The authors of the original paper recently attempted to meta-analyze all available published data, but concluded that results from such analyses are likely to be unreliable [40••] owing to inconsistencies in the definition of exposures and outcomes, study designs, statistical analyses, and presentation of results, as well as publication bias and other sources of bias and confounding inherent in epidemiology.
Interactions between the Pro12Ala variant and other lifestyle factors have also been reported. In the study by Brito et al. [34•] described above, nominal evidence of an interaction (P interaction = 0.04) between the Pro12Ala PPARG gene variant and physical activity on 2-hour glucose levels was also described [41]; in people reporting low levels of physical activity, the minor allele was associated with a lesser risk of having impaired glucose regulation (OR, 0.88, per allele; 95% CI, 0.75–1.02), whereas in persons reporting high levels of physical activity, the opposite was true (OR, 1.05, per allele; 95% CI, 0.97–1.15). The minor Pro12Ala allele was also associated with greater improvements in glucose tolerance with exercise training in HERITAGE [36•], although the study lacks a control arm, making it difficult to distinguish gene × exercise interactions from genetic effects that merely persists in the presence of exercise. A third, relatively small, family-based cross-sectional study from the United States [41] also reported tentative evidence of gene × physical activity interactions at the Pro12Ala locus in type 2 diabetes risk (P interaction = 0.02), but the direction of effect differed from those reported by Brito et al. [34•] and Ruchat et al. [36•]. Interactions between Pro12Ala and lifestyle intervention in type 2 diabetes risk have also been studied in the DPP [42] and the Finnish DPS (Diabetes Prevention Study) [43]. Neither trial reported evidence of such interactions, although in the latter, secondary analyses focused on gene × physical activity interactions on diabetes incidence revealed nominal evidence of interaction [44].
One other GWAS-derived locus, which has also been extensively studied for gene × lifestyle interactions in relation to diabetes, is FTO. A common FTO variant (rs9939609) has been consistently associated with type 2 diabetes across multiple populations, an effect mediated by increased obesity risk [45]. Soon after the discovery of FTO, two independent cross-sectional studies reported interactions between FTO gene variants and physical activity on obesity predisposition [46, 47]. Both studies indicated that the obesogenic effect of the rs9939609 minor allele is substantially diminished by physical activity. A third study conducted by us in the DPP showed that although the obesogenic allele was associated with a greater gain in subcutaneous adipose mass during the first year of the trial, the reverse was seen in persons randomized to intensive lifestyle intervention [48]. Dozens of subsequent studies have tested similar hypotheses, although collectively results were in equipoise. Thus, we collected and meta-analyzed all currently available data, published and unpublished, with the objective of confirming or refuting the hypothesis that physical activity modifies the effects of FTO variation on obesity predisposition. The analysis comprised almost 250,000 adults and children and showed that an interaction effect of this nature is indeed evident, although the magnitude of the effect varied considerably by geographic region (Kilpelainen et al., Unpublished data).
We recently conducted several large-scale meta-analyses of gene × diet interactions as part of the CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology) Consortium. These studies have, thus far, focused on fasting glucose- and insulin-associated loci. Analyses have comprised approximately 50,000 participants, primarily of European descent. In the first publication, Nettleton et al. [49••] reported a nominally significant interaction for the rs780094 GCKR variant with dietary whole grains and fasting insulin levels. None of the 15 remaining variants showed evidence of interactions on glucose or insulin concentrations and the GCKR interaction effect did not withstand correction for multiple testing. In a second study from the CHARGE consortium, we assessed the same variants for interaction with zinc intake [50••]; an SLC30A8 variant (rs11558471) yielded a nominally significant interaction with dietary and supplemental zinc intake on fasting insulin concentrations (P = 0.005). Although this interaction effect was not statistically significant after Bonferroni correction (threshold of P < 0.0025), an interaction with zinc at this locus is highly biologically plausible, as SLC30A8 encodes a zinc efflux transporter that facilities zinc accumulation in intracellular vesicles and colocalizes with insulin in the secretory pathway granules of INS-1 cells [51], thus raising the prior probability of an interaction at this locus. The almost complete absence of interaction effects at any of the remaining loci in such a large study, however, suggests that these GWAS-derived loci are, for the most part, unlikely to interact with these dietary factors in a clinically meaningful way.
