
Abstract
Obesity and type 2 diabetes mellitus (T2DM) are major drivers of cardiovascular disease (CVD). The link between environmental factors, obesity, and dysglycemia indicates that progression to diabetes with time occurs along a “continuum”, not necessarily linear, which involves different cellular mechanisms including alterations of insulin signaling, changes in glucose transport, pancreatic beta cell dysfunction, as well as the deregulation of key genes involved in oxidative stress and inflammation. The present review critically addresses key pathophysiological aspects including (i) hyperglycemia and insulin resistance as predictors of CV outcome, (ii) molecular mechanisms underpinning the progression of diabetic vascular complications despite intensive glycemic control, and (iii) stratification of CV risk, with particular emphasis on emerging biomarkers. Taken together, these important aspects may contribute to the development of promising diagnostic approaches as well as mechanism-based therapeutic strategies to reduce CVD burden in obese and diabetic subjects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Worldwide, at least 2.8 million people die each year due to complications of being overweight or obese [1]. Increased body weight leads to adverse metabolic effects on blood pressure, cholesterol, triglycerides, and insulin sensitivity [2]. Risk of coronary heart disease, ischemic stroke, and type 2 diabetes mellitus (T2DM) rises steadily with increasing waist circumference, an important hallmark of impaired glucose tolerance [3]. The worldwide prevalence of obesity has nearly doubled between 1980 and 2008. In 2008, 10 % of men and 14 % of women were obese, compared with 5 % of men and 8 % of women in 1980 [3]. Obesity and insulin resistance (IR) strongly predispose an individual to T2DM with a progressive increase of fasting glucose levels. IR is a major feature of T2DM and develops in multiple organs, including skeletal muscle, liver, adipose tissue, and the heart. The onset of hyperglycemia and diabetes is often preceded by many years of IR. Obesity plays a pivotal role in this phenomenon, providing an important link between fat accumulation and T2DM [4].
Obesity and T2DM Across the Cardiovascular Continuum
The link between environmental factors (high caloric intake, sedentary lifestyle), obesity, and subsequent dysglycemia indicates that the progression to diabetes with time occurs along a “continuum”, not necessarily linear, which involves different cellular mechanisms including alterations of insulin signaling, changes in glucose transport, pancreatic beta cell dysfunction as well as deregulation of key genes involved in oxidative stress and inflammation [5]. The progression from prediabetes to T2DM may take many years to occur, leading to different intermediate disease phenotypes with continuous changes in glucose parameters and shifts in glucose tolerance category. Although obesity is an established risk factor for T2DM, a large proportion of obese individuals do not develop diabetes [2]. Recent studies have identified connections between obesity and T2DM involving proinflammatory cytokines, insulin-related pathways, and lipid metabolism, as well as an array of cellular processes including mitochondrial dysfunction, epigenetic modifications, and endoplasmic reticulum stress [5–7]. A better understanding of these interactions may lead to the development of mechanism-based therapeutic approaches for the prevention of T2DM.
Among different specialists dealing with the diabetic disease, cardiologists are undoubtedly in a first-line position [8, 9]. Indeed, diabetes has a strong impact on atherosclerotic vascular disease [10–12]. This phenomenon is best documented in terms of its association with coronary heart disease and cardiovascular events [13]. Several studies have clearly shown that patients with diabetes are several-fold more prone to develop myocardial infarction than matched subjects without diabetes [10]. A seminal Finnish study demonstrated that diabetes increases the 7-year risk of myocardial infarction and death in older subjects [10]. The concept of diabetes as a coronary heart disease risk-equivalent emerged from this study and culminated in its coronation as a high-risk cardiovascular state requiring secondary prevention-level care. This concept has been further strengthened in the recent 2013 guidelines of the European Society of Cardiology (ESC) on the management of diabetes and CVD [14, 15••]. Notably, the risk of macrovascular complications increases with the severity of blood glucose impairment. Data from the prospective Whitehall study revealed that the risk for CVD was almost doubled in subjects with impaired compared with normal glucose tolerance [16]. Estimates predict that 40–50 % of individuals with prediabetes will develop T2DM within 10 years, highlighting the importance of early detection of abnormal glucose metabolism to prevent the progression of prediabetes to T2DM and, hence, delay the occurrence of macrovascular and microvascular complications. Although impaired glucose tolerance and diabetes are considered very high-risk conditions, we can still appreciate differences in CV outcome between these two groups. Indeed, follow-up of the Euro Heart Survey showed that 1-year survival is significantly higher in prediabetic as compared with diabetic individuals [17]. However, survival curves tend to overlap in the long term, thus strengthening the concept that all stages of glucose abnormalities are associated with increased risk of CV morbidity and mortality [18, 19].
