
Abstract
Over the last decade, as molecular platforms have permitted the characterization of the genomic landscape of pediatric central nervous system (CNS) tumors, pediatric neuro-oncology has dramatically transformed. NTRK fusions are oncogenic driver alterations that have been found in a multitude of tumor types, including pediatric CNS tumors. In recent years, NTRK inhibitors have emerged as a promising class of targeted therapies for pediatric CNS tumors with NTRK gene fusions. The use of larotrectinib and entrectinib in the relapsed setting for pediatric CNS tumors has resulted in rapid and robust responses in an important fraction of patients. These agents are well tolerated, although close to 20% of patients have spontaneous bone fractures. Given the existing data for patients with relapsed disease, clinical trials using NTRK inhibitors in the upfront setting is the next natural progression of efficacy testing and many are currently underway. There are still several challenges that need to be addressed to optimize the use of NTRK inhibitors and identify the patients with NTRK fusion-positive CNS tumors who are most likely to benefit from them. As these agents are more broadly used, resistance will become a more pervasive issue and strategies will need to be determined for this scenario. This article summarizes the current status of NTRK inhibitors for pediatric CNS tumors and discusses the opportunities and challenges of their expanding use in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Robust and rapid responses with NTRK inhibitors in pediatric CNS tumors have confirmed NTRK gene fusions as actionable, tumor-agnostic, targetable alterations. |
Continued evaluation of the safety and efficacy of NTRK inhibitors is needed across ages. |
The optimal way to utilize these agents as either monotherapy or as part of multimodal therapy still needs to be determined. |
1 Introduction
Pediatric central nervous system (CNS) tumors represent the second most common malignancy and the most common cause of cancer-related death in children [1]. As an entity, pediatric CNS tumors are composed of many different types of tumors with vastly divergent outcomes. Advances in the management of these tumors have led to improved survival, but mortality remains substantial, and a high level of morbidity continues to afflict long-term survivors.
Over the past decade, as molecular platforms have permitted the characterization of the genomic landscape of pediatric CNS tumors, pediatric neuro-oncology has dramatically transformed [2]. This new understanding of the biology of disease has impacted diagnosis, risk-stratification, and management. New targets and therapeutic vulnerabilities have been identified that are changing the way that some tumors are being treated.
One such target that has been discovered to be particularly prevalent in gliomas of very young children (< 5 years old) are oncogenic fusions involving the neurotrophic tyrosine receptor kinases (NTRK). NTRK fusions are oncogenic driver alterations that have been found in a multitude of tumor types, including infantile fibrosarcoma, secretory carcinoma, and papillary thyroid cancer, as well as CNS tumors [3]. In recent years, NTRK inhibitors have emerged as a promising class of targeted therapies for pediatric CNS tumors with NTRK gene fusions. The first NTRK inhibitors have been approved by the United States Food and Drug Administration (FDA) for patients with NTRK fusion-positive tumors, and this included pediatric patients. This review will provide an overview of NTRK inhibitors and their role in the treatment of pediatric CNS tumors.
2 NTRK as Driver of Oncogenesis
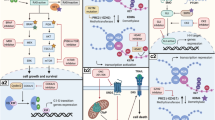
Neurotrophic tyrosine receptor kinases, including TRKA, TRKB, and TRKC, are tyrosine kinases encoded by the NTRK1, NTRK2, and NTRK3 genes, respectively (Fig. 1). All TRK proteins contain an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain. TRKA, TRKB, and TRKC are regulated by nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and NT-3 [4, 5]. The ligand binding initiates the process of oligomerization in the kinase domain, leading to activation [6]. These kinases are principally expressed in the nervous system and are active in various neuronal processes, including differentiation, survival, proliferation, and membrane trafficking [7,8,9,10]. In addition, TRK receptors are important in pain perception, thermoregulation, proprioception, appetite control, memory, and learning [11,12,13,14,15,16,17]. Furthermore, TRK receptors are expressed in skeletal tissue and are involved in chondrogenesis, osteoblastogenesis, and osteoclastogenesis, and participate in regulating tissue formation and healing in bone [18, 19].