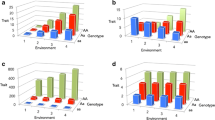
Despite the encouraging results described above, there are important caveats to using main-effect GWAS meta-analyses to screen for loci involved in GEI. The GWAS-derived type 2 diabetes–associated loci discovered so far generally convey very small, but homogeneous effects within and between populations [25•]. The same is true for most complex traits. The low variance relative to effect size is why the top-ranking GWAS variants yield very significant P values (P < 5.0 × 10−8). However, as illustrated in Fig. 1, these characteristics may also mitigate the possibility that the top-ranking GWAS loci modify the effects of the risk factors for type 2 diabetes that vary in frequency within and between the index populations. Large-scale assessments of GWAS-derived loci generally confirm that the majority of these do not modify the relationships of selected environmental risk factors with diabetes-related traits [25•]. It is worth noting though, that eventually, as GWAS meta-analysis sample sizes grow large enough, many loci involved in GEI (or gene × gene interactions) will exert sufficiently strong main effect signals to be flagged as disease-associated variants. Nevertheless, in some cases, where crossover interactions occur, the sample size will do nothing to help detect these variants, as this type of interaction can completely offset the main effect of the genotype, resulting in a corresponding P value that is close to 1.

a, Shows an example of an interaction between a biallelic variant and an environmental risk factor. Gene x environment interactions can be defined as genetic effects on [disease] traits that differ in magnitude [and sometimes direction] across environmental contexts. b, Shows the same data as in Panel A, but under the assumption that the genetic effects are equal across the spectrum of the environmental exposure (ie, no interactions exist). This assumption, which is made in most genome-wide association studies and in this example is incorrect, may cause clinically relevant genetic risk factors to be overlooked
Why Most Published Studies of GEI Are Probably False-positive
Many small-scale, possibly underpowered, interaction studies have been reported [52•]. In fairness to the studies’ investigators, it has been impossible to accurately calculate power required to detect GEIs in type 2 diabetes, primarily because to do so requires that realistic interaction effect sizes can be estimated, which has not been possible owing to the dearth of convincing published interaction results. The few genome-wide assessments of GEI that have been undertaken, of which none were published at the time of writing this review, indicate that interaction effect sizes are likely to be very small in magnitude, at least where common gene variants are concerned. Correspondingly, many published reports of gene × lifestyle interactions in type 2 diabetes or its quantitative traits may be false-positives.
False-positive accounts of GEI may be more frequent than for studies of genetic main effects. In part, this is because 1) tests of interaction are often conducted as secondary analyses, which may have involved a large number of unreported statistical comparisons, thus inflating type 1 error rates; 2) data transformation procedures can sometimes produce interaction effects that are statistically significant, but vanish when the data are reanalyzed on the normal scale (a type of “removable” interaction); 3) studies of GEI are no less prone to confounding and bias than studies focused on the genetic or environment factors alone, yet few studies of interaction have adequately controlled for confounding; and 4) a priori biologic evidence, which experimenters and reviewers often consider when determining whether a nominally significant interaction result is plausible, is often less relevant than perceived (see [53••] for further discussion of these points).
Which Way Forward for the Discovery and Translation of GEI in Type 2 Diabetes?
Studies that incorporate high-throughput, massively parallel genotyping technologies such as GWAS have been rapidly adopted as the default approach for discovering disease-associated loci for type 2 diabetes and many other complex traits. These remarkable technological developments and corresponding changes in the way geneticists from around the world work together have caused an explosion in the density of data routinely available for genetic analyses; in less than a decade, population geneticists have transitioned from analyzing datasets involving just a handful of gene variants in a few hundred individuals to those that frequently involve the analysis of millions of variants and multiple phenotypes in cohort collections totaling tens of thousands of participants or more. This quantum leap in data dimensionality has generated many novel analytical challenges.
