
Opinion statement
Primary malignant central nervous (CNS) tumors are a devastating group of diseases with urgent need for improved treatment options. Surgery, radiation, and cytotoxic chemotherapy remain the primary standard treatment modalities, with molecularly targeted therapies having proven efficacy in only small subsets of cases. Poly(ADP-ribose) polymerase (PARP) inhibitors, which have had immense success in the treatment of extracranial cancers with homologous recombination deficiency (HRD), are emerging as a potential targeted treatment for various CNS tumors. Although few primary CNS tumors display canonical BRCA gene defects, preclinical evidence suggests that PARP inhibitors may benefit certain CNS tumors with functional HRD or elevated replication stress. In addition, other preclinical studies indicate that PARP inhibitors may synergize with standard therapies used for CNS tumors including radiation and alkylating agents and may prevent or overcome drug resistance. Thus far, initial clinical trials with early-generation PARP inhibitors, typically as monotherapy or in the absence of selective biomarkers, have shown limited efficacy. However, the scientific rationale remains promising, and many clinical trials are ongoing, including investigations of more CNS penetrant or more potent inhibitors and of combination therapy with immune checkpoint inhibitors. Early phase trials are also critically focusing on determining active drug CNS penetration and identifying biomarkers of therapy response. In this review, we will discuss the preclinical evidence supporting use of PARP inhibitors in primary CNS tumors and clinical trial results to date, highlighting ongoing trials and future directions in the field that may yield important findings and potentially impact the treatment of these devastating malignancies in the coming years.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Primary malignant central nervous system (CNS) tumors constitute ~1% of new cancer diagnoses, but account for a disproportionate amount of morbidity and mortality. Relative survival has improved slowly over the past several decades, but 5-year overall survival is still only ~36% averaged across all histologies, indicating a need for additional therapeutic options [1]. Poly(ADP-ribose) polymerase (PARP) inhibitors have emerged as a potential treatment for primary CNS tumors, with investigation focused primarily on adult-type diffuse gliomas and pediatric-type diffuse high-grade gliomas (HGG), as defined by the 2021 WHO classification of tumors of the CNS [2], along with emerging preclinical studies in specific subsets of medulloblastoma and ependymoma.
Among adult-type diffuse gliomas, glioblastoma (GBM) is the most common and most aggressive subtype. Current standard of care treatment consists of maximal surgical resection, followed by chemoradiation with concurrent and adjuvant temozolomide (TMZ), with potential addition of tumor-treating fields (TTF) therapy [3, 4]. Unfortunately, with median survival of just 15 months, recurrences are expected, and second-line therapies, including bevacizumab and other alkylating agents, confer minimal therapeutic benefit. Grade 2 and 3 oligodendroglioma and astrocytoma tumors are characterized by isocitrate dehydrogenase 1 or 2 (IDH1/2) mutations in most cases, and the presence or absence of 1p/19-codeletion, respectively [2, 5]. Though less aggressive than GBM, these gliomas remain uncurable, with recurrences typically occurring over 2–10 years. Standard management consists of surgery, often followed by adjuvant radiotherapy or alkylating agent chemotherapy.
Pediatric HGG are a diverse group of highly aggressive tumors, which are increasingly classified by molecular characteristics. Diffuse midline gliomas (DMG) are characterized by histone H3 lysine 27 (H3K27) alterations, which occur in about ~80% of radiographically diagnosed diffuse intrinsic pontine gliomas (DIPG) [6]. Given their location in the brainstem, these are treated locally with radiotherapy alone, but are universally fatal with median survival of less than 1 year. Pediatric-type hemispheric gliomas are also classified molecularly, and efforts are focused on developing more subtype-specific therapies ranging from targeted chemotherapy to immunotherapy to vaccine therapy, but currently surgical resection and radiation remain the standard of care [7]. Medulloblastoma, standardly treated with surgery, craniospinal irradiation, and multiagent chemotherapy, and ependymoma, managed with surgery and focal radiotherapy, have relatively better outcomes with 5-year survival rates of ~75% and ~85% [8], but still entail considerable morbidity which could be ameliorated with more targeted therapies.
PARP is intricately involved in many aspects of the DNA damage response (DDR), and PARP inhibitors have been successful at targeting non-CNS tumors harboring DDR defects including homologous recombination deficiency (HRD). In primary CNS tumors, potential roles for PARP inhibitors arise through targeting tumor genetic defects or in generating synergistic effects with other treatment modalities including radiation, chemotherapy, or immunotherapy. Here we will review the rationale and preclinical evidence for use of PARP inhibitors in CNS malignancies and discuss completed and ongoing clinical trials testing PARP inhibitors in this setting.
Rationale and preclinical evidence for PARP inhibitor use in CNS tumors
Mechanistic basis of PARP inhibition
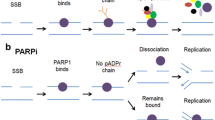
The PARP family of enzymes encompasses 17 proteins that catalyze mono- or poly-ADP-ribosylation of target proteins using NAD+ as a substrate and contribute to diverse cellular functions, including DNA damage repair, transcription, and chromatin structure modulation [9]. PARP1 is the dominant member involved in the DNA damage response, accounting for the majority of poly(ADP-ribose) (PAR) synthesis in response to genotoxic stress, with PARP2 and PARP3 playing secondary roles. PARP1 binds to DNA single-stranded breaks (SSBs), where it synthesizes long PAR chains on itself, nearby histones, and additional target proteins, leading to the recruitment, organization, and activation of proteins responsible for DNA repair. PARP1 also plays a role in base excision repair, DNA double-strand break (DSB) recognition and repair, and replication fork protection and restart [10].
PARP inhibitors are small molecule compounds that competitively bind the NAD+ binding site, blocking the catalytic activity of PARP. PARP catalytic inhibition hinders repair of DNA SSBs and base lesions, leading to collapse of replication forks during replication and generation of DSBs [11, 12]. In addition, PARP inhibitors can, to varying degrees, non-covalently “trap” PARP at damaged DNA, generating toxic PARP-DNA complexes that cause additional replication fork damage [13, 14]. PARP trapping is largely due to inhibition of its auto-PARylation activity as negatively charged PAR chains facilitate PARP release from DNA, but is influenced by differences in allosteric interactions upon PARP inhibitor binding [15,16,17].
DNA DSBs induced by either SSB repair inhibition or PARP trapping are proposed to underlie a synthetic lethal interaction between PARP inhibition and homologous recombination (HR) defects, which most commonly arise due to BRCA1 or BRCA2 mutations [11, 12, 14]. More recently, evidence has emerged that PARP plays a role in DNA replication by controlling replication fork speed and sensing unligated Okazaki fragments and that PARP inhibitors induce single-stranded DNA gaps behind the replication fork as a consequence of Okazaki fragment processing defects [18,19,20,21,22]. These ssDNA gaps may lead to toxicity in BRCA-deficient cells either directly or through the induction of DSB formation [18, 23].
Targeting CNS tumor genetic defects
PARP inhibitors have established activity in BRCA-mutant tumors, but these mutations occur infrequently in primary brain tumors [24]. A variety of other genetic alterations can cause functional HRD leading to PARP inhibitor sensitivity, also referred to as a “BRCAness” phenotype [25]. In CNS tumors, the most notable examples are IDH1/2 mutations, found in over 70% of grade II–III glioma [5]. IDH1/2 mutations generate neomorphic enzymatic activity leading to excess production of the oncometabolite 2-hydroxyglutarate (2-HG) [26]. 2-HG acts as a competitive inhibitor of the family of α-ketoglutarate-dependent dehydrogenases, which includes histone lysine demethylases and DNA demethylases, resulting in genome-wide epigenetic remodeling [27,28,29,30,31]. Inhibition of lysine demethylase KDM4B, in particular, leads to aberrant histone modifications and masking of local chromatin signaling at sites of DNA DSBs, impairing DSB repair and inducing PARP inhibitor sensitivity [32, 33••]. PARP inhibitor sensitivity is being evaluated clinically in IDH1/2-mutant glioma, as described below.