Trk proteins, binding partners, downstream pathways, and functions. NGF nerve growth factor, BDNF brain-derived neurotrophic factor, NT-4/5 neurotrophin 4/5, NT-3 neurotrophin 3. Created with BioRender.com
Although NTRK mutations, splice variants, and TRK overexpression have been described in oncogenesis, fusions involving NTRK1, NTRK2, or NTRK3 are most common [9]. Normally, the 3′ sequences of NTRK1, NTRK2, or NTRK3, including the kinase domain, and the 5′ sequences of genes, create these fusions. The products are oncoproteins that can be constitutively active without ligand binding, ultimately leading to the upregulation of downstream pathways. These fusion proteins can signal through the same pathways that unaltered TRK proteins do upon ligand binding [20, 21]. It is important to note that NTRK fusions rarely co-occur with other alterations [22]. Furthermore, NTRK fusion proteins induce high-grade astrocytomas in transduced primary astrocytes, confirming these fusions to be the primary oncogenic stimulus [23].
NTRK fusions exist in cancer in three contexts: (1) tumors with a high frequency of these fusions where they are almost pathognomonic; (2) tumors with a lower frequency, but NTRK fusions occur consistently; and (3) tumors where NTRK fusions are sporadic. For pediatric tumors, congenital mesoblastic nephroma and infantile fibrosarcoma are examples of the first category. Pertinent to CNS tumors, infantile hemispheric gliomas are an example of the second category, with NTRK fusion frequency of about 20% [24, 25]. For the third category, there are now several examples of NTRK fusions in a variety of histologic subtypes due to the increase in large-scale sequencing platforms that have led to the identification of genomic alterations across many tumors.
In the evaluation of a large pediatric cancer database, St. Jude’s PeCan, NTRK fusions were found in 0.34% of pediatric cancers [26]. In this study, 10 of 12 identified fusions were in CNS tumors: 7 high-grade glioma and 3 low-grade glioma. Likewise, a study looking at NTRK fusions in 1347 pediatric tumors found a frequency of 2.22% for all patients and 2.07% for patients with CNS tumors [27]. Specific NTRK fusions discovered in pediatric CNS tumors are described in Fig. 2.

3 NTRK Inhibitors
With the discovery of NTRK fusions across multiple cancers and the existence of tyrosine kinase inhibitors (TKIs) with activity against TRKA, TRKB, and TRKC, these agents are being tested in clinical trials with increasing frequency. Entrectinib, a multi-kinase inhibitor with activity against TRKs, and larotrectinib, a selective TRK inhibitor, have the most robust data in NTRK-fused pediatric CNS tumors. These agents have IC50 values in the low nanomolar range in tested cells, including lymphoma and sarcoma cell lines [3, 28, 29]. While a number of other multikinase inhibitors assert activity against TRKA/B/C (i.e., crizotinib, ponatinib, and cabozantinib), these do so with a higher IC50, and clinical activity has not been consistently reported [3, 30].
Early phase clinical trials of entrectinib and larotrectinib in both pediatric and adult patients demonstrated promising results for NTRK fusion-positive tumors in a tumor-agnostic manner. Furthermore, the responses were quick and durable in patients in the relapsed setting. Additionally, efficacy has been observed in patients with primary CNS tumors and CNS metastases, indicating adequate penetration of the blood-brain barrier.
In 2018, larotrectinib received accelerated approval by the FDA for adult and pediatric patients with solid tumors that have an NTRK gene fusion [31]. Subsequently in 2019, entrectinib received FDA approval for the treatment of adults with ROS1-positive, metastatic non-small cell lung cancer, and ≥ 12-year-old patients with solid tumors with an NTRK gene fusion [32]. In October of 2023, entrectinib’s approval was expanded to include patients older than 1 month of age [33].
3.1 Larotrectinib
Larotrectinib is a highly specific inhibitor of TRKA, TRKB, and TRKC. Its efficacy has been tested in several clinical trials. A pooled analysis of three phase I/II trials using larotrectinib (NCT02122913, NCT02637687, NCT02576431) that included 159 patients with solid non-CNS tumors showed an objective response rate (ORR) of 79% [34]. This response was independent of tumor histology.