A major weakness of the standard approaches used to analyze GWAS data is that the possibility of interactions between loci and other risk factors is usually completely ignored [54•]. If, as many agree, most complex diseases emerge as a consequence of gene × environment and gene × gene interactions, this implies that there is a great deal more, very valuable information about the genetics of human disease remaining to be discovered than has been so far. However, it is worth pointing out that such discoveries are unlikely to explain any of the “missing heritability” in type 2 diabetes (ie, the discrepancy between the estimated heritability of the disease and the variance in the disease explained by specific gene variants), as the commonly cited heritability estimates were not derived in a way that accommodates the presence of interactions.
As discussed below, several novel approaches have recently been proposed that will facilitate the analysis of interactions within high-density datasets.
Variance Prioritization
As described earlier in this review, the approach used to identify disease-associated variants in conventional main-effect GWAS experiments may bias against the discovery of loci that exert their effects through interactions with other risk factors. A characteristic of loci that exert effects via interactions is that the variance associated with their main effects tend to be relatively high. Pare et al. [55••] recently proposed a method that exploits this characteristic to prioritize variants that are most likely to convey their effects through interactions. An advantage of this approach is that it does not require knowledge of the interacting variables, which distinguishes it from almost all other interaction analysis methods. The authors applied their method to data from the Women’s Genome Health Study, a study of more than 25,000 US women [56], and identified interactions between variants at LEPR (rs12753193) and BMI on C-reactive protein levels, ICAM1 (rs1799969) and smoking on soluble intercellular adhesion molecule-1(ICAM-1) levels, and PNPLA3 (rs738409) and BMI on soluble ICAM-1 levels [55••].
Selective Sampling Methods
Numerous approaches have been described for selectively sampling populations to minimize sample size requirements in studies of GEI (see [52•] for overview of several established methods). A recent development in sample selection strategies, geared specifically toward GWAS, was recently described by Boks et al.. Provided that interaction effects are approximately linear, the genetic effect on disease risk scales proportionately across the spectrum of the environmental exposure; thus it should be possible to select out subgroups of the population that differ substantially in their exposure to the environmental risk factor, undertake GWAS (or similar) within these groups, and then test whether the magnitude of the genetic effect estimates differ by environmental exposure level. Boks et al. [57] estimate that by selecting these subgroups from the top and bottom 10% of the environmental exposure distribution, the required sample size can be reduced by approximately 70% compared with conventional approaches, which would substantially reduce costs in GWAS-type experiments. The authors highlight that this approach depends on the initial sampling frame being large enough to yield sufficiently extreme environmental exposure differences between the two subgroups, which is fundamental to the success of the approach. Thus, the approach is unlikely to work where the sampling frame is small or where the spectrum of environmental exposure is narrow. An additional caveat to this approach is that, by design, the environmental exposure distribution and all of its correlates within the selected subcohort will differ substantially from those of the background population, which will limit how the dataset can be used for subsequent analyses.
The Joint Meta-analysis Method
A major challenge to studying GEI is that the sample size requirements are often considerably larger than in main-effect genetic association studies [58]. Because interaction analyses generally require information on environmental exposures, the ability to amass sufficiently large cohort collections is very difficult, particularly when seeking cohorts with standardized environmental exposure data. This challenge can become unmanageable when conducting genome-wide interaction studies, in which more than a million interaction hypothesis tests are performed, because the available cohorts are generally too small to yield adequate statistical power. Prompted by this realization, a number of novel innovative analytical methods to maximize statistical power in genome-wide interaction analyses have been developed.