Several other recurrent CNS tumor genetic changes may confer PARP inhibitor sensitivity via defects in HR or through effects on replication, though have yet to enter the clinical arena. First, upregulation of enhancer of zeste homolog inhibitory protein (EZHIP), a protein that drives H3K27 hypomethylation by inhibiting polycomb repressive complex 2 (PRC2), characterizes group A posterior fossa ependymoma (PFA), the most common and aggressive subtype, as well as DIPG/DMG with H3K27 trimethylation loss but without H3K27M mutations [34,35,36]. In addition to mimicking the effects of H3K27M mutations, EZHIP overexpression has been shown to suppress HR repair by blocking BRCA2-PALB2 interaction, leading to PARP inhibitor hypersensitivity [37•]. Second, ATRX mutations, which frequently co-occur in IDH1/2-mutant glioma and pediatric-type diffuse hemispheric glioma, H3 G34-mutant, have been linked to increased replication stress [38]. Independently of IDH1/2 mutations, ATRX loss increases PARP inhibitor sensitivity and is a marker for synergy between PARP and ATR inhibitors [38]. Finally, amplification or upregulation of the MYCN oncogene occurs in subsets of sonic hedgehog (SHH)–mediated and group 4 medulloblastoma, portends poor prognosis, and is a driver of replication stress, which can be enhanced by PARP inhibitors, leading to mitotic catastrophe [39]. In MYCN-amplified medulloblastoma models, PARP is highly expressed in tumor compared to normal cerebellum, and PARP inhibitors in combination with low-dose CHK1 inhibitor are effective in vitro and in vivo [40]. Although some of the cancers described here have been enrolled in clinical trials testing PARP inhibitors, the underlying genetic defects remain to be directly incorporated into molecularly targeted trial design.
Synergy with radiation and DNA-damaging agents
PARP inhibitors can increase sensitivity to many of the standard DNA-damaging treatments used in CNS tumors including ionizing radiation. In glioma cells, PARP inhibitor–mediated radiosensitization is replication-dependent and likely occurs due to replication collapse at unrepaired SSBs or PARP-DNA trapped complexes [41]. Importantly, normal brain tissue is largely nonreplicating and thus relatively protected from enhanced radiation-induced cytotoxicity mediated by PARP inhibition. The ability of various PARP inhibitors to potentiate radiation sensitivity has been shown in models of glioblastoma, pediatric high-grade astrocytoma, DIPG, medulloblastoma, and ependymoma [42,43,44,45,46]. In a systematic review, the median dose enhancement ratio generated by PARP inhibitors was 1.3 [47], though this conceivable may be further enhanced in tumors with intrinsic DDR defects. Moreover, PARP inhibitors can radiosensitize glioblastoma stem cells, which have upregulated DDR and may mediate therapeutic resistance [48, 49].
PARP inhibitors also potentiate the activity of other DNA-damaging agents, an effect which in CNS tumors has primarily been studied with TMZ. TMZ is a monofunctional alkylating agent that methylates DNA bases at different sites. Silencing of the DNA direct repair gene O6-methylguanine-DNA methyltransferase (MGMT), which occurs in approximately two-thirds of lower grade glioma and half of GBM, confers sensitivity to TMZ as unrepaired O6-methylguanine lesions trigger cell death in a mismatch repair (MMR)–dependent manner [50,51,52]. However, intrinsic resistance to TMZ exists in MGMT-expressing tumors and acquired resistance inevitably develops in MGMT-silenced tumors, most commonly due to inactivation of MMR [53,54,55,56,57]. As TMZ also generates substantial N7-methylguanine and N3-methyladenine lesions, which are processed by BER, PARP inhibition has been postulated as a way to re-sensitize resistant cells to TMZ [58]. PARP inhibitors may also promote TMZ sensitivity by blocking PARP-mediated PARylation of MGMT, which has been reported to promote repair of O6-methylguanine lesions [59]. In addition, PARP enzymes other than PARP1 may play a role as PARP inhibitors can restore sensitivity even upon PARP1 knockout in certain MMR-deficient models [60].
In vitro, PARP inhibitors reliably restore the activity of TMZ in MGMT-expressing or MMR-deficient glioma or medulloblastoma cells, while generally having a limited effect on MGMT-deficient cells that are already highly TMZ-sensitive [58,59,60,61,62,63]. In contrast, in vivo studies using patient-derived xenograft models have suggested that the sensitizing effects of PARP inhibitors are limited to those with intrinsic TMZ sensitivity, possibly because PARP inhibitor concentrations needed to induce re-sensitization are difficult to achieve clinically, at least with certain PARP inhibitors [64,65,66]. Co-treatment with PARP inhibitors in TMZ-sensitive cells has also been shown to prevent the emergence of TMZ resistance [67]. In light of these preclinical findings, PARP inhibitors have been investigated in human trials in both TMZ-naive and TMZ-resistant tumors, as discussed below.
Combination with immunotherapy
Tumors exploit inhibitory immune checkpoints, such as cytotoxic T lymphocyte–associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), to suppress T cell effector function and escape immune surveillance [68]. Antibodies targeting CTLA-4 and PD-1/PD-L1 pathways block the interaction of inhibitory molecules with their ligand on tumor cells or antigen-presenting cells, thereby reinvigorating the anti-tumor immune response [68]. Immune checkpoint blockade (ICB) has shown substantial clinical efficacy in patients with various solid tumors but only subsets of patients respond [68]. Putative predictive biomarkers of ICB response include high tumor mutational burden (TMB), increased number of tumor-infiltrating lymphocytes (TILs), an inflammatory gene signature, positive PD-L1 expression, and MMR deficiency/microsatellite instability [69,70,71,72]. GBM is considered to have an immunologically “cold” tumor microenvironment, with low TMB and multiple immunosuppressive mechanisms [73,74,75]. Consequently, ICB has been largely ineffective in clinical trials of adult GBM, with two randomized controlled trials failing to show a benefit with nivolumab over bevacizumab in recurrent GBM or with the addition of nivolumab to RT and TMZ in newly diagnosed GBM [73, 76, 77]. Nevertheless, observed responses with ICB in pediatric patients with recurrent hypermutant GBM harboring germline MMR deficiencies suggest it may still hold promise for the treatment of primary CNS tumors [78, 79]. These findings also beget the prospect of targeting DNA damage repair pathways in order to sensitize gliomas to ICB.
In other solid tumors, PARP inhibition has been shown to enhance tumor immunogenicity and response to ICB through DNA damage–induced activation of immune recognition pathways and increased neoantigen formation [80,81,82]. In the setting of HRD, PARP inhibition creates DSBs resulting in cytosolic dsDNA fragments that are detected by cGMP-AMP synthase (cGAS), ultimately leading to activation of stimulator of interferon genes (STING) and production of type I interferons (IFNs) [82,83,84,85]. The enhanced expression of proinflammatory cytokines and chemokines serves to increase recruitment of TILs [83, 86,87,88]. Through DNA damage–mediated generation of type I IFNs and inhibition of GSK3β, PARP inhibitors have also been shown to induce a compensatory increase in the expression of PD-L1 [70, 82, 84,85,86, 88, 89]. The genomic instability induced by PARP inhibition may also serve to increase TMB, thereby leading to more tumor-specific neoantigens that can be recognized by cytotoxic T cells, as has been seen in the setting of MMR-deficient tumors [71, 90,91,92]. Moreover, via release of type I INFs and other proinflammatory cytokines, PARP inhibition has been shown to enhance antigen presentation by increasing expression of major histocompatibility complex class I (MHC I) on tumor cells and promoting the recruitment and activation of antigen-presenting cells [83, 87, 88, 93]. Of note, while TMB has emerged as a biomarker of ICB response in multiple tumor types, treatment-induced hypermutation has not been consistently associated with ICB response in GBM [72, 91, 94, 95].
