
Abstract
In order to address the need for production of parthenocarpic fruits, embryogenic cultures of 'Brewster' ('Chen Tze') litchi derived from leaves of a mature tree were transformed with the PISTILLATA (PI) cDNA in antisense orientation through Agrobacterium-mediated transformation. In four independently transformed lines, transgene integration was confirmed by conventional and quantitative PCR. The four transgenic lines differed with respect to the relative copy number of the transgene and expression levels, and there was no correlation between the two traits. The expression of the litchi PI homolog was detected in transformed and non-transformed embryogenic cultures. Although expression levels were low overall, they were significantly higher in wild-type plants compared to lines that were transformed. This result suggests that post-transcriptional silencing of the litchi PI homolog induced by an antisense oriented transgene could be a successful strategy; however, silencing in floral primordia to produce parthenocarpic fruits can only be confirmed in mature plants after several years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Litchi (Litchi chinensis Sonn., family Sapindaceae) is a subtropical fruit tree native to South-East Asia. The fruit is a drupe, in which the edible part is the aril. Normal fruits contain a single large seed that, along with the pericarp, reduces the flesh content within the fruit by 50%–70% (Menzel et al. 2005). Two different types of seedless litchi fruit occur: aborted-seeded fruit and parthenocarpic fruit. Aborted-seeded fruits, known as “chicken tongue”, arise from abnormal ovules or as a result of incomplete fertilization, whereas parthenocarpic seedless fruits develop when the ovary has not been fertilized (Huang 2005). The occurrence of aborted-seeded fruits is cultivar dependent and can vary from season to season (Wu et al. 2007). Fruits with aborted seeds are preferred by consumers, therefore demanding a higher price in the market (Subhadrabandhu and Stern 2005). There are a few cultivars that produce nearly seedless fruit, although the fruits are smaller and usually weigh <10 g (Menzel 2002). Most 'Hexiachuan' fruits are parthenocarpic, although they have a small size and are of low quality (Wu et al. 2007). The expansion of litchi into other regions of the world has been limited by its poor production due to irregular flowering and unpredictable weather that limits pollination during blooming (Huang et al. 2001; Litz and Raharjo 2005). It has been suggested that induction of seedless litchi fruit could resolve this (Huang 2001).
Conventional breeding of litchi has several limitations due to its long juvenile period (7–8 yr), the length of time before horticultural traits can be evaluated, low fertility, and high levels of heterozygosity common to most tropical and subtropical perennial fruit crops (Gomez-Lim and Litz 2004). Most new cultivars are selected from superior seedlings obtained after open pollination. Cross-breeding is not commonly used because the genetic background of most litchi germplasm is unclear (Wu et al. 2007). Different strategies have been attempted in order to induce both aborted-seeded fruits and parthenocarpic fruits, by application of growth regulators that inhibit zygotic embryo development (Huang and Stern 2005).
The application of biotechnology approaches for improvement of existing litchi cultivars has been impeded by the absence of a de novo regeneration pathway from mature phase or elite cultivars. Raharjo and Litz (2007) described the induction of embryogenic cultures from leaves of mature phase 'Brewster' ('Chen Tze') trees, a prerequisite for utilizing genetic transformation and in vitro mutagenesis to improve this and other superior cultivars. In this study, we have addressed the genetic transformation of 'Brewster' ('Chen Tze') that ultimately could result in parthenocarpy.
Floral regulatory genes were initially studied in Antirrhinum majus and Arabidopsis thaliana, and their functions were explained by the ABC model (Schwarz-Sommer et al. 1992; Weigel and Meyerowitz 1994). Although the two dicotyledonous species are distantly related, the set of regulatory genes in both species is almost identical (Samach et al. 1997). Homologous genes have been isolated from a wide range of plant species. In every case, the set of floral regulatory genes belongs to the well-known MADS-box family, whose members contain a highly conserved DNA-binding region, i.e., the MADS-box domain (Riechmann and Meyerowitz 1997). In A. thaliana, PISTILLATA (PI) is a B class gene that acts in whorls 2 and 3 during floral organ development. Hence, the PI mutations convert petals to sepals and stamens to carpels (Bowman et al. 1991; Goto and Meyerowitz 1994). Some parthenocarpic fruits, e.g., apple (Malus domestica), have been shown to result from natural mutations in a homolog of the PI gene, demonstrating the functional similarity between the homeotic genes of a perennial woody tree species and those of an annual plant for controlling floral organ development (Yao et al. 2001).