Manning et al. [59••] recently described one such approach, termed the joint meta-analysis (JMA) method, that incorporates the analysis of interaction and marginal genetic effects into a single test. JMA, which builds on previously described methods [60•, 61, 62], appears considerably more powerful than conventional interaction tests [59••]. As proof of concept, Manning et al. [59••] applied the JMA method alongside other existing approaches to data from five population cohort collections comprising almost 20,000 nondiabetic individuals to test for interactions between 60 PPARG tag SNPs and BMI on fasting insulin concentrations. The JMA approach was the most powerful of the selected methods and identified three variants that interacted with BMI at an experiment-wide significance threshold of P < 1.67 × 10−5. For the top-ranked variant (rs1801282 Pro12Ala), the JMA interaction P value was 8.29 × 10−9, which compared with a P value of 1.15 × 10−6 for Kraft et al.’s [61] 2 d.f. method. P values for the remaining approaches all exceeded the experiment-wide significance threshold.
The JMA method has since been used in several unpublished GWAS meta-analyses and has led to the discovery of novel loci that influence glucose homeostasis via interactions with BMI (Manning et al., Personal communication).
Nested Case-cohort Studies
The ideal epidemiologic study design for the discovery of GEI in type 2 diabetes is one in which environmental exposure status is ascertained prior to the development of disease, thus mitigating the effects of disease-labeling bias, a type of bias that influences the way in which people report disease-associated behaviors such as habitual dietary patterns and physical activity levels [63]. We recently conducted such a study using data from nine European countries called the InterAct Project [10••]. InterAct is a European Union financed nested case-cohort study comprised of 12,403 verified incident cases of type 2 diabetes, which occurred during 4 million person-years of follow-up, and a subcohort of 16,154 nondiabetic individuals for comparison. Relative to all existing studies of gene × lifestyle interactions in diabetes, InterAct has a high level of statistical power to test interaction hypotheses and its results are likely less prone to confounding and bias than many previous studies; as such, the study offers a unique opportunity to verify or refute prior reports of interaction. InterAct is also designed for the unbiased discovery of interaction effects using GWAS and Cardio-Metabochip (Illumina Inc. San Diego, CA) arrays.
Genotype-based Recall Experiments
A much discussed, yet rarely implemented approach to the analysis of gene × intervention interactions, termed genotype-based recall, involves the selection of participants with contrasting genetic risk profiles who are subsequently recruited into a randomized controlled trial [64]. The approach maximizes statistical power by manipulating allele frequency distributions in the target population, so that two equally sized groups of participants are sampled from the tails of a genetic risk score distribution or so that homozygous carriers of a rare allele are compared with an equal number of homozygous carriers of the common allele. Genetic risk scores can be computed in various ways, but usually this process involves weighting a series of risk alleles for a specific disease trait by effect estimates derived from an independent and well-powered dataset, and subsequently summing the weighted risk alleles together to form a continuously distributed score [65]. Genetic risk scores published to date have focused on characterizing the main genetic effects of loci. Nevertheless, once a series of replicated interaction loci are known, it will be perfectly feasible to weight these variants by genetic and environmental coefficients to construct GEI risk scores.
The genotype-based recall strategy is probably most powerful when very rare genotypes are targeted, thus maximizing the genetic difference between groups. However, the more infrequent the genotypes of interest, the larger the sampling frame required to identify these persons, with sampling frames greater than 100,000 individuals for high-fidelity selection schema (ie, those in which the high- and low-risk subgroups carry all or none of the risk alleles, respectively) for risk scores comprised of the top five type 2 diabetes variants. A second caveat to the genotype-based recall approach is that trials undertaken in this way clearly have a very specific data structure, rendering them of limited use for secondary analyses that are not focused on the same genotype(s). It is these limitations that perhaps explain why the genotype-based recall method has not been widely implemented to date.