The first clinical trial testing larotrectinib in the pediatric patient population was the SCOUT trial (NCT02637687). SCOUT was a phase I/II trial for patients between 1 month and 21 years of age with locally advanced or metastatic solid tumors or CNS tumors. In 2018, Laetsch et al. reported on phase I [35]. The maximum tolerated dose was not reached, and 100 mg/m2 was established as the recommended phase II dose (RP2D). Of 15 patients with TRK fusion-positive cancers, 14 (93%) had objective responses, including 4 complete responses. Among enrolled patients with TRK fusion-positive cancers, most had infantile fibrosarcoma or other soft tissue sarcomas, but none had CNS tumors.
Doz et al. reported on the efficacy and safety of larotrectinib in patients with CNS tumors with TRK fusions who were included in SCOUT and the NAVIGATE clinical trials [36]. In this study, 33 patients with TRK fusion-positive CNS tumors were included. The ORR was 30% for all patients, although 82% of patients with measurable disease had a response. The responses to larotrectinib were rapid, with a median time to response of 1.9 months (range 1.0–3.8 months). Treatment-related adverse events were reported for 20 patients, with grade 3/4 in 3 patients. A summary of the efficacy and side effects of this analysis is included in Table 1 and Fig. 3.

3.2 Entrectinib
Entrectinib is a CNS-penetrant oral inhibitor of TRKA, TRKB, TRKC, ROS1, and ALK. In three phase I/II clinical trials (STARTRK-1, STARTRK-2, ALKA-372-001), entrectinib’s efficacy was evaluated in adult patients (age > 18 years) with metastatic or locally advanced solid tumors with NTRK fusions. In a pooled analysis of 54 patients, 31 (57%) had an objective response, including 4 complete responses [37]. Weight gain and neutrophil count decrease were the most common grade 3 or 4 treatment-related adverse events.
The STARTRK-NG trial (NCT02650401) is the first clinical trial evaluating entrectinib specifically in the pediatric population. STARTRK-NG is an ongoing phase I/II trial for patients less than 22 years of age with progressive or recurrent solid tumors, including CNS tumors. The trial included a dose escalation cohort for all patients, regardless of fusion status, followed by an expansion cohort for those with fusion-positive tumors and target diseases. In phase II, enrolled patients with intracranial or extracranial solid tumors harboring target fusions or neuroblastoma were enrolled at the RP2D. In 2022, Desai et al. reported on the primary endpoints, RP2D, and objective response rate (ORR) [38]. In phase II, 16 patients with CNS tumors with fusions were evaluable and responses were observed in 8 patients, yielding an ORR of 50% (Table1).
In STARTRK-NG, all patients reported ≥ 1 adverse event and close to three-quarters experienced grade 3/4 events. The most common treatment-related AEs included weight gain, anemia, increased blood creatinine, and nausea (Fig. 3). Fractures occurred in 13 (21%) patients, with 3 patients discontinuing entrectinib due to them.
3.3 Second-Generation NTRK Inhibitors
While the results for entrectinib and larotrectinib have been promising, next-generation TRK inhibitors are already being explored. These newer TRK inhibitors are designed to address on-target resistance mutations (see below). These agents have smaller molecular weight and more compact macrocyclic structures, which permits them to evade steric impedance in the kinase domain, allowing them to engage the ATP-binding pocket [39]. These drugs have increased activity against TRKA/B/C when compared with larotrectinib or entrectinib [40, 41]. Repotrectinib, selectinib, and talectinib are currently being studied in clinical trials. Limited data exist on the use of second-generation NTRK inhibitors for pediatric CNS tumors. Clinical trials using first- and second-generation NTRK inhibitors in pediatric CNS tumors are included in Table 2.
4 Resistance to NTRK Inhibitors
Despite the success of NTRK inhibitors, reports of resistance have emerged. Two mechanisms of resistance to these agents have been described: mutations of the TRK kinase domain (on-target resistance) or off-target mechanisms. Off-target mechanisms of resistance depend on the activation of alternative cellular pathways without direct alteration of the TRK proteins. Both mechanisms enable the tumor to evade inhibition of TRK, despite continued dependence on TRK signaling [3].
For first-generation TRK inhibitors, acquired on-target resistance is caused by mutations within the ATP binding pocket of the kinase domain [42]. These mutations cause resistance by interfering with the binding of the inhibitor, altering the ATP-binding affinity or changing conformation of the kinase domain [43, 44]. These mutations have been identified in patients with NTRK fusion-positive tumors that have progressed during treatment with larotrectinib or entrectinib.