Different diabetes-related conditions contribute to enhancing cardiovascular risk. Among them, IR and hyperglycemia are major drivers of atherothrombotic events leading to poor cardiovascular outcome [20]. Recent meta-analyses have shown that elevated insulin and glucose concentrations are associated with an increased CVD risk, regardless of diabetes [21–23]. A pooled analysis of 65 trials examined the impact of the validated and frequently used marker Homeostasis Model Assessment IR (HOMA-IR) on cardiovascular outcomes including coronary heart disease, stroke, or combined CVD [24]. Interestingly, the pooled relative risk of CHD was 1.52 (1.31, 1.76; 62.4 %) for glucose, 1.12 (0.92, 1.37; 41.0 %) for insulin and 1.64 (1.35, 2.00; 0 %) for HOMA-IR [24]. The high predictive value of HOMA-IR is due to the fact that such an index incorporates both glucose and insulin concentrations and is more strongly associated with CVD than glucose or insulin concentrations alone. These data suggest that hyperglycemia and IR are powerful predictors of cardiovascular events and their combination exerts a detrimental, synergic effect [25]. This concept is also outlined by the notion that patients with the combination of T2DM and visceral obesity display worse myocardial function than patients having T2DM or obesity alone [26]. Other studies have reported significant associations between HOMA-IR and post-procedural myocardial injury and clinical outcome after a percutaneous coronary intervention (PCI) with drug-eluting stents [27, 28]. A recent study showed that post-procedural troponin T and creatine kinase-myocardial band levels progressively rose across tertiles of HOMA-IR in 516 patients undergoing PCI [28]. During a median follow-up of 623 days, patients with the highest tertiles of HOMA-IR had the highest risk of cardiovascular events. The Cox proportional hazard models identified HOMA-IR as independently associated with worse clinical outcome after adjustment for clinical and procedural factors [HR 1.98 (CI 95 % 1.510–2.608)] [28]. These data clearly suggest that preventing features of T2DM may strongly reduce the burden of cardiovascular disease (CVD) [29]. The prevalence of impaired glucose tolerance is extremely high among patients admitted for an acute coronary syndrome. International surveys have demonstrated that dysglycemia is more common than normoglycemia in CVD patients admitted to the hospital, and the oral glucose tolerance test (OGTT) is able to detect glucometabolic alterations in 55–60 % of patients with overt CVD [30]. In this regard, the recent European guidelines strength the concept that early detection of glucose perturbations by OGTT in patients with coronary artery disease (CAD) offers an opportunity to prevent the development of DM by means of lifestyle programs and/or pharmacological treatments [15••].
Mechanisms of Atherosclerotic Vascular Disease in Patients with Obesity and T2DM
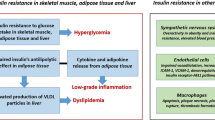
In the diabetic vasculature, hyperglycemia and IR trigger an array of signaling pathways and gene-activating events favoring the atherosclerotic process [25] (Fig. 1). Although a large number of studies have characterized the mechanisms of diabetic vascular disease, the individual contributions of hyperglycemia and IR remain largely unknown. Indeed, factors increasing CV risk tend to cluster together in the diabetic patient. IR is believed to be a pathophysiological disturbance that underlies many of the risk factors, but it is not clear whether IR is a CV risk factor per se [4]. Likewise, it is hard to appreciate the detrimental effects of chronic hyperglycemia across the spectrum of many other cardiovascular risk factors concurring in the diabetic patient. Indeed, intensive treatment of hyperglycemia failed to improve cardiovascular outcome [31], whereas a systematic, multifactorial treatment significantly reduced cardiovascular mortality [32]. Accordingly, the ORIGIN trial failed to show that early implementation of insulin-based regimens reduces macrovascular complications [33]. In line with these findings, the new guidelines do not recommend very tight glucose control if the goal is to reduce macrovascular complications [15••]. Taken together, these disappointing results have recently contributed to the emerging skepticism of clinicians toward the importance of hyperglycemia as a CV risk factor. A possible interpretation is that glucose levels may represent a marker instead of a predictor of CVD. This might contribute to an explanation of why the normalization of glycemia does not reduce CVD burden. However, the scenario is much more complex, since an array of experimental and clinical studies clearly shows that glucose levels and impaired insulin signaling are potent drivers of the atherosclerotic process, even in the absence of concomitant risk factors such as hypertension, obesity, and dyslipidemia [12]. Hence, the major challenge to curing diabetes is to unravel the intricate networks linking different risk factors with atherosclerotic disease and, hence, to develop mechanism-based therapeutic approaches in this setting.