One way to maximize the possibility that genotype-based recall experiments succeed might involve the collection of muscle, adipose, or other tissue samples from participants in the two genotype groups prior to randomizing them into a trial. These tissue samples could be treated in vitro with a range of compounds (eg, insulin-sensitizing drugs, specific nutrients, or exercise-mimicking agonists such as AICAR) to determine whether interactions between the compounds and the selected genotypes influence relevant phenotypes, such as glucose disposal rates and insulin sensitivity. Although there are clear limitations to this approach, it might help refine the choice of interventions for subsequent in vivo experimental studies and thus maximize the likelihood that they succeed in demonstrating interactions.
Conclusions
Many hope that genetic information will one day help improve the prevention and management of type 2 diabetes. However, unless genetic risk is substantial and can be modified through therapeutic intervention or interventions can be targeted toward those who, because of their genetic constitution, are most likely to respond well and/or lack susceptibility to their side effects, genetics will be of limited use for the primary or secondary prevention of diabetes. With this in mind, it is difficult to perceive how in the absence of appropriately designed studies that seek to discover and elucidate the nature of gene × environment (or treatment) interactions the full potential of genetics to improve human health and reduce the burden of diabetes will be realized. Nevertheless, merely discovering statistically reliable examples of GEI will not suffice, as epidemiologic observations of interactions will also need to be translated into clinical settings; the combination of genome-wide interaction analyses set within nested case-cohort studies to identify interactions and genotype-based recall clinical trials to translate those findings into the intervention setting may be the optimal combination of methods to achieve this end.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Internation Diabetes Federation: http://www.diabetesatlas.com/content/epidemiology-and-morbidity. 2011.
Fox CS, Coady S, Sorlie PD, D’Agostino Sr RB, Pencina MJ, Vasan RS, et al. Increasing cardiovascular disease burden due to diabetes mellitus: the Framingham Heart Study. Circulation. 2007;115:1544–50.
Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34.
Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403.
Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–50.
Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, Brenneman AT, et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet. 2009;374:1677–86.
Baron AL. Facet number in Drosophila melanogaster as influenced by certain genetic and environmental factors. J Exp Zool. 1935;70:461–90.
Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–56.
Nobel Foundation: http://nobelprize.org/nobel_prizes/medicine/laureates/1965/. 1965.
•• The InterAct Consortium. Design and cohort description of the InterAct Project: an examination of the interaction of genetic and lifestyle factors on the incidence of type 2 diabetes in the EPIC Study. Diabetologia. 2011;54(9):2272–82. The InterAct Study is a European-based nested case-cohort study designed for the detection of gene x lifestyle interactions in type 2 diabetes. The study is the largest and most comprehensive of its kind to date involving around 12,500 incident cases and 16,000 non-cases selected from a background population of around a third of a million people.
Prentice AM. Early influences on human energy regulation: thrifty genotypes and thrifty phenotypes. Physiol Behav. 2005;86:640–5.
• Speakman JR. Thrifty genes for obesity, an attractive but flawed idea, and an alternative perspective: the ‘drifty gene’ hypothesis. Int J Obes. 2008;32:1611–7. This is an opinion piece arguing against the selection of so-called thrifty genes in the human gene pool.
Bouchard C, Rankinen T. Individual differences in response to regular physical activity. Med Sci Sports Exerc. 2001;33:S446–51. discussion S452-443.
Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D’Agostino Sr RB. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med. 2007;167:1068–74.
Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–32.
Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–19.
de Miguel-Yanes JM, Shrader P, Pencina MJ, Fox CS, Manning AK, Grant RW, et al. Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care. 2011;34:121–5.
Lu Q, Elston RC. Using the optimal receiver operating characteristic curve to design a predictive genetic test, exemplified with type 2 diabetes. Am J Hum Genet. 2008;82:641–51.
Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature. 2008;454:463–9.
Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801.
Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobelet C, et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes. 2003;52:2874–81.
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–71.
Franks PW, Loos RJ. PGC-1alpha gene and physical activity in type 2 diabetes mellitus. Exerc Sport Sci Rev. 2006;34:171–5.