The off-target mechanisms of resistance are secondary to alterations involving other receptor tyrosine kinases or downstream pathway mediators besides the TRK proteins. Frequently, the MAP kinase pathway is involved, including alterations of BRAF, KRAS, ERBB2, and MET [45]. In this context, combination therapy could provide an avenue for disease control [45].
The frequency of resistance to NTRK inhibitors is largely unknown, and factors associated to resistance are yet to be determined. Fewer reports of resistance to first-generation NTRK inhibitors have been observed in children [46]. It is uncertain whether this is due to pediatric tumors having a lower tendency to develop resistance or a bias due to a smaller number of pediatric patients receiving these agents. In patient with resistance to first-generation NTRK inhibitors, responses to second-generation agents have been reported [47, 48]. Hence, it is increasingly important to study second-generation NTRK inhibitors in the context of acquired resistance [43, 49].
5 Opportunities and Challenges
The robust and rapid responses using NTRK inhibitors in the relapsed setting for pediatric CNS tumors sets the stage for their broader use. Cytoreduction with these inhibitors has important therapeutic implications and opens opportunities to optimize cancer-directed therapy for patients with CNS tumors with NTRK fusions. Broadly, these agents have the capacity to improve outcomes and decrease acute treatment-related morbidity, such as surgical morbidity and the risk of infection with chemotherapy-induced neutropenia. Treatment with inhibitors could decrease the long-term neurocognitive impact of historical treatment approaches and increase survival rates.
Especially for infants with hemispheric gliomas, where NTRK fusions are frequent and tumors can be large and hemorrhagic, the upfront use of NTRK inhibitors can reduce the risks of surgical complications that can be common [25], if initial neurosurgical procedures are limited to biopsies or minimal debulking. Furthermore, in these patients the long-term neurocognitive outcomes are poor [25], and there is a possibility that these agents could decrease the use of radiotherapy and cytotoxic chemotherapy and limit the long-term impact of these treatments.
However, there are still several challenges that need to be addressed to optimize the use of these inhibitors and identify the patients with NTRK fusion-positive CNS tumors who are most likely to benefit from them [50]. Firstly, the efficacy of these inhibitors depends on the identification of the fusions, which relies on the availability and optimization of robust molecular testing and neuropathologic evaluation. Choosing the right assay for NTRK fusion detection hinges on the type of tumor and the genes involved and requires considering elements, including the availability of material, the accessibility of assays, and the necessity for simultaneous comprehensive genomic and histopathologic testing [51]. Specifically for the use of immunohistochemistry, positive staining can be seen in neuronal tissue, limiting its use in CNS tumors [51]. Although fluorescence in situ hybridization (FISH) testing often serves as the primary method for identifying chromosomal rearrangements, identifying NTRK fusions using FISH requires considerable expertise and is limited by the large NTRK proteins and multiple fusion partners [42]. DNA sequencing, RNA sequencing, and reverse transcription-polymerase chain reaction (RT-PCR) can be employed but tend to be limited by availability.
Furthermore, most of the existing data on the efficacy of NTRK inhibitors for pediatric CNS tumors are in the setting of relapsed or recurrent tumors. Since rapid and sustained responses have been seen in a substantial portion of patients, testing of NTRK inhibitors in the upfront setting is the next natural progression of efficacy testing. Fortunately, there are open clinical trials that are already testing NTRK inhibitors in this context (NCT04655404).
Relevant to frontline therapy, there is much uncertainty on how NTRK inhibitors should be optimally used in conjunction with other treatment modalities, such as cytotoxic chemotherapy or radiotherapy. Although rapid and profound responses are seen after the initiation of NTRK inhibitors, there is growing experience that shows tumors recur after these agents are stopped. Nonetheless, these tumors can respond once again after rechallenging with an NTRK inhibitor [52]. With this, the duration of treatment remains unknown and must be explored in future clinical trials. Furthermore, other treatment modalities may be needed to achieve disease control without long-term use of these NTRK inhibitors. Combination therapies added to NTRK inhibitors, such as other targeted agents or more traditional chemotherapy, are being explored to see if these may decrease the recurrences.