As described previously, IDH1/2 mutations confer a “BRCAness” phenotype and sensitivity to PARP inhibitors, making these tumors prime candidates in which to explore PARP inhibitor and immunotherapy combinations. However, in addition to inducing HR defects, 2-HG accumulation has also been shown to impair T cell recruitment via decreased tumor cell production of CXCL10 and impaired T cell activity [96, 97]. In turn, inhibition of mutant IDH1/2 results in increased PD-L1 levels, improved T cell infiltration, and enhanced response to ICB in mouse models [96, 98]. Additional studies are needed to elucidate the interplay between DNA damage–mediated immune activation, IDH1/2-induced BRCAness, and 2-HG-mediated immunosuppression.
PARP inhibitors under clinical investigation in CNS tumors
No PARP inhibitors are yet approved for treatment of CNS tumors, but those currently under clinical investigation in the brain tumor setting include the following: three of the four PARP inhibitors that are currently FDA-approved for other indications (olaparib, niraparib, and talazoparib), the highly investigated PARP inhibitor veliparib, two PARP inhibitors with clinical approval in China for ovarian cancer (pamiparib and fuzuloparib), and two novel compounds designed for CNS penetration and PARP1 selectivity (AZD9574 and NMS-293) (Table 1).
The intrinsic potency of different PARP inhibitors correlates most closely with their PARP trapping potential, which itself is dependent on both their catalytic inhibition and allosteric retention type [13, 16, 17]. Talazoparib is the strongest PARP trapping inhibitor, followed by olaparib, niraparib, and pamiparib all with similar trapping potencies, followed by veliparib which has significantly less trapping [13, 16, 99, 100]. NMS-293 has been reported to be non-PARP trapping [101], while data for fuzuloparib and AZD9574 are not yet available. Non-PARP trappers such as veliparib have limited single agent potency and are typically investigated in combination with cytotoxic agents. All the PARP inhibitors listed have approximately equipotent activity against PARP1 and PARP2, except for the next-generation compounds AZD9574 and NMS-293 which have >8000-fold and >200-fold selectivity for PARP-1 respectively [102, 103]. It is thought that PARP-2 inhibition may contribute to hematologic toxicity and so PARP-1 selectivity may confer an improved toxicity profile while maintaining tumor control efficacy [104].
The various PARP inhibitors also differ in their pharmacokinetic and pharmacodynamic properties, with penetration of the blood–brain barrier being a key parameter for CNS tumor efficacy. Olaparib and talazoparib are known substrates of the P-glycoprotein efflux pump and have poor distribution to brain tissue in the presence of an intact blood–brain barrier [105••, 106, 107]. However, as described below, phase 0 clinical trial data suggests olaparib may sufficiently penetrate tumors with a disrupted blood–brain barrier [105••]. Niraparib and pamiparib have moderately improved brain-to-plasma (B:P) ratios (0.1 and 0.2), veliparib and AZD9574 have high intact brain penetration (B:P ~0.3-1), and NMS-293 may have substantially higher CNS accumulation (B:P ~4-10) [66, 101, 107,108,109].
Clinical trials of PARP inhibitors in CNS tumors
IDH1/2-mutant glioma PARP inhibitor trials
Based on the preclinical studies demonstrating that IDH1/2 mutations confer susceptibility to PARP inhibitors, several clinical trials have been initiated testing PARP inhibitors in IDH1/2-mutant gliomas (Table 2). Two clinical trials (OLAGLI and ETCTN10129) testing standard dose olaparib as monotherapy have recently reported results [110, 111]. The OLAGLI trial enrolled 35 patients with recurrent high-grade IDH-mutant glioma after radiotherapy and at least one line of alkylating chemotherapy while ETCTN10129 enrolled 15 patients in the glioma cohort with recurrent IDH1/2-mutant contrast-enhancing glioma. In both studies, pre-specified primary endpoints of 6-month progression-free survival (PFS-6) or overall response rate (ORR) by RANO criteria were not met, although there was some demonstration of benefit in these heavily pre-treated populations, with prolonged SD seen in subsets of patients.
Ongoing studies in IDH1/2-mutant glioma are investigating PARP inhibitors with improved CNS penetration or in combination with alkylating chemotherapy, with an emphasis on correlative studies to determine intratumoral drug activity. ABTC-1801 is testing the combination of pamiparib and TMZ in recurrent IDH1/2-mutant glioma. Preliminary phase 0 data demonstrated mean unbound concentrations of pamiparib of >20-fold the in vitro IC50 for PARP inhibition in both enhancing and non-enhancing tumors. In the phase I component of the study, the regimen of pamiparib 60 mg twice daily and low-dose metronomic TMZ (20 mg daily) was found to have tolerable hematologic toxicity and will be used as the recommended phase II dose (RP2D) [112•]. The PNOC017 trial is similarly testing pamiparib in combination with TMZ in newly diagnosed or recurrent IDH1/2-mutant glioma in young adults, also with a phase 0 component to evaluate intratumoral drug concentration. Talazoparib is being tested in combination with carboplatin in recurrent HGG with IDH1/2 mutations or other DDR deficiencies, using low-dose single-fraction whole brain radiotherapy to improve drug brain penetration (TAC-GReD trial). AZD9574 is being tested in recurrent IDH1/2-mutant non-enhancing glioma in module 2 of the CERTIS1 trial (NCT05406700). Finally, two phase 0 trials (NCT05406700 and NCT05076513) are investigating brain penetration of niraparib and biomarkers of drug response in IDH1/2-mutant glioma.
Glioblastoma trials of PARP inhibitors in combination with TMZ and/or radiotherapy
The preclinical findings suggesting that PARP inhibition can induce radiosensitization and mitigate TMZ resistance in glioblastoma models have been a predominant focus of PARP inhibitor trials in CNS tumors, with multiple different PARP inhibitors being tested in a variety of different regimens. Veliparib was the first PARP inhibitor to be combined clinically with radiotherapy and TMZ in the treatment of newly diagnosed GBM, but was found to have unacceptable dose-limiting hematologic toxicities in combination with concurrent chemoradiation even when given at a low dose of 10 mg twice daily (4 of 12 patients with dose-limiting thrombocytopenia) and even with de-escalation to every other week administration of veliparib (3 of 6 patients with dose-limiting hematological toxicity) (NCT00770471) [113]. Three subsequent trials have thus combined veliparib with radiotherapy or TMZ independently but overall have yielded negative results. Veliparib in combination with TMZ was investigated in a phase I/II trial in patients with recurrent GBM previously treated with TMZ but failed to improve 6-month PFS (NCT01026493) [114]. More recently, the randomized phase II VERTU trial investigated veliparib and radiotherapy followed by adjuvant veliparib and TMZ, with standard concurrent radiotherapy and TMZ followed by adjuvant TMZ as control arm, in patients with newly diagnosed MGMT promoter-unmethylated GBM [115•]. There were similar toxicity and health-related QOL outcomes between arms, but no improvement in survival compared to historical benchmarks. Of note, the trial was non-comparative in design and insufficiently powered to detect moderate differences in survival. Finally, the complementary Alliance A07112 trial, a large randomized placebo-controlled trial investigating the addition of veliparib to adjuvant TMZ in newly diagnosed MGMT promoter-methylated GBM, found no significant improvement in OS or PFS with veliparib [116•]. An unplanned analysis suggested that concurrent veliparib may limit the emergence of TMZ resistance in subsets of cancers, but this remains to be further investigated. Correlative translational studies from both the VERTU and Alliance trials, including expression or polymorphisms of DNA repair genes, have yet to be reported.