Barroso I, Luan J, Sandhu MS, Franks PW, Crowley V, Schafer AJ, et al. Meta-analysis of the Gly482Ser variant in PPARGC1A in type 2 diabetes and related phenotypes. Diabetologia. 2006;49:501–5.
• Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89. This is a recent report from the DIAGRAM+ consortium summarizing the findings from meta-analyses of GWAS data. The report outlines association signals for more than 30 replicated loci.
Franks PW, Barroso I, Luan J, Ekelund U, Crowley VEF, Sandhu MS, et al. Wareham NJ PGC-1a genotype modifies the association of volitional energy expenditure with VO2max. Med Sci Sports Exerc. 2003;35(12):1998–2004.
Ridderstråle M, Johansson LE, Rastam L, Lindblad U. Increased risk of obesity associated with the variant allele of the PPARGC1A Gly482Ser polymorphism in physically inactive elderly men. Diabetologia. 2006;49(3):496–500.
Franks PW, Ekelund U, Brage S, Luan J, Schafer AJ, O’Rahilly S, et al. Wareham NJ PPARGC1A coding variation may initiate impaired NEFA clearance during glucose challenge. Diabetologia. 2007;50(3):569–73.
Nelson TL, Fingerlin TE, Moss L, Barmada MM, Ferrell RE, Norris JM. The peroxisome proliferator-activated receptor gamma coactivator-1 alpha gene (PGC-1alpha) is not associated with type 2 diabetes mellitus or body mass index among Hispanic and non Hispanic Whites from Colorado. Exp Clin Endocrinol Diabetes. 2007;115(4):268–75.
Stefan N, Thamer C, Staiger H, Machicao F, Machann J, Schick F, et al. Genetic variations in PPARD and PPARGC1A determine mitochondrial function and change in aerobic physical fitness and insulin sensitivity during lifestyle intervention. J Clin Endocrinol Metab. 2007;92(5):1827–33.
Brito EC, Vimaleswaran VK, Brage S, Andersen LB, Sardinha LB, Wareham NJ, et al. PPARGC1A sequence variation and cardiovascular risk-factor levels: a study of main genetic effects and gene x environment interactions in children from the European Youth Heart Study. Diabetologia. 2009;52(4):609–13.
Bray MS, Hagberg JM, Perusse L, Rankinen T, Roth SM, Wolfarth B, et al. The human gene map for performance and health-related fitness phenotypes: the 2006–2007 update. Med Sci Sports Exerc. 2009;41:35–73.
Kooperberg C, Leblanc M. Increasing the power of identifying gene x gene interactions in genome-wide association studies. Genet Epidemiol. 2008;32:255–63.
• Brito EC, Lyssenko V, Renstrom F, Berglund G, Nilsson PM, Groop L, et al. Previously associated type 2 diabetes variants may interact with physical activity to modify the risk of impaired glucose regulation and type 2 diabetes: a study of 16,003 Swedish adults. Diabetes. 2009;58:1411–8. This is a comprehensive assessment of genetic variation at the PPARGC1A locus in relation to cardiovascular and metabolic quantitative traits and their interactions with physical activity in more than 2000 European school children.
Moore AF, Jablonski KA, McAteer JB, Saxena R, Pollin TI, Franks PW, et al. Extension of type 2 diabetes genome-wide association scan results in the diabetes prevention program. Diabetes. 2008;57:2503–10.
• Ruchat SM, Rankinen T, Weisnagel SJ, Rice T, Rao DC, Bergman RN, et al. Improvements in glucose homeostasis in response to regular exercise are influenced by the PPARG Pro12Ala variant: results from the HERITAGE Family Study. Diabetologia. 2010;53:679–89. This is an analysis of eight variants in seven type 2 diabetes genes in relation to a range of quantitative traits derived from an intravenous glucose tolerance test before and after 20 weeks of aerobic conditioning exercise.