The antisense strategy has been one of the most referenced approaches for gene silencing not only for the study of gene function but for specific breeding purposes as well. Within fruit crops, it has been applied to apple (Kotoda et al. 2006), pear (Gao et al. 2007), plum (Callahan and Scorza 2007), strawberry (Jiménez-Bermúdez et al. 2002), citrus (Wong et al. 2001), mango (Cruz-Hernández et al. 1997), and papaya (Magdalita et al. 2002). By transforming litchi with the A. thaliana PI cDNA in the antisense orientation, we expect to suppress the homologous gene in litchi (PI-Lch) and ultimately obtain parthenocarpic fruit development.
The goals of this study were to describe a procedure for genetic transformation of an elite litchi cultivar 'Brewster' ('Chen Tze') and to use a gene that could address the major breeding limitation of seedlessness, which would involve interfering with stamen development and forcing fruit production without pollination. For these purposes, four different transgenic litchi lines that resulted from Agrobacterium-mediated transformation of embryogenic cultures were studied. A relative quantification of the transgene was performed, along with the study of its expression and its ability to silence the putative endogenous homologous gene in litchi.
Materials and methods
Plant material.
An embryogenic culture of 'Brewster' ('Chen Tze') L. chinensis Sonn. was induced from leaflets of young vegetative flushes of mature, field-grown trees from the tropical and subtropical fruit collection of the University of Florida Tropical Research and Education Center, Florida, USA. The 'Chen Tze' litchi is a traditional cultivar of South China that has been vegetatively propagated for several centuries; it was introduced into Florida as air layered plants in the early twentieth century and renamed 'Brewster'. Therefore, the leaf explants were from several hundred-year-old trees. The leaflets were surface-sterilized with 0.02% (w/v) sodium hypochlorite for 10 min and washed with three rinses of sterile, deionized water. Intact sterile leaflets were plated on induction medium in 125 × 15 mm sterile disposable Petri dishes. Induction medium consisted of B5 (Gamborg et al. 1968) major salts and Murashige and Skoog (MS; Murashige and Skoog 1962) minor salts and organic components with 100 mg L−1 myo-inositol, 400 mg L−1 glutamine, 200 mg L−1 casein hydrolysate, 30 g L−1 sucrose, and with 4.52 mM 2,4-dichlorophenoxyacetic acid (2,4-D), 9.30 μM kinetin, and 3 g L−1 Gel-GroTM (ICN Biochemicals, Santa Ana, CA) gellan gum (Raharjo and Litz 2007). The kinetin was filter-sterilized and added to autoclaved medium. The Petri dishes were sealed with Parafilm® (Pechiney Plastic Packaging Company, Chicago, IL).
Embryogenic cultures were maintained in liquid medium containing half-strength MS major salts and full-strength MS minor salts and organic components, 30 g L−1 sucrose, 4.2 μM 2,4-D and 0.91 μM zeatin. The zeatin was filter-sterilized and added to autoclaved maintenance medium. Embryogenic suspensions consisting of 200 mg of proembryogenic masses (PEMs) were inoculated into 40 mL of maintenance medium in 125-mL Erlenmeyer flasks. The flasks were sealed with sterile, heavy-duty aluminum foil and Parafilm®. The cultures were maintained at 100 rpm on a rotary shaker and were subcultured at 2-week intervals into fresh medium. At each subculture, suspensions were passed through sterile, nylon filtration fabric (1.6 mm opening), and an inoculum consisting of about 200 mg of the larger fraction size (>1.6 mm opening) was added to each 125-mL Erlenmeyer flask containing 40 mL fresh liquid maintenance medium. Growth conditions for induction and maintenance were darkness at 25°C.
The pH of all media was adjusted to 5.7. Media were sterilized by autoclaving at 1.1 kg cm2 at 121°C for 20 min.