With the first clinical trials using TRK inhibitors occurring over the preceding years, robust data on the long-term toxicities of these agents do not currently exist. Specifically for young children, given the physiologic function of TRK signaling in CNS development and function, the long-term effects of TRK inhibitors on neurologic function must be comprehensively evaluated. Hence, comprehensive evaluation, including neuro-cognitive testing, is imperative in children in whom these agents are used [3]. Furthermore, the management of other side effects that are secondary to on-target inhibition of TRK, such as weight gain, must be optimized. Finally, further data are needed on the frequency of fractures in patients receiving entrectinib and possible interventions to mitigate these events. On the basis of available data, the frequency of bone fractures is high and can lead to the discontinuation of these agents [38]. TRK receptors are involved in bone homeostasis but the exact mechanism that leads to high fracture rates has not been fully elucidated. Futhermore, additional data are needed to investigate the patients that are at highest risk for fractures and strategies to mitigate the risk of fractures occurring.
As these agents are used more pervasively, resistance will likely become more prevalent. With this, the newer generation of agents for on-target mutations may be the best avenue for alternative treatment. For the off-target mutations, the combination of more than one agent may allow for better disease control. In this context, repeat molecular profiling, include sequencing and expression analysis, is essential to define treatment strategies. Furthermore, the expanding role of cell-free DNA (cfDNA) in pediatric CNS tumors could be relevant for the monitoring of resistance to these agents. Initial reports have recently been published on cfDNA testing as a strategy to identify resistance mechanisms in adult malignancies [53].
An important point to consider for the future of these agents is their global availability. Understanding that close to 90% of children with cancer live in low- and middle-income countries (LMICs) [1], access to novel agents is limited by their availability and cost. Nonetheless, in settings with limited resources, NTRK inhibitors could be highly beneficial due to their potential to decrease the need for hospitalization and their lack of important impact on patient immunity. Furthermore, the significant cytoreduction seen can lead to less complicated neurosurgical procedures, an important element to consider due to the limited pediatric neurosurgical capacity in many countries [54]. As mentioned, medication access must be developed in conjunction with the availability comprehensive diagnostics to identify the patients who would benefit from targeted therapy. Strategies to expand access to these novel agents in resource-limited settings are needed and must rely on a multisectoral dialogue at the national, regional, and global levels. St. Jude Children’s Research Hospital is soon to launch an international, multi-site phase II trial (GLOBOTRK, NCT06528691) that will enroll patients in the USA and five LMICs to evaluate the efficacy of entrectinib as frontline treatment in infants less than 3 years of age with CNS tumors with NTRK and ROS1 fusions. This trial will be a proof-of-principle that clinical trials with molecularly defined treatment for children with CNS tumors can be run in LMICs.
6 Conclusions
The presence of NTRK fusions defines a unique molecular subgroup of pediatric CNS tumors where targeted therapy will play a principal role in their treatment in the future. The high response rates achieved with TRK inhibitors across a wide range of tumor types, including CNS tumors, has confirmed NTRK gene fusions as actionable, tumor-agnostic, targetable alterations. Moreover, the frequency and diversity of reported NTRK fusions in pediatric cancers is increasing, making testing more pertinent, particularly in tumors in which these alterations are more common. It is important to continue to evaluate the safety and efficacy of NTRK inhibitors and continue to explore the optimal way to utilize these agents as either monotherapy or as part of multimodal therapy.
References
Steliarova-Foucher E, Colombet M, Ries LAG, Moreno F, Dolya A, Bray F, et al. International incidence of childhood cancer, 2001–10: a population-based registry study. Lancet Oncol. 2017;18(6):719–31.
Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, Gottardo NG. Pediatric brain tumors: innovative genomic information is transforming the diagnostic and clinical landscape. J Clin Oncol. 2015;33(27):2986–98.
Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731–47.
Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65(5):895–903.
Davies AM, Horton A, Burton LE, Schmelzer C, Vandlen R, Rosenthal A. Neurotrophin-4/5 is a mammalian-specific survival factor for distinct populations of sensory neurons. J Neurosci. 1993;13(11):4961–7.