Luan J, Browne PO, Harding AH, Halsall DJ, O’Rahilly S, Chatterjee VK, et al. Evidence for gene-nutrient interaction at the PPARgamma locus. Diabetes. 2001;50:686–9.
Memisoglu A, Hu FB, Hankinson SE, Manson JE, De Vivo I, Willett WC, et al. Interaction between a peroxisome proliferator-activated receptor gamma gene polymorphism and dietary fat intake in relation to body mass. Hum Mol Genet. 2003;12:2923–9.
Franks PW, Mesa JL, Harding AH, Wareham NJ. Gene-lifestyle interaction on risk of type 2 diabetes. Nutr Metab Cardiovasc Dis. 2007;17:104–24.
•• Palla L, Higgins JP, Wareham NJ, Sharp SJ. Challenges in the use of literature-based meta-analysis to examine gene-environment interactions. Am J Epidemiol. 2010;171:1225–32. This is a paper that discusses the practical and methodologic limitations of conducting literature-based meta-analyses of GEI studies.
Nelson TL, Fingerlin TE, Moss LK, Barmada MM, Ferrell RE, Norris JM. Association of the peroxisome proliferator-activated receptor gamma gene with type 2 diabetes mellitus varies by physical activity among non-Hispanic whites from Colorado. Metabolism: clinical and experimental. 2007;56:388–93.
Florez JC, Jablonski KA, Sun MW, Bayley N, Kahn SE, Shamoon H, et al. Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab. 2007;92:1502–9.
Lindi VI, Uusitupa MI, Lindstrom J, et al. Association of the Pro12Ala polymorphism in the PPAR-gamma2 gene with 3-year incidence of type 2 diabetes and body weight change in the Finnish Diabetes Prevention Study. Diabetes. 2002;51:2581–6.
Kilpelainen TO, Lakka TA, Laaksonen DE, et al. SNPs in PPARG associate with type 2 diabetes and interact with physical activity. Med Sci Sports Exerc. 2008;40:25–33.
Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94.
Andreasen CH, Stender-Petersen KL, Mogensen MS, et al. Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes. 2008;57:95–101.
Rampersaud E, Mitchell BD, Pollin TI, et al. Physical activity and the association of common FTO gene variants with body mass index and obesity. Arch Intern Med. 2008;168:1791–7.
Franks PW, Jablonski KA, Delahanty LM, McAteer JB, Kahn SE, Knowler WC, et al. Assessing gene-treatment interactions at the FTO and INSIG2 loci on obesity-related traits in the Diabetes Prevention Program. Diabetologia. 2008;51:2214–23.
•• Nettleton JA, McKeown NM, Kanoni S, et al. Interactions of dietary whole-grain intake with fasting glucose- and insulin-related genetic loci in individuals of European descent: a meta-analysis of 14 cohort studies. Diabetes Care. 2010;33:2684–91. This is a large-scale meta-analysis of data on gene x nutrient (dietary whole grains) interactions in relation to fasting insulin and glucose concentrations. The study identified a variant at the GCKR locus that showed tentative evidence of gene x nutrient interactions.
•• Kanoni S, Nettleton JA, Hivert MF, et al. Total zinc intake may modify the glucose-raising effect of a zinc transporter (SLC30A8) variant: a 14-cohort meta-analysis. diabetes 2011 (epublished Aug 1). This is a large-scale meta-analysis of data on gene x zinc (dietary and supplemental) interactions in relation to fasting insulin and glucose concentrations. The study identified a variant at the SLC30A8 locus, a gene encoding a zinc transporter, that showed tentative evidence of gene x nutrient interactions.
Rutter GA. Think zinc: new roles for zinc in the control of insulin secretion. Islets. 2010;2:49–50.
• Franks PW, Brito EC. Interaction between exercise and genetics in type 2 diabetes mellitus: an epidemiological perspective. In: Roth SM, Pescatello L, editors. Exercise genomics: molecular & translational medicine. Springer Science: Clifton; 2011. This is a systematic review of literature on gene x exercise interactions related to type 2 diabetes.