Bacterial strain and plasmid.
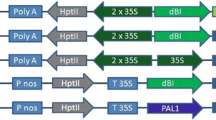
The disarmed Agrobacterium tumefaciens strain EHA105 with the binary vector pCambia3301 (http://www.cambia.org/daisy/cambia/2071/version/1/part/4/data/pCAMBIA3301.pdf?branch=main&language=default; Cambia, Canberra, Australia) was used for genetic transformation (Fig. 1). The transferred DNA region contains the bar expression cassette providing resistance to phosphinothricin (Block et al. 1987) driven by the Cauliflower Mosaic Virus (CaMV) 35S promoter, the PISTILLATA cDNA sequence (Goto and Meyerowitz 1994) in reverse orientation flanked by the CaMV 35S promoter and the rbcS terminator sequences, and the GUS (uidA gene) expression cassette (Jefferson 1987) as a reporter gene. The vector was introduced into A. tumefaciens strain EHA105 by electroporation. The strain EHA105 containing pCAMBIA3301/B-PISTILLATA was grown in 1.0 mL LB broth at 27°C for 48 h. A single colony of Agrobacterium strain EHA105/pCAMBIA3301/B-PISTILLATA was inoculated into 5 mL of LB medium (pH 7.0) supplemented with 50 mg L−1 kanamycin and 20 mg L−1 rifampicin at 28°C at 200 rpm in darkness for 16 h. The culture was diluted to 20 mL with fresh LB medium modified by the addition of 50 mg L−1 kanamycin, 20 mg L−1 rifampicin, and 120 μM acetosyringone, and incubated at 125 rpm in darkness at 27°C. After 8 h, 0.8 mL of acetosyringone-activated Agrobacterium suspension (OD600 = 0.2) was added to each flask containing litchi embryogenic cultures.

pCAMBIA3301/B-PISTILLATA. The transferred DNA region contains the bar expression cassette providing phosphinothricin resistance and driven by the CaMV 35S promoter, the PISTILLATA sequence flanked by the CaMV 35S promoter and the rbcS terminator sequences, and the GUS (uidA) expression cassette as a reporter gene.
Inhibition of PEM growth by phosphinothricin.
The concentration of phosphinothricin that inhibited growth of litchi embryogenic suspension cultures was determined. The inoculum consisted of embryogenic 'Brewster' cultures from five flasks (40 mL liquid maintenance media in 125-mL Erlenmeyer flasks). Six concentrations of phosphinothricin were tested in addition to the control: 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0 mg L−1 with five replications for each treatment and the control. Filter-sterilized phosphinothricin was added to 40 mL liquid maintenance medium in 125-mL Erlenmeyer flasks, and 1.0 mL of settled cell volume (SCV) of PEMs was inoculated into each flask. The flasks were capped with aluminum foil and sealed with Parafilm®. Cultures were maintained on a rotary shaker at 125 rpm at 27°C under diffuse light. The SCV of PEMs was measured after 5 wk using the method described above. For SCV measurement, PEMs were decanted into sterile graduated plastic centrifuge tubes and the SCV was measured after about 1 min. Data were analyzed by ANOVA at a 1% confidence level (SAS Institute, Cary, NC).
Co-cultivation with Agrobacterium.
Approximately 4.0 g of embryogenic 'Brewster' ('Chen Tze') cultures were sieved through sterile nylon filtration fabric (mesh size 1.8 mm), after which 0.25 g of the filtrate was added to 125-mL flasks containing 40 mL of maintenance medium and 15 mg carborundum. After 1 wk, the flasks were homogenized using a vortex 30 s and 0.8 mL of log phase acetosyringone-activated EHA105/pCAMBIA3301/B-PISTILLATA culture was added to each flask. The PEMs and EHA105/pCAMBIA3301/B-PISTILLATA cultures were co-cultivated at 27°C for 3 d at 100 rpm in darkness.