Nakagawara A, Liu XG, Ikegaki N, White PS, Yamashiro DJ, Nycum LM, et al. Cloning and chromosomal localization of the human TRK-B tyrosine kinase receptor gene (NTRK2). Genomics. 1995;25(2):538–46.
Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans Roy Soc Lond B Biol Sci. 2006;361(1473):1545–64.
Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736.
Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5(1):25–34.
Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309.
Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13(4):485–8.
Greco A, Villa R, Fusetti L, Orlandi R, Pierotti MA. The Gly571Arg mutation, associated with the autonomic and sensory disorder congenital insensitivity to pain with anhidrosis, causes the inactivation of the NTRK1/nerve growth factor receptor. J Cell Physiol. 2000;182(1):127–33.
Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7(11):1187–9.
Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6(7):736–42.
Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, et al. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368(6468):246–9.
Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, et al. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75(1):113–22.
Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. 1994;25(11):1386–403.
Sun W, et al. Neuro-bone tissue engineering: emerging mechanisms, potential strategies, and current challenges. Bone Res. 2023;11(1):65.
Su YW, Zhou XF, Foster BK, Grills BL, Xu J, Xian CJ. Roles of neurotrophins in skeletal tissue formation and healing. J Cell Physiol. 2018;233(3):2133–45.
Jin W, Yun C, Hobbie A, Martin MJ, Sorensen PH, Kim SJ. Cellular transformation and activation of the phosphoinositide-3-kinase-Akt cascade by the ETV6-NTRK3 chimeric tyrosine kinase requires c-Src. Cancer Res. 2007;67(7):3192–200.
Ranzi V, Meakin SO, Miranda C, Mondellini P, Pierotti MA, Greco A. The signaling adapters fibroblast growth factor receptor substrate 2 and 3 are activated by the thyroid TRK oncoproteins. Endocrinology. 2003;144(3):922–8.
Westphalen CB, Krebs MG, Le Tourneau C, Sokol ES, Maund SL, Wilson TR, et al. Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population. NPJ Precis Oncol. 2021;5(1):69.
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–50.
Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. 2019;10(1):4343.
Chiang J, Bagchi A, Li X, Dhanda SK, Huang J, Pinto SN, et al, High-grade glioma in infants and young children is histologically, molecularly, and clinically diverse—results from the SJYC07 trial and institutional experience. Neuro Oncol. 2023;26(1):178-190.
Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol. 2018;2018.
Zhao X, Kotch C, Fox E, Surrey LF, Wertheim GB, Baloch ZW, et al. NTRK fusions identified in pediatric tumors: the frequency, fusion partners, and clinical outcome. JCO Precis Oncol. 2021;1.
de la Cruz CC, Hunsaker T, Vazvaei F, Draganov D, Yu L, Merchant M. Determination of the efficacious entrectinib exposures required for pathway inhibition and anti-tumor activity in a subcutaneous and intracranial TPM3-NTRK1mutant tumor model. Cancer Res. 2019;79:3894.
Menichincheri M, Ardini E, Magnaghi P, Avanzi N, Banfi P, Bossi R, et al. Discovery of entrectinib: a new 3-aminoindazole as a potent anaplastic lymphoma kinase (ALK), c-ros oncogene 1 kinase (ROS1), and pan-tropomyosin receptor kinases (Pan-TRKs) inhibitor. J Med Chem. 2016;59(7):3392–408.
Fuse MJ, Okada K, Oh-Hara T, Ogura H, Fujita N, Katayama R. Mechanisms of resistance to NTRK inhibitors and therapeutic strategies in NTRK1-rearranged cancers. Mol Cancer Ther. 2017;16(10):2130–43.
US Food & Drug Administration. FDA approves larotrectinib for solid tumors with NTRK gene fusions. https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions. Accesed 23 July 2024.
US Food & Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf. Accesed 23 July 2024.
US Food & Drug Administration. FDA expands pediatric indication for entrectinib and approves new pellet formulation. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-expands-pediatric-indication-entrectinib-and-approves-new-pellet-formulation#:~:text=FDA%20expands%20pediatric%20indication%20for%20entrectinib%20and%20approves%20new%20pellet%20formulation,-Share&text=On%20October%2020%2C%202023%2C%20the,(Rozlytrek%2C%20Genentech%20Inc. Accesed 23 July 2024.
Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21(4):531–40.
Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705–14.