Olaparib has been tested in several GBM trials, starting with the OPARATIC trial, which evaluated the pharmacokinetics, safety, and tolerability of olaparib and TMZ for recurrent GBM. As with veliparib, olaparib exacerbated TMZ-related hematological toxicity, but dose reduction of olaparib to 150 mg 3 days/week was determined to be tolerable with daily TMZ 75 mg/m2 [105••]. Olaparib was detected in all tumor core and margin specimens at doses sufficient for in vitro radiosensitization, despite failing to cross the blood–brain barrier in preclinical models. The PARADIGM study performed dose escalation of olaparib with radiotherapy in patients with newly diagnosed GBM who were not eligible for standard chemoradiation. In this setting, olaparib was well tolerated at doses up to 200 mg twice daily, which notably are higher than those achievable in patients receiving radiotherapy to extracranial sites [117]. Ongoing trials are testing olaparib with both TMZ and radiotherapy to determine optimal dosing. PARADIGM-2 is a phase I study stratified by MGMT promoter methylation status evaluating concurrent and adjuvant olaparib with radiotherapy and TMZ in the MGMT-methylated cohort and with radiotherapy alone in the MGMT-unmethylated cohort [118]. Contemporaneously, the OLA-TMZ-RTE study is dose-escalating olaparib in combination with the conventional Stupp regimen [3] in newly diagnosed unresectable or partially resectable GBM [119]. Both PARADIGM-2 and OLA-TMZ-RTE have planned correlative studies of candidate predictive biomarkers including analysis of DNA repair pathways.
Pamiparib, niraparib, fuzuloparib, and NMS-293 are currently in early phase studies. Pamiparib was studied in combination with radiotherapy and/or TMZ in a phase I/II trial in newly diagnosed or recurrent GBM (NCT03150862). Pamiparib at a dose of 60 mg twice daily was generally well tolerated and resulted in a modified disease control rate of 69.8% in the upfront setting and an ORR of 9.1% in the recurrent setting, supporting further evaluation of these combinations [120, 121]. An ongoing phase 0 study with exploratory phase II component is comparing pharmacokinetics and intratumoral drug exposure of pamiparib versus olaparib, with patients displaying a pharmacokinetic response going on to receive concurrent PARP inhibitor, radiotherapy, and TMZ (NCT04614909). A phase I study of niraparib in advanced cancer established a maximum tolerated dose (MTD) of 40 mg daily with TMZ and showed activity in one subject with GBM (NCT01294735) [122], while two ongoing phase II studies are investigating full-dose niraparib alone or in combination with radiotherapy for recurrent GBM (NCT05297864, NCT04715620). Finally, fuzuloparib and NMS-293 are being tested in combination with TMZ in recurrent GBM in phase II and I/II trials (NCT04552977, NCT04910022).
PARP inhibitor-immunotherapy trials
To date, the majority of clinical data regarding the efficacy of combined PARP inhibition and ICB has come from trials in patients with extracranial solid tumors. Early results have indicated the combination of PARP inhibition and ICB is generally well tolerated with evidence of anti-tumor activity in a subset of patients with germline BRCA1/2 mutations and relapsed ovarian carcinoma and HER2-negative metastatic breast cancer [123,124,125,126].
There is emerging data on combined PARP inhibition and ICB in glioma, with several clinical trials currently in progress. In a phase 2 basket trial of olaparib in combination with durvalumab in IDH-mutant solid tumors (NCT03991832), early interim results from the glioma arm (N = 9) showed the combination is generally well tolerated although there was limited anti-tumor activity with only 1 patient demonstrating a partial response and a median PFS of 2.5 months [127]. A non-randomized phase II trial is investigating the combination of pembrolizumab with olaparib and TMZ in patients with recurrent gliomas (NCT05188508). This trial includes patients with grade II and III IDH-mutated gliomas as well as IDH wild-type gliomas with genetic mutations in HR genes. A recently initiated randomized phase II trial is studying pembrolizumab, olaparib, and TMZ in recurrent GBM (NCT05463848). This trial includes a surgical “window-of-opportunity” in which a cohort of patients will receive treatment before and after surgical resection allowing for evaluation of the immunomodulatory effects in the tumor microenvironment.
Pediatric brain tumor PARP inhibitor trials
A Pediatric Brain Tumor Consortium (PBTC) phase I trial of veliparib and TMZ was performed to evaluate the pharmacokinetics and MTD of veliparib in combination with TMZ in recurrent pediatric brain tumors (PBTC-027). As in the adult setting, dose reductions were required of TMZ due to high hematological toxicity, and the RP2D was veliparib 25 mg/m2 twice daily and TMZ 135 mg/m2 daily [128]. This study was followed by the PBTC-033 phase I/II trial in upfront DIPG in which veliparib 65 mg/m2 twice daily was combined with radiotherapy followed by adjuvant veliparib and TMZ at the previously established RP2D. The treatment was tolerated, but the study was closed at interim analysis due to lack of survival benefit compared with historical controls [129]. This regimen is now being tested in newly diagnosed pediatric HGG without H3K27M mutations in ACNS1721, with the hypothesis that drug entry and efficacy may differ between brainstem and hemispheric HGG. Planned exploratory analyses will investigate associations of tumor genomic, transcriptomic, and epigenetic alterations and germline alterations in HRD and energy metabolism genes with treatment response and outcome. Finally, talazoparib was investigated in combination with TMZ in a large study of recurrent pediatric solid tumors including CNS tumors (ADVL1411). Results revealed promising activity in CNS tumors with 1 PR and 5 SD, including 3 prolonged SD, in 13 subjects with CNS malignancies, which may prompt further study particularly given the relatively poor blood–brain barrier penetration of talazoparib and the low dose of TMZ used in the study [130].
Toxicity considerations
PARP inhibitors are generally well tolerated as monotherapy, with fatigue, gastrointestinal symptoms (nausea, vomiting, diarrhea), and hematological changes (anemia, thrombocytopenia, and neutropenia) being the most common side effects. Adverse event rates are comparable across the different approved PARP inhibitors [131]. In CNS trials, PARP inhibitors are also relatively well tolerated in combination with partial brain radiotherapy at doses comparable to standard monotherapy regimens [115•, 117]. In contrast, concurrent PARP inhibitor and alkylating chemotherapy exacerbate hematological toxicity, requiring significant dose reductions to 20–25% of standard monotherapy doses and/or intermittent dosing strategies. Thus, one critical issue being addressed in early phase studies is whether dose reductions necessary for co-treatment with TMZ will be sufficient to yield active intratumoral drug concentrations and ultimately clinical benefit.
Future perspectives
PARP inhibitors have revolutionized cancer therapy for BRCA-deficient cancers and confer benefit in tumors harboring other HR defects or more generally displaying platinum sensitivity. In addition, this potential is now recognized to expand to other BRCAness phenotypes and to open the door for novel combination strategies in HR-proficient cancers. In CNS tumors, there is strong preclinical evidence that PARP inhibitors can target tumors with altered pathways related to HR or replication stress and can synergize with standard brain cancer therapies. However, early results from clinical trials testing monotherapy olaparib in IDH1/2-mutant tumors and veliparib combined with radiotherapy or TMZ in GBM and pediatric HGG have been disappointing. These results suggest that PARP inhibitors alone in non-BRCA-mutant tumors may be insufficient and that clinically active drug delivery, which must include penetration of both enhancing and non-enhancing tumor, remains a challenge in malignant brain tumors. A variety of next-generation PARP inhibitors with potential for greater efficacy (with enhanced PARP trapping or PARP1 selectivity) or improved brain penetration are currently under investigation. In addition, advances in drug delivery across the blood–brain barrier, for example, using nanoparticles or convention-enhanced delivery, may allow for more effective PARP inhibitor treatment strategies [132,133,134]. Finally, accumulating preclinical data also suggests a role for combining PARP inhibitors with other DNA damage response modulators, for example, ATR or CHK1 inhibitors [38, 40, 135]. Altogether, the use of PARP inhibitors in the treatment of primary CNS tumors remains promising and carefully designed trials incorporating validated biomarkers and tissue endpoints will be critical for their success.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Miller KD, Ostrom QT, Kruchko C, Patil N, Tihan T, Cioffi G, et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J Clin. 2021;71(5):381–406. https://doi.org/10.3322/caac.21693.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231–51. https://doi.org/10.1093/neuonc/noab106.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. https://doi.org/10.1056/NEJMoa043330.