Schematic representing the detrimental effects of endothelial insulin resistance and hyperglycemia. Inhibitory IRS-1 phosphorylation by protein kinase C impairs downstream targets PI3K and Akt leading to eNOS dysfunction and reduced synthesis of NO. This chain of events blunts NO-mediated capillary recruitment and impairs insulin delivery in hormone-sensitive organs leading to systemic insulin resistance. On the other hand, hyperglycemia causes PKC-dependent activation of NADPH oxidase and mitochondrial adaptor p66Shc, leading to ROS generation, NF-kB activation, and the upregulation of inflammatory molecules. Transient hyperglycemic spikes as well as ROS trigger epigenetic changes, which are responsible for persistent vascular dysfunction despite the restoration of normoglycemia. Such an oxidative and inflammatory milieu triggers important precursors of vascular damage including circulating cytokines, microRNAs, microparticles, and AGEs, which may serve as important CVD biomarkers in obese and diabetic subjects. ROS reactive oxygen species, PKC protein kinase C, IRS-1 insulin receptor substrate-1, AGEs advanced glycation end products, IL-6 interleukin 6, NO nitric oxide
The “Bad Legacy Effect” of Hyperglycemia
High glucose levels favor the imbalance between endothelial nitric oxide (NO) availability and accumulation of reactive oxygen species (ROS) [12]. The generation of ROS rapidly inactivates NO to form peroxynitrite (ONOO-), a powerful oxidant triggering protein nitrosylation and dysfunction of key enzymes implicated in endothelial homeostasis [34]. In patients with diabetes, hyperglycemia leads to the accumulation of mitochondrial ROS and subsequent activation of important biochemical pathways including advanced glycation end products, protein kinase C (PKC), nuclear factor-kB (NF-kB), polyol, and hexosamine flux [35]. A recent study showed that PKC is highly activated in endothelial cells isolated from diabetic subjects and correlates with oxidative stress, impaired insulin signaling and, most importantly, endothelial dysfunction, as assessed by flow-mediated vasodilation [36•]. In the diabetic endothelium, PKC leads to increased ROS generation via activation of the adaptor p66Shc and NADPH oxidase signaling (Fig. 1) [12, 37]. The mitochondrial adaptor p66Shc functions as a redox enzyme implicated in mitochondrial ROS generation and translation of oxidative signals into apoptosis [38–41]. We have reported that diabetic p66 Shc-/-mice are protected against hyperglycemia-induced endothelial dysfunction and oxidative stress [42]. The relevance of p66Shc in the clinical setting of diabetes is supported by the notion that p66Shc gene expression is increased in peripheral blood mononuclear cells obtained from patients with T2DM and correlates with oxidative stress [43]. We have recently demonstrated that hyperglycemia-induced p66Shc upregulation is not reverted by intensive glycemic control in diabetic mice, thus contributing to persistent oxidative stress and vascular dysfunction [44•]. Interestingly enough, in-vivo silencing of p66Shc, performed at the time of normoglycemia restoration, suppressed persistent endothelial dysfunction, suggesting that p66Shc is an important source of free radicals involved in the “bad legacy effect” of hyperglycemia [44•]. This latter phenomenon, also known as hyperglycemic memory, might represent an important determinant of residual vascular risk in diabetes and is becoming the focus of many ongoing investigations [45, 46]. Understanding the mechanisms underpinning hyperglycemic memory may help to unravel why intensive glycemic control does not exert any beneficial effect on macrovascular complications in patients with T2DM. In this context, we have also reported that alterations of chromatin, known as epigenetic changes, are responsible for persistent p66Shc overexpression during subsequent normoglycemia [44•] (Fig. 1). Epigenetic alterations, namely methylation and acetylation of DNA/histone complexes, are emerging as important modulators of gene expression in diabetic vascular disease [47–50].
Glucose Fluctuations
An important breakthrough in the etiologic pathway linking hyperglycemia and vascular damage is the demonstration that glucose fluctuations rather than constant high glucose are able to maintain the activation of molecular machineries involved in oxidative stress and inflammation and, hence, to trigger atherosclerotic disease [48, 51]. A recent study demonstrated that transient hyperglycemic spikes activate epigenetic changes responsible for long-lasting activation of the transcription factor NF-kB and subsequent upregulation of inflammatory adhesion molecules [52••]. The clinical relevance of these findings is supported by the notion that, although HbA1c is reduced to target levels, blood glucose concentrations in patients with diabetes always fluctuate from hyperglycemic peaks to glucose nadirs [53]. Moreover, current evidence suggest that HbA1c explains <25 % of the variation in the risk of developing diabetic complications. Indeed, HbA1c does not correlate with glycemic variability when adjusted for mean blood glucose [54]. Collectively, these data suggest that targeting transient spikes of hyperglycemia in addition to HbA1c may suppress detrimental processes responsible for the progression of vascular complications in T2DM (Fig. 1).