The PEMs were then transferred to 40 mL liquid maintenance medium supplemented with filter-sterilized 200 mg L−1 cefotaxime and 500 mg L−1 carbenicillin in 125-mL Erlenmeyer flasks at 27°C and 125–130 rpm to eliminate A. tumefaciens. After 1 wk, the PEMs were transferred into fresh liquid maintenance medium supplemented with filter-sterilized 200 mg L−1 cefotaxime, 500 mg L−1 carbenicillin, and 3.0 mg L−1 phosphinothricin, and subcultured at 2-wk intervals in liquid medium of the same formulation for 3 mo.
Growth of putative transgenic embryogenic cultures on selection medium.
The growth of putatively transformed embryogenic cultures and non-transformed cultures on semi-solid selection maintenance medium with 2.0 mg L−1 phosphinothricin was compared. Each sample consisted of 150 ± 5 mg PEMs. There were four replicates for each treatment and the experiment was repeated twice. Weight measurements and subcultures were performed at 2-wk intervals for 8 wk.
Development of transformed somatic embryos.
Somatic embryo development was initiated from putatively transformed embryogenic cultures from liquid maintenance selection medium according to Raharjo and Litz (2007). Each suspension culture was passed through sterile, nylon filtration fabric 2 wk after subculture, and the filtrate was transferred to fresh medium. Approximately 200 mg of PEMs were spread evenly on the surface of semi-solid MS medium in 100 × 20 mm Petri dishes containing 45 g L−1 sucrose, 10% (v/v) filter-sterilized coconut water (CW), and 3 g L−1 gellan gum. CW was obtained from freshly harvested immature coconuts and was filter-sterilized before adding to autoclaved medium. The cultures were incubated in darkness at 25°C. Germination/conversion occurred following the transfer of mature, white-opaque somatic embryos ≥5 mm in diameter to plant recovery medium, consisting of semi-solid MS medium with 30 mg L−1 sucrose, 14.4 μM gibberellic acid (GA3), and 3 g L−1 gellan gum in 100 × 20 mm Petri dishes containing 50 mL of medium (Raharjo and Litz 2007). The GA3 was filter-sterilized and added to autoclaved medium. Following root and shoot emergence, plantlets were transferred individually into sterile glass containers (90 × 55 mm) containing 60 mL semi-solid MS basal medium with 30 g L−1 sucrose, 100 mg L−1 activated charcoal, and 4 g L−1 Agargel (Sigma, St. Louis, MO). Growing conditions were 25°C with a 16-h photoperiod (60–80 μmol s−1 m−2) provided by cool white fluorescent lights.
GUS assay.
Phosphinothricin-resistant PEMs, somatic embryos, and leaves of regenerated plants were evaluated for GUS (β-glucuronidase) activity. Tissue samples were washed with sterile deionized water and treated with X-Gluc (5-bromo-4-chloro-3-indolyl B-D glucuronide; Sigma) according to Jefferson (1987). The tissues were cleared with methanol/acetone (3:1) and stored in 10% (v/v) glycerol.
DNA and RNA analyses.
In longan (Dimocarpus longan Lour.), a closely related member of the Sapindaceae family, a PISTILLATA-like gene (LVFS-199) expressed in floral buds has been described (Matsumoto 2006). The coding sequence of LVFS-199 (GenBank Acc DT368224) has a 96% identity with the one defined as “Litchi chinensis MAD box protein 1 (MAD1) mRNA” in GenBank (AY705793). The litchi sequence is referred as PI-Lch.
Three independent purifications of genomic DNA and RNA were extracted from each putatively transformed line and non-transformed PEMs. Nucleic acids were isolated from 50 mg tissue samples using the E.Z.N.A.® SP Plant DNA Kit and the E.Z.N.A.® Plant RNA Kit (Omega Bio-Tek, Norcross, GA). The concentration of DNA samples was determined with a spectrophotometer and then equalized to 5 ng/μL with 0.5× TE buffer. RNA samples were further purified by DNase I digestion, acid phenol/chloroform extraction, and ethanol precipitation (Nolan et al. 2006). RNA integrity was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Only RNA samples with a RNA integrity number (RIN) above 5 were used for expression analyses. cDNA was synthesized from 5 μg of RNA with the Transcriptor First Strand cDNA Synthesis Kit (Roche, Penzberg, Germany) using anchored oligo(dT)18 primers. cDNA samples were diluted to a final volume of 400 μL using 0.5× TE buffer.