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318(23):2306–16. https://doi.org/10.1001/jama.2017.18718.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. https://doi.org/10.1056/NEJMoa0808710.
Srikanthan D, Taccone MS, Van Ommeren R, Ishida J, Krumholtz SL, Rutka JT. Diffuse intrinsic pontine glioma: current insights and future directions. Chin Neurosurg J. 2021;7(1):6. https://doi.org/10.1186/s41016-020-00218-w.
Chatwin HV, Cruz Cruz J, Green AL. Pediatric high-grade glioma: moving toward subtype-specific multimodal therapy. FEBS J. 2021;288(21):6127–41. https://doi.org/10.1111/febs.15739.
Low JT, Ostrom QT, Cioffi G, Neff C, Waite KA, Kruchko C, et al. Primary brain and other central nervous system tumors in the United States (2014-2018): a summary of the CBTRUS statistical report for clinicians. Neurooncol Pract. 2022;9(3):165–82. https://doi.org/10.1093/nop/npac015.
Richard IA, Burgess JT, O’Byrne KJ, Bolderson E. Beyond PARP1: the potential of other members of the poly (ADP-ribose) polymerase family in DNA repair and cancer therapeutics. Front Cell Dev Biol. 2021;9:801200. https://doi.org/10.3389/fcell.2021.801200.
Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18(10):610–21. https://doi.org/10.1038/nrm.2017.53.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. https://doi.org/10.1038/nature03443.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. https://doi.org/10.1038/nature03445.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99. https://doi.org/10.1158/0008-5472.CAN-12-2753.
Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362ps17. https://doi.org/10.1126/scitranslmed.aaf9246.
Chen HD, Chen CH, Wang YT, Guo N, Tian YN, Huan XJ, et al. Increased PARP1-DNA binding due to autoPARylation inhibition of PARP1 on DNA rather than PARP1-DNA trapping is correlated with PARP1 inhibitor’s cytotoxicity. Int J Cancer. 2019;145(3):714–27. https://doi.org/10.1002/ijc.32131.
Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, DiGiammarino EL, et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol Cancer Res. 2015;13(11):1465–77. https://doi.org/10.1158/1541-7786.MCR-15-0191-T.
Zandarashvili L, Langelier MF, Velagapudi UK, Hancock MA, Steffen JD, Billur R, et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science. 2020;368(6486). https://doi.org/10.1126/science.aax6367.
Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell. 2021;81(15):3128–44 e7. https://doi.org/10.1016/j.molcel.2021.06.011.
Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW. The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol Cell. 2018;71(2):319–31 e3. https://doi.org/10.1016/j.molcel.2018.06.004.
Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018;559(7713):279-84. https://doi.org/10.1038/s41586-018-0261-5.
Panzarino NJ, Krais JJ, Cong K, Peng M, Mosqueda M, Nayak SU, et al. Replication gaps underlie BRCA deficiency and therapy response. Cancer Res. 2021;81(5):1388-97. https://doi.org/10.1158/0008-5472.CAN-20-1602.
Vaitsiankova A, Burdova K, Sobol M, Gautam A, Benada O, Hanzlikova H, et al. PARP inhibition impedes the maturation of nascent DNA strands during DNA replication. Nat Struct Mol Biol. 2022;29(4):329-38. https://doi.org/10.1038/s41594-022-00747-1.
Simoneau A, Xiong R, Zou L. The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev. 2021;35(17-18):1271–89. https://doi.org/10.1101/gad.348479.121.
Sokol ES, Pavlick D, Khiabanian H, Frampton GM, Ross JS, Gregg JP, et al. Pan-cancer analysis of BRCA1 and BRCA2 genomic alterations and their association with genomic instability as measured by genome-wide loss of heterozygosity. JCO Precis Oncol. 2020;4:442–65. https://doi.org/10.1200/po.19.00345.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20. https://doi.org/10.1038/nrc.2015.21.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44. https://doi.org/10.1038/nature08617.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. https://doi.org/10.1016/j.ccr.2010.12.014.
Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–9. https://doi.org/10.1038/embor.2011.43.
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8. https://doi.org/10.1038/nature10860.
Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. 2018;50(1):62–72. https://doi.org/10.1038/s41588-017-0001-z.
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–83. https://doi.org/10.1038/nature10866.
Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9(375):eaal2463. https://doi.org/10.1126/scitranslmed.aal2463.
Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature. 2020;582(7813):586-91. https://doi.org/10.1038/s41586-020-2363-0. Described the mechanism by which 2-hydroxyglutarate produced by IDH1/2-mutant cells suppresses homologous recombination and leads to PARP inhibitor sensitivity, underlying the basis of multiple clinical trials testing PARP inhibitors in IDH1/2-mutant glioma.
Antin C, Tauziede-Espariat A, Debily MA, Castel D, Grill J, Pages M, et al. EZHIP is a specific diagnostic biomarker for posterior fossa ependymomas, group PFA and diffuse midline gliomas H3-WT with EZHIP overexpression. Acta Neuropathol Commun. 2020;8(1):183. https://doi.org/10.1186/s40478-020-01056-8.
Castel D, Kergrohen T, Tauziede-Espariat A, Mackay A, Ghermaoui S, Lechapt E, et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 2020;139(6):1109–13. https://doi.org/10.1007/s00401-020-02142-w.
Hubner JM, Muller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. 2019;21(7):878–89. https://doi.org/10.1093/neuonc/noz058.
Han J, Yu M, Bai Y, Yu J, Jin F, Li C, et al. Elevated CXorf67 expression in PFA ependymomas suppresses DNA repair and sensitizes to PARP inhibitors. Cancer Cell. 2020;38(6):844-56 e7. https://doi.org/10.1016/j.ccell.2020.10.009. Demonstrated that overexpression of EZHIP suppresses homologous recombination and leads to PARP inhibitor sensitivity, suggesting that PARP inhibitors may be efficacious in subsets of ependymoma and DIPG characterized by EZHIP overexpression.
Garbarino J, Eckroate J, Sundaram RK, Jensen RB, Bindra RS. Loss of ATRX confers DNA repair defects and PARP inhibitor sensitivity. Transl Oncol. 2021;14(9):101147. https://doi.org/10.1016/j.tranon.2021.101147.
Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488(7409):49–56. https://doi.org/10.1038/nature11327.
Di Giulio S, Colicchia V, Pastorino F, Pedretti F, Fabretti F, Nicolis di Robilant V, et al. A combination of PARP and CHK1 inhibitors efficiently antagonizes MYCN-driven tumors. Oncogene. 2021;40(43):6143–52. https://doi.org/10.1038/s41388-021-02003-0.
Dungey FA, Loser DA, Chalmers AJ. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys. 2008;72(4):1188–97. https://doi.org/10.1016/j.ijrobp.2008.07.031.
Buck J, Dyer PJC, Hii H, Carline B, Kuchibhotla M, Byrne J, et al. Veliparib is an effective radiosensitizing agent in a preclinical model of medulloblastoma. Front Mol Biosci. 2021;8:633344. https://doi.org/10.3389/fmolb.2021.633344.
Chornenkyy Y, Agnihotri S, Yu M, Buczkowicz P, Rakopoulos P, Golbourn B, et al. Poly-ADP-ribose polymerase as a therapeutic target in pediatric diffuse intrinsic pontine glioma and pediatric high-grade astrocytoma. Mol Cancer Ther. 2015;14(11):2560–8. https://doi.org/10.1158/1535-7163.MCT-15-0282.