Doz F, van Tilburg CM, Geoerger B, Højgaard M, Øra I, Boni V, et al. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol. 2022;24(6):997–1007.
Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):271–82.
Desai AV, Robinson GW, Gauvain K, Basu EM, Macy ME, Maese L, et al. Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK, ROS1, or ALK aberrations (STARTRK-NG). Neuro Oncol. 2022;24(10):1776–89.
Qin, H, Patel MR. The challenge and opportunity of NTRK inhibitors in non-small cell lung cancer. Int J Mol Sci. 2022;23(6):2916.
Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019;30(Suppl 8):viii23–30.
Murray BW, Rogers E, Zhai D, Deng W, Chen X, Sprengeler PA, et al. Molecular characteristics of repotrectinib that enable potent inhibition of TRK fusion proteins and resistant mutations. Mol Cancer Ther. 2021;20(12):2446–56.
Laetsch TW, Hong DS. tropomyosin receptor kinase inhibitors for the treatment of TRK fusion cancer. Clin Cancer Res. 2021;27(18):4974–82.
Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016;6(1):36–44.
Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017;7(9):963–72.
Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med. 2019;25(9):1422–7.
Blauel ER, Laetsch TW. The promise of TRK inhibitors in pediatric cancers with NTRK fusions. Cancer Genet. 2022;262–263:71–9.
Hemming ML, Nathenson MJ, Lin JR, Mei S, Du Z, Malik K, et al. Response and mechanisms of resistance to larotrectinib and selitrectinib in metastatic undifferentiated sarcoma harboring oncogenic fusion of NTRK1. JCO Precis Oncol. 2020;4:79–90.
Chen MF, Yang SR, Shia J, Girshman J, Punn S, Wilhelm C, et al. Response to repotrectinib after development of NTRK resistance mutations on first- and second-generation TRK inhibitors. JCO Precis Oncol. 2023;7: e2200697.
Drilon A, Li G, Dogan S, Gounder M, Shen R, Arcila M, et al. What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann Oncol. 2016;27(5):920–6.
Laetsch TW. Improving outcomes in pediatric NTRK gene fusion-positive solid tumors: importance of genomic testing and targeted therapy with the TRK inhibitor larotrectinib. Clin Adv Hematol Oncol. 2024;22 Suppl 3(4):1–11.
Solomon JP, Linkov I, Rosado A, Mullaney K, Rosen EY, Frosina D, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33(1):38–46.
Bagchi A, Orr BA, Campagne O, Dhanda S, Nair S, Tran Q, et al. Lorlatinib in a child with ALK-fusion-positive high-grade glioma. N Engl J Med. 2021;385(8):761–3.
Sivaprakasam K, Harada G, Choudhury NJ, Savin C, Shah RH, Berger MF et al. ctDNA analysis of NTRK fusion and mechanisms of acquired resistance to TRK inhibitors. J Clin Oncol. 2023;41(16).
Roach JT, Qaddoumi I, Baticulon RE, Figaji A, Campos DA, Arredondo L, et al. Pediatric neurosurgical capacity for the care of children with CNS tumors worldwide: a cross-sectional assessment. JCO Glob Oncol. 2023;9: e2200402.
Lopez GY, Perry A, Harding B, Li M, Santi M. CDKN2A/B loss is associated with anaplastic transformation in a case of NTRK2 fusion-positive pilocytic astrocytoma. Neuropathol Appl Neurobiol. 2019;45(2):174–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
National Cancer Institute (P30CA021765) and American Lebanese Syrian Associated Charities.
Conflict of interest
M.M. and G.W.R. declare that they have no conflicts of interest that might be relevant to the contents of this manuscript. D.C.M. receives research support from Roche/Genentech.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
D.C.M. drafted the manuscript. D.M.C. and M.M. prepared the tables and figures. All authors contributed to the interpretation of the findings, the editing of the article, and the approval of the final submitted version. All authors accept responsibility for the decision to submit for publication.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moreira, D.C., Mikkelsen, M. & Robinson, G.W. Current Landscape of NTRK Inhibition for Pediatric CNS Tumors. CNS Drugs (2024). https://doi.org/10.1007/s40263-024-01121-z
Accepted:
Published:
DOI: https://doi.org/10.1007/s40263-024-01121-z