•• Franks PW, Nettleton JA. Invited commentary: gene X lifestyle interactions and complex disease traits—inferring cause and effect from observational data, sine qua non. Am J Epidemiol. 2010;172:992–7; discussion 998–999. This is an editorial commentary discussing causal inference in epidemiologic studies of gene x lifestyle interactions.
• Moore JH, Asselbergs FW, Williams SM. Bioinformatics challenges for genome-wide association studies. Bioinformatics. 2010;26:445–55. This is an overview of the limitations of GWAS, including discussion of why such studies are poorly suited to the discovery of GEIs.
•• Pare G, Cook NR, Ridker PM, Chasman DI. On the use of variance per genotype as a tool to identify quantitative trait interaction effects: a report from the Women’s Genome Health Study. PLoS Genet. 2010;6:e1000981. This is a paper that describes and tests an agnostic method for the detection of GEIs. The authors identified several novel interactions using this method in the Women’s Genome Health Study.
Ridker PM, Chasman DI, Zee RY, Parker A, Rose L, Cook NR, et al. Rationale, design, and methodology of the Women’s Genome Health Study: a genome-wide association study of more than 25,000 initially healthy american women. Clin Chem. 2008;54:249–55.
Boks MP, Schipper M, Schubart CD, Sommer IE, Kahn RS, Ophoff RA. Investigating gene environment interaction in complex diseases: increasing power by selective sampling for environmental exposure. Int J Epidemiol. 2007;36:1363–9.
Wong MY, Day NE, Luan JA, Wareham NJ. Estimation of magnitude in gene-environment interactions in the presence of measurement error. Stat Med. 2004;23:987–98.
•• Manning AK, LaValley M, Liu CT, Rice K, An P, Liu Y, et al. Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP x environment regression coefficients. Genet Epidemiol. 2011;35:11–8. This is a paper describing and applying an adaptation of the 2 d.f. method for jointly studying interactions (between a genetic and environmental risk factor) and genetic main effects. The method, known as JMA, along with others were tested using data from multiple cohorts in which PPARG had been tagged and these variants tested for interactions with BMI on fasting insulin levels.
• Chatterjee N, Wacholder S. Invited commentary: efficient testing of gene-environment interaction. Am J Epidemiol. 2009;169:231–3; discussion 234–235. This is a paper describing methods upon which Manning et al. [59••] builds.
Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Hum Hered. 2007;63:111–9.
Becker BJ, Wu MJ. The synthesis of regression slopes in meta-analysis. Stat Sci. 2007;22:414–29.
Wareham NJ, Franks PW, Harding AH. Establishing the role of gene-environment interactions in the etiology of type 2 diabetes. Endocrinol Metab Clin North Am. 2002;31:553–66.
Franks PW. The interaction of genetic factors and physical activity energy expenditure in the aetiology of the metabolic syndrome. In: Department of community medicine. Cambridge, Cambridge University; 2003, p. 490.
Renström F, Payne F, Nordström A, Brito EC, Rolandsson O, Hallmans G, et al. the GIANT consortium. Replication and extension of genome-wide association study results for obesity in 4,923 adults from Northern Sweden. Hum Mol Genet. 2009;18(8):1489–96.
Acknowledgments
The author’s research is supported by grants from the Swedish Diabetes Association, Novo Nordisk, the Swedish Heart-Lung Foundation, the Swedish Research Council, the European Union, and the National Institutes of Health. The author thanks Dr. F. Renström for her careful critique of this manuscript prior to publication.
Disclosure
Conflicts of interest: P.W. Franks: has served on the boards for INSERM, LifeGene SAB, and the German National Cohort; has received honoraria from Novo Nordisk; and has received payment for multiple book chapters and review articles.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Franks, P.W. Gene × Environment Interactions in Type 2 Diabetes. Curr Diab Rep 11, 552–561 (2011). https://doi.org/10.1007/s11892-011-0224-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11892-011-0224-9