Jue TR, Nozue K, Lester AJ, Joshi S, Schroder LB, Whittaker SP, et al. Veliparib in combination with radiotherapy for the treatment of MGMT unmethylated glioblastoma. J Transl Med. 2017;15(1):61. https://doi.org/10.1186/s12967-017-1164-1.
Russo AL, Kwon HC, Burgan WE, Carter D, Beam K, Weizheng X, et al. In vitro and in vivo radiosensitization of glioblastoma cells by the poly (ADP-ribose) polymerase inhibitor E7016. Clin Cancer Res. 2009;15(2):607–12. https://doi.org/10.1158/1078-0432.CCR-08-2079.
van Vuurden DG, Hulleman E, Meijer OL, Wedekind LE, Kool M, Witt H, et al. PARP inhibition sensitizes childhood high grade glioma, medulloblastoma and ependymoma to radiation. Oncotarget. 2011;2(12):984-96. https://doi.org/10.18632/oncotarget.362.
Lesueur P, Chevalier F, Austry JB, Waissi W, Burckel H, Noel G, et al. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: a systematic review of pre-clinical and clinical human studies. Oncotarget. 2017;8(40):69105-24. https://doi.org/10.18632/oncotarget.19079.
Lesueur P, Chevalier F, El-Habr EA, Junier MP, Chneiweiss H, Castera L, et al. Radiosensitization effect of talazoparib, a PARP inhibitor, on glioblastoma stem cells exposed to low and high linear energy transfer radiation. Sci Rep. 2018;8(1):3664. https://doi.org/10.1038/s41598-018-22022-4.
Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014;21(2):258–69. https://doi.org/10.1038/cdd.2013.136.
Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12(2):104–20. https://doi.org/10.1038/nrc3185.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. https://doi.org/10.1056/NEJMoa043331.
Bell EH, Zhang P, Fisher BJ, Macdonald DR, McElroy JP, Lesser GJ, et al. Association of MGMT promoter methylation status with survival outcomes in patients with high-risk glioma treated with radiotherapy and temozolomide: an analysis from the NRG Oncology/RTOG 0424 Trial. JAMA Oncol. 2018;4(10):1405–9. https://doi.org/10.1001/jamaoncol.2018.1977.
Cahill DP, Codd PJ, Batchelor TT, Curry WT, Louis DN. MSH6 inactivation and emergent temozolomide resistance in human glioblastomas. Clin Neurosurg. 2008;55:165–71.
Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13(7):2038–45. https://doi.org/10.1158/1078-0432.CCR-06-2149.
Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15(14):4622–9. https://doi.org/10.1158/1078-0432.CCR-08-3012.
Yoshimoto K, Mizoguchi M, Hata N, Murata H, Hatae R, Amano T, et al. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front Oncol. 2012;2:186. https://doi.org/10.3389/fonc.2012.00186.
Yu Y, Villanueva-Meyer J, Grimmer MR, Hilz S, Solomon DA, Choi S, et al. Temozolomide-induced hypermutation is associated with distant recurrence and reduced survival after high-grade transformation of low-grade IDH-mutant gliomas. Neuro Oncol. 2021;23(11):1872–84. https://doi.org/10.1093/neuonc/noab081.
Curtin NJ, Wang LZ, Yiakouvaki A, Kyle S, Arris CA, Canan-Koch S, et al. Novel poly(ADP-ribose) polymerase-1 inhibitor, AG14361, restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin Cancer Res. 2004;10(3):881–9. https://doi.org/10.1158/1078-0432.ccr-1144-3.
Wu S, Li X, Gao F, de Groot JF, Koul D, Yung WKA. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021;23(6):920–31. https://doi.org/10.1093/neuonc/noab003.
Higuchi F, Nagashima H, Ning J, Koerner MVA, Wakimoto H, Cahill DP. Restoration of temozolomide sensitivity by PARP inhibitors in mismatch repair deficient glioblastoma is independent of base excision repair. Clin Cancer Res. 2020;26(7):1690–9. https://doi.org/10.1158/1078-0432.CCR-19-2000.
Daniel RA, Rozanska AL, Mulligan EA, Drew Y, Thomas HD, Castelbuono DJ, et al. Central nervous system penetration and enhancement of temozolomide activity in childhood medulloblastoma models by poly(ADP-ribose) polymerase inhibitor AG-014699. Br J Cancer. 2010;103(10):1588–96. https://doi.org/10.1038/sj.bjc.6605946.
Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014;349(3):408–16. https://doi.org/10.1124/jpet.113.210146.
Yuan AL, Ricks CB, Bohm AK, Lun X, Maxwell L, Safdar S, et al. ABT-888 restores sensitivity in temozolomide resistant glioma cells and xenografts. PLoS ONE. 2018;13(8):e0202860. https://doi.org/10.1371/journal.pone.0202860.
Clarke MJ, Mulligan EA, Grogan PT, Mladek AC, Carlson BL, Schroeder MA, et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol Cancer Ther. 2009;8(2):407–14. https://doi.org/10.1158/1535-7163.MCT-08-0854.
Gupta SK, Mladek AC, Carlson BL, Boakye-Agyeman F, Bakken KK, Kizilbash SH, et al. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin Cancer Res. 2014;20(14):3730–41. https://doi.org/10.1158/1078-0432.CCR-13-3446.
Gupta SK, Kizilbash SH, Carlson BL, Mladek AC, Boakye-Agyeman F, Bakken KK, et al. Delineation of MGMT hypermethylation as a biomarker for veliparib-mediated temozolomide-sensitizing therapy of glioblastoma. J Natl Cancer Inst. 2016;108(5). https://doi.org/10.1093/jnci/djv369.
Yuan AL, Meode M, Tan M, Maxwell L, Bering EA, Pedersen H, et al. PARP inhibition suppresses the emergence of temozolomide resistance in a model system. J Neurooncol. 2020;148(3):463–72. https://doi.org/10.1007/s11060-020-03561-1.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5. https://doi.org/10.1126/science.aar4060.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–40. https://doi.org/10.1172/JCI91190.
Lagos GG, Izar B, Rizvi NA. Beyond tumor PD-L1: emerging genomic biomarkers for checkpoint inhibitor immunotherapy. Am Soc Clin Oncol Educ Book. 2020;40:1–11. https://doi.org/10.1200/EDBK_289967.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13. https://doi.org/10.1126/science.aan6733.
Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633–41. https://doi.org/10.1200/JCO.2017.75.3384.
de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020;22(4):539–49. https://doi.org/10.1093/neuonc/noz185.
Kelly WJ, Giles AJ, Gilbert M. T lymphocyte-targeted immune checkpoint modulation in glioma. J Immunother Cancer. 2020;8(1). https://doi.org/10.1136/jitc-2019-000379.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20(1):12–25. https://doi.org/10.1038/s41568-019-0224-7.
Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase 3 trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022. https://doi.org/10.1093/neuonc/noac116.
Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase 3 trial. Neuro Oncol. 2022. https://doi.org/10.1093/neuonc/noac099.
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206–11. https://doi.org/10.1200/JCO.2016.66.6552.
Das A, Sudhaman S, Morgenstern D, Coblentz A, Chung J, Stone SC, et al. Genomic predictors of response to PD-1 inhibition in children with germline DNA replication repair deficiency. Nat Med. 2022;28(1):125–35. https://doi.org/10.1038/s41591-021-01581-6.
Bever KM, Le DT. DNA repair defects and implications for immunotherapy. J Clin Invest. 2018;128(10):4236–42. https://doi.org/10.1172/JCI122010.
Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11(7):1018–30. https://doi.org/10.1016/j.celrep.2015.04.031.
Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–42. https://doi.org/10.1016/j.immuni.2014.10.017.
Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018;25(11):2972–80 e5. https://doi.org/10.1016/j.celrep.2018.11.054.
Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019;29(5):1236–48 e7. https://doi.org/10.1016/j.celrep.2019.09.065.
Stewart RA, Pilie PG, Yap TA. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res. 2018;78(24):6717–25. https://doi.org/10.1158/0008-5472.CAN-18-2652.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–20. https://doi.org/10.1158/1078-0432.CCR-16-3215.
Pantelidou C, Sonzogni O, De Oliveria TM, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8(+) T-cell Recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9(6):722–37. https://doi.org/10.1158/2159-8290.CD-18-1218.
Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79(2):311–9. https://doi.org/10.1158/0008-5472.CAN-18-1003.
Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. 2017;109(1). https://doi.org/10.1093/jnci/djw199.
Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552(7683):116–20. https://doi.org/10.1038/nature24673.
Hodi FS, Wolchok JD, Schadendorf D, Larkin J, Long GV, Qian X, et al. TMB and inflammatory gene expression associated with clinical outcomes following immunotherapy in advanced melanoma. Cancer Immunol Res. 2021;9(10):1202–13. https://doi.org/10.1158/2326-6066.CIR-20-0983.
Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, et al. DNA sensing in mismatch repair-deficient tumor cells is essential for anti-tumor immunity. Cancer Cell. 2021;39(1):96–108 e6. https://doi.org/10.1016/j.ccell.2020.11.006.
Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. 2019;129(3):1211–28. https://doi.org/10.1172/JCI123319.
Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020;580(7804):517–23. https://doi.org/10.1038/s41586-020-2209-9.
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–9. https://doi.org/10.1038/s41591-019-0349-y.
Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. 2018;24(8):1192–203. https://doi.org/10.1038/s41591-018-0095-6.
Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127(4):1425–37. https://doi.org/10.1172/JCI90644.
Kadiyala P, Carney SV, Gauss JC, Garcia-Fabiani MB, Haase S, Alghamri MS, et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J Clin Invest. 2021;131(4). https://doi.org/10.1172/JCI139542.
Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–43. https://doi.org/10.1158/1535-7163.MCT-13-0803.
Wang H, Ren B, Liu Y, Jiang B, Guo Y, Wei M, et al. Discovery of pamiparib (BGB-290), a potent and selective poly (ADP-ribose) polymerase (PARP) inhibitor in clinical development. J Med Chem. 2020;63(24):15541–63. https://doi.org/10.1021/acs.jmedchem.0c01346.
Montagnoli A, Papeo G, Rainoldi S, Caprera F, Ciomei M, Felder E, et al. Abstract 4843: NMS-P293, a PARP-1 selective inhibitor with no trapping activity and high CNS penetration, possesses potent in vivo efficacy and represents a novel therapeutic option for brain localized metastases and glioblastoma. Cancer Res. 2018;78(13_Supplement):4843-. https://doi.org/10.1158/1538-7445.Am2018-4843.
Montagnoli A, Rainoldi S, Ciavolella A, Ballinari D, Caprera F, Ceriani L, et al. Abstract 1223: NMS-P293, a novel potent and selective PARP-1 inhibitor with high antitumor efficacy and tolerability. Cancer Res. 2016;76(14_Supplement):1223-. https://doi.org/10.1158/1538-7445.Am2016-1223.
Jamal K, Staniszewska A, Gordon J, Wen S, McGrath F, Dowdell G, et al. Abstract 2609: AZD9574 is a novel, brain penetrant PARP-1 selective inhibitor with activity in an orthotopic, intracranial xenograft model with aberrant DNA repair. Cancer Res. 2022;82(12_Supplement):2609-. https://doi.org/10.1158/1538-7445.Am2022-2609.
Ngoi NYL, Leo E, O’Connor MJ, Yap TA. Development of next-generation poly(ADP-ribose) polymerase 1-selective inhibitors. Cancer J. 2021;27(6):521–8. https://doi.org/10.1097/PPO.0000000000000556.
Hanna C, Kurian KM, Williams K, Watts C, Jackson A, Carruthers R, et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: results of the phase I OPARATIC trial. Neuro Oncol. 2020;22(12):1840-50. https://doi.org/10.1093/neuonc/noaa104. Demonstrated that olaparib penetrates the core and margin regions of recurrent glioblastoma at concentrations sufficient for radiosensitization, despite having minimal penetration of an intact blood–brain barrier. Also established a regimen for combination of olaparib with TMZ using reduced-dose olaparib three times per week and continuous low-dose TMZ.
Kizilbash SH, Gupta SK, Chang K, Kawashima R, Parrish KE, Carlson BL, et al. Restricted delivery of talazoparib across the blood-brain barrier limits the sensitizing effects of PARP inhibition on temozolomide therapy in glioblastoma. Mol Cancer Ther. 2017;16(12):2735–46. https://doi.org/10.1158/1535-7163.MCT-17-0365.
Xiong Y, Guo Y, Liu Y, Wang H, Gong W, Liu Y, et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia. 2020;22(9):431–40. https://doi.org/10.1016/j.neo.2020.06.009.
Li X, Delzer J, Voorman R, de Morais SM, Lao Y. Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metab Dispos. 2011;39(7):1161-9. https://doi.org/10.1124/dmd.110.037820.
Pike A, Balazs A, Cselényi Z, Degorce SL, Ghosh A, Hande SM, et al. Abstract 5076: evaluation of the CNS penetration of a next generation PARP inhibitor, AZD9574, in cynomolgus monkey using positron emission tomography. Cancer Res. 2022;82(12_Supplement):5076-. https://doi.org/10.1158/1538-7445.Am2022-5076.
Ducray F, Sanson M, Chinot OL, Fontanilles M, Rivoirard R, Thomas-Maisonneuve L, et al. Olaparib in recurrent IDH-mutant high-grade glioma (OLAGLI). J Clin Oncol. 2021;39(15_suppl):2007-. https://doi.org/10.1200/JCO.2021.39.15_suppl.2007.
Fanucci K, Pilat MJP, Shah R, Boerner SA, Li J, Durecki DE, et al. Multicenter phase 2 trial of the PARP inhibitor (PARPi) olaparib in recurrent IDH1 and IDH2-mutant contrast-enhancing glioma. J Clin Oncol. 2022;40(16_suppl):2035-. https://doi.org/10.1200/JCO.2022.40.16_suppl.2035.
Schiff D, Bindra R, Li J, Ye X, Ellingson B, Walbert T, et al. CTNI-18. Phase I and preliminary phase 0 results of ABTC 1801: a multi-arm clinical trial of the PARP inhibitor pamiparib (BGB290) with very low dose metronomic temozolomide in recurrent IDH mutant gliomas. Neuro Oncol. 2021;23(Supplement_6):vi63-vi. https://doi.org/10.1093/neuonc/noab196.243. Abstract reporting preliminary results that pamiparib penetrates both enhancing and non-enhancing brain tumors at pharmacologically active concentrations. Also established a regimen for combination of pamiparib and low-dose metronomic TMZ that is under investigation in the phase II study.
Kleinberg L, Supko JG, Mikkelsen T, Blakeley JON, Stevens G, Ye X, et al. Phase I adult brain tumor consortium (ABTC) trial of ABT-888 (veliparib), temozolomide (TMZ), and radiotherapy (RT) for newly diagnosed glioblastoma multiforme (GBM) including pharmacokinetic (PK) data. J Clin Oncol. 2013;31(15_suppl):2065-. https://doi.org/10.1200/jco.2013.31.15_suppl.2065.
Robins HI, Zhang P, Gilbert MR, Chakravarti A, de Groot JF, Grimm SA, et al. A randomized phase I/II study of ABT-888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma: an NRG oncology RTOG group study. J Neurooncol. 2016;126(2):309–16. https://doi.org/10.1007/s11060-015-1966-z.