Endothelial Insulin Resistance
The onset of hyperglycemia and diabetes is often preceded by many years of IR [12]. The impact of IR as an individual CV risk factor in patients with diabetes has emerged only recently [2, 55, 56]. Indeed, over the last decade IR has been regarded as the consequence of visceral obesity, without any active role in the etiology of diabetic cardiovascular complications [57]. After many years of seminal research in this area we may conclude that it is rather naïve to regard IR as the epiphenomenon of obesity and metabolic syndrome. Against this, several experimental studies have shown that the loss of insulin signaling in the endothelium leads to vascular dysfunction, expression of adhesion molecules, and atherosclerotic lesions in mice [58••, 59–62]. Although IR has been attributed to adipocyte-derived inflammation, recent evidence is overturning the “adipocentric paradigm” [57]. Indeed in obesity, inflammation and macrophage activation seem to primarily occur in non-adipose tissue. This concept is supported by the notion that suppression of inflammation in the vasculature prevents IR in other organs and prolongs lifespan [58••]. Transgenic mice with endothelium-specific overexpression of the inhibitory NF-kB subunit IkBα were protected against the development of IR. In these mice, obesity-induced macrophage infiltration of adipose tissue and plasma oxidative stress markers were reduced, whereas blood flow, muscle mitochondrial content, and locomotor activity were increased, confirming the pivotal role of the transcription factor NF-kB in oxidative stress, vascular dysfunction, and inflammation [58••]. Another study confirmed these findings, showing that genetic disruption of the insulin receptor substrate 2 (IRS-2) in endothelial cells reduces glucose uptake by skeletal muscle [61]. These novel findings strengthen the central role of endothelium in obesity-induced IR, suggesting that a blockade of vascular inflammation and oxidative stress may be a promising approach to prevent metabolic disorders. Consistently, pharmacological improvement in insulin sensitivity in patients with T2DM and metabolic syndrome is associated with the restoration of flow-mediated vasodilation [63, 64]. In other words, the maintenance of endothelial homeostasis warrants physiological nitric oxide release with subsequent capillary recruitment and appropriate insulin delivery within hormone-sensitive organs [65].
Stratification of Cardiovascular Risk
In Europe alone, about 63 million people are affected by prediabetes and 53 million by diabetes, and these numbers will grow exponentially over the coming decades [15••]. The most powerful strategy for reducing cardiovascular mortality is represented by early diagnosis and, hence, treatment of vascular complications. At present, we are still lacking cost-effective markers able to identify atherosclerotic disease at an early stage. The issue of risk stratification deserves attention because not every obese/diabetic subject carries the same degree of inflammation and oxidative stress. The diversity of metabolic phenotypes with different outcomes underscores the need for cardiovascular risk stratification within such a heterogeneous population. Despite diabetes being associated with a significant atherosclerotic burden, the role of vascular imaging in this setting remains poorly defined [9]. Coronary artery calcium imaging has been found to be superior to established risk factors for predicting silent myocardial ischemia and short-term outcome in a small cohort of high-risk DM subjects [66]. This is a rather expensive tool that may not be sustainable in developing countries. Moreover, the benefits of myocardial revascularization in asymptomatic patients remain to be determined [14, 67]. The issue with the emerging biomarkers is that they hardly perform beyond traditional cardiovascular risk scores. The Atherosclerotic Risk in Communities (ARIC) study prospectively evaluated whether adding C-reactive protein or 18 other novel risk factors individually to a basic risk model would improve prediction of incident CAD in middle-aged men and women [68]. Unfortunately, none of these risk markers predicted CVD, regardless of the risk score. Besides these disappointing results, current European guidelines confirm that albuminuria remains the most powerful predictor of incident CV events and heart failure in T2DM patients and recommend the estimation of urinary albumin excretion rates when performing risk stratification in DM subjects (class I, level B) [15••].
Novel Biomarkers
Circulating molecules, such as proinflammatory and anti-inflammatory cytokines, are being considered as potential cardiovascular biomarkers in diabetes [69]. A case-control study, within the prospective, population-based EPIC (European Prospective Investigation into Cancer and Nutrition) study, has demonstrated that a combined elevation of IL-1β and IL-6 was independently associated with an increased risk of T2DM, suggesting the importance of low-grade inflammation in the pathogenesis of diabetes [70]. A cross-sectional analysis performed in patients with and without type 1 diabetes (T1DM) showed that IL-6 and fibrinogen levels were significantly elevated in T1DM patients, regardless of adiposity and glycemic control [71]. Another study showed that IL-6 is significantly increased in diabetics undergoing PCI with a peri-interventional hyperglycemic state, and inversely correlates with responsiveness to clopidogrel and aspirin [72]. Increased oxidative stress in the vasculature is a major contributor of endothelial dysfunction in DM via the generation of superoxide and subsequent impairment of NO bioavailability [12]. In this regard, a previous work examined glomerular and cortical eNOS expression in renal biopsies of patients with diabetic nephropathy and noticed a strong correlation between eNOS activity and degree of proteinuria, which is indicative of glomerular endothelial dysfunction [73]. Moreover, circulating markers of oxidative stress, including F2 isoprostanes and antibodies against oxLDL, are increased in humans with diabetes, obesity, and IR [74]. The analysis from the community-based Framingham Offspring Study found an increased prevalence of IR across 8-epi-prostaglandin F2α tertiles [74]. Thus, systemic oxidative stress seems to be related to IR in prediabetic subjects. Advanced glycation end products (AGEs) have been linked to the atherosclerotic process and are emerging as a novel signature of atherosclerotic disease [75]. Measuring AGEs in the skin using auto-fluorescence may provide important information on risk stratification in diabetic patients. In a study involving 972 diabetic patients, the addition of skin AGEs to the UKPDS risk engine resulted in the re-classification of 27 % of the patients from the low- to the high-risk group [76]. Indeed, the 10-year cardiovascular event rate was higher in patients with a UKPDS score >10 % when skin AGEs were above the median (56 vs. 39 %). Novel markers in diabetes certainly include microRNAs (miRs), a newly identified class of small, non-coding RNAs that are emerging as key players in the pathogenesis of hyperglycemia-induced vascular damage [77, 78]. These small non-coding RNAs orchestrate different aspects of diabetic vascular disease by regulating gene expression at the post-transcriptional level. Microarray profiling has shown an altered profile of miRs expression in subjects with T2DM [79•]. In this study, diabetic patients displayed a significant deregulation of miRs involved in angiogenesis, vascular repair, and endothelial homeostasis. Among other miRs, miR-126, an important pro-angiogenic effector [80], was significantly downregulated in plasma samples of 822 patients from the Brunick cohort [79•]. Closely related to the miRs are the microparticles (MPs). The latter are shed membrane particles of <1 mm in diameter, thought to be budded into the circulation from endothelial cells (EMPs) and various blood cells, including platelets, leukocytes, and erythrocytes [81]. A recent study showed that MP characteristics are associated with the type of vascular complication involved in DM and might serve as a biomarker for the pro-coagulant state and vascular pathology in patients with DM [82]. Moreover, plasma EMPs have been associated with the presence of hypertension and arterial stiffness in diabetic patients [83], whereas another study suggested that EMPs could be used as surrogate markers of unstable plaques and might help to improve cardiovascular prediction in DM patients [84]. Taken together, these new findings indicate that microRNAs and microparticles may represent a novel diagnostic opportunity for the early identification of obese and diabetic subjects at high cardiovascular risk.
Conclusions
In the present review, we have delineated the major mechanisms as well as the connections linking obesity and T2DM. These interactions are complex and the relative importance of IR and hyperglycemia remain undefined as to the stratification of cardiovascular risk in these heterogeneous populations. Further genetic and epigenetic studies may help to elucidate additional common pathophysiological pathways for obesity and diabetes and identify new promising treatment targets to reduce CVD in this setting.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world–a growing challenge. N Engl J Med. 2007;356:213–5.
Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, et al. Obesity and type 2 diabetes: what can be unified and what needs to be individualized? J Clin Endocrinol Metab. 2011;96:1654–63.
Despres JP. Body fat distribution and risk of cardiovascular disease: an update. Circulation. 2012;126:1301–13.
Eckel RH. The complex metabolic mechanisms relating obesity to hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2011;31:1946–8.
Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–6.
Arsenault BJ, Beaumont EP, Despres JP, Larose E. Mapping body fat distribution: a key step towards the identification of the vulnerable patient? Ann Med. 2012;44:758–72.
Despres JP. Intra-abdominal obesity: an untreated risk factor for Type 2 diabetes and cardiovascular disease. J Endocrinol Investig. 2006;29:77–82.
Ryden L, Mellbin L. Joint ESC/EASD guidelines on diabetes, where are we now and where should we go? Curr Vasc Pharmacol. 2012;10:690–2.
Paneni F. 2013 ESC/EASD guidelines on the management of diabetes and cardiovascular disease: established knowledge and evidence gaps. Diabetes Vasc Dis Res. 2014;11:5–10.
Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34.
Gu K, Cowie CC, Harris MI. Diabetes and decline in heart disease mortality in US adults. JAMA. 1999;281:1291–7.
Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013;34:2436–43.
Beckman JA, Paneni F, Cosentino F, Creager MA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Eur Heart J. 2013;34:2444–52.
Anselmino M, Ryden L. Strategies to enhance cardiovascular disease prevention in patients with diabetes. Curr Opin Cardiol. 2009;24:461–7.
Authors/Task Force M, Ryden L, Grant PJ, Anker SD, Berne C, Cosentino F, et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: the Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur Heart J. 2013;34:3035–87. The recent ESC/EASD Guidelines on the management of diabetes and CVD represent an important document providing a systematic approach to diagnose and treat the combination of DM and CVD. The evidence-based strategy promoted by the ESC/EASD Guidelines will be invaluable for a consistent improvement of CV outcome in DM subjects, thus strengthening the importance of appropriate diagnostic and therapeutic algorithms to achieve the best care for patients in an individualized setting.