Sim HW, McDonald KL, Lwin Z, Barnes EH, Rosenthal M, Foote MC, et al. A randomized phase II trial of veliparib, radiotherapy, and temozolomide in patients with unmethylated MGMT glioblastoma: the VERTU study. Neuro Oncol. 2021;23(10):1736-49. https://doi.org/10.1093/neuonc/noab111. A randomized trial which showed that the addition of veliparib to radiotherapy and temozolomide was safe and tolerable, but did not prolong progression-free or overall survival in unmethylated MGMT GBM patients.
Sarkaria JN, Ballman KV, Kizilbash SH, Sulman EP, Giannini C, Mashru SH, et al. Randomized phase II/III trial of veliparib or placebo in combination with adjuvant temozolomide in newly diagnosed glioblastoma (GBM) patients with MGMT promoter hypermethylation (Alliance A071102). J Clin Oncol. 2022;40(16_suppl):2001-. https://doi.org/10.1200/JCO.2022.40.16_suppl.2001. Abstract reporting results of a large randomized controlled trial which showed that the addition of veliparib to adjuvant TMZ did not significantly improve survival in newly diagnosed, MGMT hypermethylated GBM patients. Unplanned exploratory analysis was consistent with potential for veliparib to limit development of TMZ resistance in subset of patients.
Chalmers A, Stobo J, Short SC, Herbert C, Saran F, Morris A, et al. ACTR-22. Results of phase I of the PARADIGM trial: a phase I dose escalation study of olaparib in combination with short course radiotherapy in elderly patients with newly diagnosed glioblastoma (GBM). Neuro Oncol. 2017;19(suppl_6):vi5-vi. https://doi.org/10.1093/neuonc/nox168.017.
Fulton B, Short SC, James A, Nowicki S, McBain C, Jefferies S, et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin Transl Radiat Oncol. 2018;8:12–6. https://doi.org/10.1016/j.ctro.2017.11.003.
Lesueur P, Lequesne J, Grellard JM, Dugue A, Coquan E, Brachet PE, et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer. 2019;19(1):198. https://doi.org/10.1186/s12885-019-5413-y.
Piotrowski A, Puduvalli V, Wen P, Campian J, Colman H, Pearlman M, et al. ACTR-39. Pamiparib in combination with radiation therapy (RT) and/or temozolomide (TMZ) in patients with newly diagnosed or recurrent/refractory (R/R) glioblastoma (GBM); phase 1b/2 study update. Neuro Oncol. 2019;21(Supplement_6):vi21-vi2. https://doi.org/10.1093/neuonc/noz175.081.
Piotrowski A, Puduvalli V, Wen P, Colman H, Campian J, Pearlman M, et al. CTNI-38. Pamiparib in combination with radiation therapy (RT) and/or temozolomide (TMZ) in patients with newly diagnosed (ND) or recurrent/refractory (R/R) glioblastoma (GBM); phase 1b/2 study update. Neuro Oncol. 2020;22(Supplement_2):ii51-ii. https://doi.org/10.1093/neuonc/noaa215.205.
Kurzrock R, Galanis E, Johnson DR, Kansra V, Wilcoxen K, Mcclure T, et al. A phase I study of niraparib in combination with temozolomide (TMZ) in patients with advanced cancer. J Clin Oncol. 2014;32(15_suppl):2092-. https://doi.org/10.1200/jco.2014.32.15_suppl.2092.
Domchek S, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Phase II study of olaparib (O) and durvalumab (D) (MEDIOLA): updated results in patients (pts) with germline BRCA-mutated (gBRCAm) metastatic breast cancer (MBC). Ann Oncol. 2019;30. https://doi.org/10.1093/annonc/mdz253.017.
Drew Y, Kaufman B, Banerjee S, Lortholary A, Hong SH, Park YH, et al. Phase II study of olaparib + durvalumab (MEDIOLA): updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Ann Oncol. 2019;30:v485–v6. https://doi.org/10.1093/annonc/mdz253.016.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5(8):1141–9. https://doi.org/10.1001/jamaoncol.2019.1048.
Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol. 2019;5(8):1132–40. https://doi.org/10.1001/jamaoncol.2019.1029.
Ramos R, Climans SA, Adile A, Ghiassi P, Baker S, Phillips MJ, et al. Combination olaparib and durvalumab for patients with recurrent IDH-mutated gliomas. J Clin Oncol. 2021;39(15_suppl):e14026-e. https://doi.org/10.1200/JCO.2021.39.15_suppl.e14026.
Su JM, Thompson P, Adesina A, Li XN, Kilburn L, Onar-Thomas A, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro Oncol. 2014;16(12):1661–8. https://doi.org/10.1093/neuonc/nou103.
Baxter PA, Su JM, Onar-Thomas A, Billups CA, Li XN, Poussaint TY, et al. A phase I/II study of veliparib (ABT-888) with radiation and temozolomide in newly diagnosed diffuse pontine glioma: a Pediatric Brain Tumor Consortium study. Neuro Oncol. 2020;22(6):875–85. https://doi.org/10.1093/neuonc/noaa016.
Schafer ES, Rau RE, Berg SL, Liu X, Minard CG, Bishop AJR, et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: a Children’s Oncology Group Phase 1 Consortium study (ADVL1411). Pediatr Blood Cancer. 2020;67(2):e28073. https://doi.org/10.1002/pbc.28073.
Cai Z, Liu C, Chang C, Shen C, Yin Y, Yin X, et al. Comparative safety and tolerability of approved PARP inhibitors in cancer: a systematic review and network meta-analysis. Pharmacol Res. 2021;172:105808. https://doi.org/10.1016/j.phrs.2021.105808.
Chen EM, Quijano AR, Seo YE, Jackson C, Josowitz AD, Noorbakhsh S, et al. Biodegradable PEG-poly(omega-pentadecalactone-co-p-dioxanone) nanoparticles for enhanced and sustained drug delivery to treat brain tumors. Biomaterials. 2018;178:193–203. https://doi.org/10.1016/j.biomaterials.2018.06.024.
King AR, Corso CD, Chen EM, Song E, Bongiorni P, Chen Z, et al. Local DNA repair inhibition for sustained radiosensitization of high-grade gliomas. Mol Cancer Ther. 2017;16(8):1456–69. https://doi.org/10.1158/1535-7163.MCT-16-0788.
Vogelbaum MA, Aghi MK. Convection-enhanced delivery for the treatment of glioblastoma. Neuro Oncol. 2015;17 Suppl 2:ii3-ii8. https://doi.org/10.1093/neuonc/nou354.
Sule A, Van Doorn J, Sundaram RK, Ganesa S, Vasquez JC, Bindra RS. Targeting IDH1/2 mutant cancers with combinations of ATR and PARP inhibitors. NAR Cancer. 2021;3(2):zcab018. https://doi.org/10.1093/narcan/zcab018.
Rudolph J, Jung K, Luger K. Inhibitors of PARP: number crunching and structure gazing. Proc Natl Acad Sci U S A. 2022;119(11):e2121979119. https://doi.org/10.1073/pnas.2121979119.
Wang L, Yang C, Xie C, Jiang J, Gao M, Fu L, et al. Pharmacologic characterization of fluzoparib, a novel poly(ADP-ribose) polymerase inhibitor undergoing clinical trials. Cancer Sci. 2019;110(3):1064–75. https://doi.org/10.1111/cas.13947.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
S. Gueble and J. Vasquez declare that they have no conflicts of interest. R. Bindra reports grants and personal fees from Modifi Bio, outside the submitted work. In addition, R. Bindra has a patent 62/344,678 pending to Yale.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neuro-oncology
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gueble, S.E., Vasquez, J.C. & Bindra, R.S. The Role of PARP Inhibitors in Patients with Primary Malignant Central Nervous System Tumors. Curr. Treat. Options in Oncol. 23, 1566–1589 (2022). https://doi.org/10.1007/s11864-022-01024-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-022-01024-5