Fuller JH, Shipley MJ, Rose G, Jarrett RJ, Keen H. Coronary-heart-disease risk and impaired glucose tolerance. The Whitehall study. Lancet. 1980;1:1373–6.
Lenzen M, Ryden L, Ohrvik J, Bartnik M, Malmberg K, Scholte Op Reimer W, et al. Diabetes known or newly detected, but not impaired glucose regulation, has a negative influence on 1-year outcome in patients with coronary artery disease: a report from the Euro Heart Survey on diabetes and the heart. Eur Heart J. 2006;27:2969–74.
Tominaga M, Eguchi H, Manaka H, Igarashi K, Kato T, Sekikawa A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The Funagata Diabetes Study. Diabetes Care. 1999;22:920–4.
The DECODE study group. European Diabetes Epidemiology Group. Diabetes Epidemiology: Collaborative Analysis of Diagnostic Criteria in Europe. Glucose tolerance and mortality: comparison of WHO and American Diabetes Association diagnostic criteria. Lancet. 1999;354:617–21.
Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
Emerging Risk Factors C, Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–22.
Faerch K, Vaag A, Holst JJ, Hansen T, Jorgensen T, Borch-Johnsen K. Natural history of insulin sensitivity and insulin secretion in the progression from normal glucose tolerance to impaired fasting glycemia and impaired glucose tolerance: the Inter99 study. Diabetes Care. 2009;32:439–44.
Kim SH, Reaven GM. Isolated impaired fasting glucose and peripheral insulin sensitivity: not a simple relationship. Diabetes Care. 2008;31:347–52.
Gast KB, Tjeerdema N, Stijnen T, Smit JW, Dekkers OM. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: meta-analysis. PLoS ONE. 2012;7:e52036.
Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–85.
Paneni F, Gregori M, Tocci G, Palano F, Ciavarella GM, Pignatelli G, et al. Do diabetes, metabolic syndrome or their association equally affect biventricular function? A tissue Doppler study. Hypertens Res. 2013;36:36–42.
Kumar R, Lee TT, Jeremias A, Ruisi CP, Sylvia B, Magallon J, et al. Comparison of outcomes using sirolimus-eluting stenting in diabetic versus nondiabetic patients with comparison of insulin versus non-insulin therapy in the diabetic patients. Am J Cardiol. 2007;100:1187–91.
Uetani T, Amano T, Harada K, Kitagawa K, Kunimura A, Shimbo Y, et al. Impact of insulin resistance on post-procedural myocardial injury and clinical outcomes in patients who underwent elective coronary interventions with drug-eluting stents. JACC Cardiovasc Interv. 2012;5:1159–67.
Kim SH, Reaven GM. Insulin resistance and hyperinsulinemia: you can't have one without the other. Diabetes Care. 2008;31:1433–8.
Bartnik M, Ryden L, Ferrari R, Malmberg K, Pyorala K, Simoons M, et al. The prevalence of abnormal glucose regulation in patients with coronary artery disease across Europe. The Euro Heart Survey on diabetes and the heart. Eur Heart J. 2004;25:1880–90.
Control G, Turnbull FM, Abraira C, Anderson RJ, Byington RP, Chalmers JP, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. 2009;52:2288–98.
Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580–91.
Gerstein HC, Bosch J, Dagenais GR, Diaz R, Jung H, Maggioni AP, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. 2012;367:319–28.
Cosentino F, Luscher TF. Tetrahydrobiopterin and endothelial nitric oxide synthase activity. Cardiovasc Res. 1999;43:274–8.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70.
Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, et al. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. This study, performed in primary human endothelial cells isolated from T2DM patients, is the first to provide clear evidence concerning the activation of PKC-related pathways in the diabetic endothelium. These findings have important implications for mechanism-based therapeutic approaches to prevent vascular disease burden in diabetic patients.
Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–31.
Cosentino F, Francia P, Camici GG, Pelicci PG, Luscher TF, Volpe M. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol. 2008;28:622–8.
Paneni F, Cosentino F, Marrara F, Palano F, Capretti G, Gregori M, et al. The clinical relevance of dysfunctional HDL in patients with coronary artery disease: a 3-year follow-up study. Int J Cardiol. 2012;158:158–60.
Migliaccio E, Giorgio M, Pelicci PG. Apoptosis and aging: role of p66Shc redox protein. Antioxid Redox Signal. 2006;8:600–8.
Trinei M, Migliaccio E, Bernardi P, Paolucci F, Pelicci P, Giorgio M. p66Shc, mitochondria, and the generation of reactive oxygen species. Methods Enzymol. 2013;528:99–110.
Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, et al. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci U S A. 2007;104:5217–22.
Pagnin E, Fadini G, de Toni R, Tiengo A, Calo L, Avogaro A. Diabetes induces p66shc gene expression in human peripheral blood mononuclear cells: relationship to oxidative stress. J Clin Endocrinol Metab. 2005;90:1130–6.
Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, et al. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012;111:278–89. Our recent work provides mechanistic insights for the persistent of vascular dysfunction despite optimal glycemic control with insulin. We demonstrated that epigenetic changes of p66(Shc) promoter, namely DNA hypomethylation and increased histone 3 acetylation, drive persistent oxidative stress and endothelial dysfunction during subsequent normoglycemia. These data suggest that chromatin alterations may contribute to an explanation of the residual vascular risk in diabetic patients.
Paneni F, Volpe M, Luscher TF, Cosentino F. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: bringing all the strands together. Diabetes. 2013;62:1800–7.
Ceriello A. The emerging challenge in diabetes: the “metabolic memory”. Vascu Pharmacol. 2012;57:133–8.
El-Osta A. Glycemic memory. Curr Opin Lipidol. 2012;23:24–9.
Cooper ME, El-Osta A. Epigenetics: mechanisms and implications for diabetic complications. Circ Res. 2010;107:1403–13.
Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145–56.
Paneni F, Costantino S, Volpe M, Luscher TF, Cosentino F. Epigenetic signatures and vascular risk in type 2 diabetes: a clinical perspective. Atherosclerosis. 2013;230:191–7.
Ceriello A, Esposito K, Piconi L, Ihnat MA, Thorpe JE, Testa R, et al. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes. 2008;57:1349–54.
El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–17. El-Osta et al. demonstrate for the first time that transient spikes of hyperglycemia activate persistent epigenetic signatures in the human endothelium leading to NF-kB upregulation and subsequent inflammation. This work indicates that glycemic flucutations rather than costant high glucose is a detrimental process triggering vascular damage in diabetic patients.
Picconi F, Di Flaviani A, Malandrucco I, Giordani I, Frontoni S. Impact of glycemic variability on cardiovascular outcomes beyond glycated hemoglobin. Evidence and clinical perspectives. Nutr Metab Cardiovasc Dis. 2012;22:691–6.
Bazinet M, Hamdy S, Begin L, Aprikian A, Fair W, Wright G. Monoclonal-antibody pd-41 recognizes a prostate-cancer associated antigen whose expression increases in metastases and following hormonal-therapy. Int J Oncol. 1995;7:1421–5.
Bigornia SJ, Farb MG, Tiwari S, Karki S, Hamburg NM, Vita JA, et al. Insulin status and vascular responses to weight loss in obesity. J Am Coll Cardiol. 2013;62:2297–305.
Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–904.
Kim JK. Endothelial nuclear factor kappaB in obesity and aging: is endothelial nuclear factor kappaB a master regulator of inflammation and insulin resistance? Circulation. 2012;125:1081–3.
Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, et al. Blockade of the nuclear factor-kappaB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012;125:1122–33. The article shows that the transcription factor NF-kB is critically involved in endothelial IR. Suppression of NF-kB signaling in the endothelium results in improved insulin signaling in other organs as well as improved lifespan in mice. These novel findings suggest that NF-kB is a key molecular intermediate linking metabolic disease, inflammation, and aging.
Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010;11:379–89.
Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116:1071–80.
Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307.
Li Q, Park K, Li C, Rask-Madsen C, Mima A, Qi W, et al. Induction of vascular insulin resistance and endothelin-1 expression and acceleration of atherosclerosis by the overexpression of protein kinase C-beta isoform in the endothelium. Circ Res. 2013;113:418–27.
Vitale C, Mercuro G, Cornoldi A, Fini M, Volterrani M, Rosano GM. Metformin improves endothelial function in patients with metabolic syndrome. J Intern Med. 2005;258:250–6.
Naka KK, Papathanassiou K, Bechlioulis A, Pappas K, Kazakos N, Kanioglou C, et al. Rosiglitazone improves endothelial function in patients with type 2 diabetes treated with insulin. Diabetes Vasc Dis Res. 2011;8:195–201.
Avogaro A, de Kreutzenberg SV, Federici M, Fadini GP. The endothelium abridges insulin resistance to premature aging. J Am Heart Assoc. 2013;2:e000262.
Anand DV, Lim E, Hopkins D, Corder R, Shaw LJ, Sharp P, et al. Risk stratification in uncomplicated type 2 diabetes: prospective evaluation of the combined use of coronary artery calcium imaging and selective myocardial perfusion scintigraphy. Eur Heart J. 2006;27:713–21.
Roffi M, Angiolillo DJ, Kappetein AP. Current concepts on coronary revascularization in diabetic patients. Eur Heart J. 2011;32:2748–57.
Folsom AR, Chambless LE, Ballantyne CM, Coresh J, Heiss G, Wu KK, et al. An assessment of incremental coronary risk prediction using C-reactive protein and other novel risk markers: the atherosclerosis risk in communities study. Arch Intern Med. 2006;166:1368–73.
Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, et al. Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol. 2013;62:667–76.
Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–7.
Snell-Bergeon JK, West NA, Mayer-Davis EJ, Liese AD, Marcovina SM, D'Agostino Jr RB, et al. Inflammatory markers are increased in youth with type 1 diabetes: the SEARCH Case-Control study. J Clin Endocrinol Metab. 2010;95:2868–76.
Geisler T, Mueller K, Aichele S, Bigalke B, Stellos K, Htun P, et al. Impact of inflammatory state and metabolic control on responsiveness to dual antiplatelet therapy in type 2 diabetics after PCI: prognostic relevance of residual platelet aggregability in diabetics undergoing coronary interventions. Clin Res Cardiol. 2010;99:743–52.
Hohenstein B, Hugo CP, Hausknecht B, Boehmer KP, Riess RH, Schmieder RE. Analysis of NO-synthase expression and clinical risk factors in human diabetic nephropathy. Nephrol Dial Transplant. 2008;23:1346–54.
Meigs JB, Larson MG, Fox CS, Keaney Jr JF, Vasan RS, Benjamin EJ. Association of oxidative stress, insulin resistance, and diabetes risk phenotypes: the Framingham Offspring Study. Diabetes Care. 2007;30:2529–35.
Paneni F, Cosentino F. Advanced glycation endproducts and plaque instability: a link beyond diabetes. Eur Heart J. 2013. doi:10.1093/eurheartj/eht454.
Meerwaldt R, Graaff R, Oomen PH, Links TP, Jager JJ, Alderson NL, et al. Simple non-invasive assessment of advanced glycation endproduct accumulation. Diabetologia. 2004;47:1324–30.
Shantikumar S, Caporali A, Emanueli C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res. 2012;93:583–93.
Zampetaki A, Mayr M. MicroRNAs in vascular and metabolic disease. Circ Res. 2012;110:508–22.
Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. 2010;107:810–7. This comprehensive analysis shows an array of deregulated miRs in diabetic patients, thus shedding some light on potential biomarkers in this arena. Among other miRs, miR-126, an important pro-angiogenic precursor, was significantly downregulated in plasma samples from T2DM patients.
Mocharla P, Briand S, Giannotti G, Dorries C, Jakob P, Paneni F, et al. AngiomiR-126 expression and secretion from circulating CD34(+) and CD14(+) PBMCs: role for proangiogenic effects and alterations in type 2 diabetics. Blood. 2013;121:226–36.
Shantsila E, Kamphuisen PW, Lip GY. Circulating microparticles in cardiovascular disease: implications for atherogenesis and atherothrombosis. J Thromb Haemost. 2010;8:2358–68.
Tsimerman G, Roguin A, Bachar A, Melamed E, Brenner B, Aharon A. Involvement of microparticles in diabetic vascular complications. Thromb Haemost. 2011;106:310–21.
Chen Y, Feng B, Li X, Ni Y, Luo Y. Plasma endothelial microparticles and their correlation with the presence of hypertension and arterial stiffness in patients with type 2 diabetes. J Clin Hypertens (Greenwich). 2012;14:455–60.
Bernard S, Loffroy R, Serusclat A, Boussel L, Bonnefoy E, Thevenon C, et al. Increased levels of endothelial microparticles CD144 (VE-Cadherin) positives in type 2 diabetic patients with coronary noncalcified plaques evaluated by multidetector computed tomography (MDCT). Atherosclerosis. 2009;203:429–35.
Acknowledgments
Research discussed in this manuscript was supported by the Swiss Heart Foundation and the Italian Ministry of Education, University and Research, PRIN 2010-2011 (to F.C.). F.P was the recipient of a Ph.D. program in Experimental Medicine at the University of Rome “Sapienza”.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Francesco Paneni and Sarah Costantino declare that they have no conflicts of interest.
Francesco Cosentino reports grants from the Swiss Heart Foundation, the Italian Ministry of Education, University and Research, PRIN 2010-2011, personal fees from Roche, personal fees from Bristol Myers Squibb, personal fees from MSD, personal fees from Abbott, and personal fees from Astra Zeneca.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Cardiovascular Disease and Stroke
Rights and permissions
About this article

Cite this article
Paneni, F., Costantino, S. & Cosentino, F. Insulin Resistance, Diabetes, and Cardiovascular Risk. Curr Atheroscler Rep 16, 419 (2014). https://doi.org/10.1007/s11883-014-0419-z
Published:
DOI: https://doi.org/10.1007/s11883-014-0419-z