Standard PCR was used to verify the presence of the recombinant DNA construct in PEMs. Three primer pairs were employed for the detection of the bar gene, uidA gene, and the antisense A. thaliana PISTILLATA sequence (Table 1). Each amplification reaction was carried out in a final volume of 25 μL, containing 100 ng of gDNA and using the ReadyMixTM Taq PCR Reaction Mix with MgCl2 (Sigma-Aldrich) according to manufacturer’s instructions. pCambia3301 DNA was included as a positive control. PCRs were incubated in the GeneAmp® PCR System 2700 (Applied Biosystems, Foster City, CA) with the following temperature profile: an initial step of 3 min at 94°C followed by 35 cycles of 1 min at 94°C, 1 min at 60°C, and 1 min at 72°C, then a final step of 5 min at 72°C. Amplicons were separated on a 0.8% agarose gel in TBE buffer and stained with ethidium bromide.
Quantitative PCR (qPCR) was used for transgene copy number estimation and gene expression analysis in PEMs. Amplification reactions were run in triplicate on the LightCycler® 480 platform (Roche), in a total volume of 20 μL containing 6 μL of gDNA or cDNA template, 2 μM of each amplification primer (Table 1), and 10 μL of 2× LightCycler 480 SYBR Green I Master (Roche). Amplified sequences for both PI and PI-Lch (156 bp and 171 bp, respectively) were included within the C-terminal domain, which shows the highest variability among the MADS-box family members. The cycling conditions were as follows: an initial denaturation step of 95°C for 10 min to activate the Taq DNA polymerase, followed by 45 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. The amplification process was followed by a melting curve analysis, ranging from 60 to 95°C, with a ramp rate of 0.02°C/s and 25 fluorescence acquisitions per degree Celsius. The quantification cycle (C q) values were determined with the LightCycler 480 Software 1.5, previous manual setting of the fluorescence threshold. In order to control a possible gDNA contamination of cDNA samples, each reverse transcription-quantitative PCR (RT-qPCR) assay was accompanied by a RT-minus amplification reaction and only differences ≥10 cycles between the respective Cq values were accepted for further analyses. Besides calculation of the melting temperature of each amplicon, amplification specificity was confirmed by agarose electrophoresis in a random subset of amplification reactions and sequencing of some selected amplicons (BigDye® Terminator v3.1 Cycle Sequencing Kit and 3500 Series Genetic Analyzer; Applied Biosystems).
Relative quantities of the transgene or the targeted mRNAs, and the corresponding significance levels (p value), were calculated from C q data following an efficiency-correction model (Pfaffl 2001) as implemented in the 2008 version of REST software (Pfaffl et al. 2002). The PCR efficiency of each amplification reaction was estimated with the DART-PCR workbook (Peirson et al. 2003). The SSR locus named LMLY11, a single copy sequence of the litchi genome (Viruel and Hormaza 2004), was used as reference gene to control gDNA input during transgene copy number estimation. Normalization factors for mRNA level quantification were calculated as the geometric mean of the expression levels of several control genes, whose suitability as internal references was assessed with the geNorm software (Vandesompele et al. 2002). These reference genes were selected among those proposed by Zhong et al. (2011).
Results
Verification of transformation.
Embryogenic 'Brewster' ('Chen Tze') litchi cultures were transformed with a DNA construct containing the bar gene, the uidA gene, and the A. thaliana PI cDNA in antisense. The growth of embryogenic litchi suspension cultures was completely suppressed by 1.0–2.0 mg L−1 phosphinothricin (Fig. 2), and the effect was highly significant (p < 0.01). Therefore, 2.0 mg L−1 phosphinothricin was used to select for transformed embryogenic cultures that contained the bar gene.

Effect of phosphinothricin on inhibition of growth of 'Brewster' ('Chen Tze') litchi embryogenic suspension cultures.
Three months after co-culturing 'Brewster' ('Chen Tze') embryogenic cultures with EHA105/pCAMBIA3301/B-PISTILLATA and with continuous selection in the presence of 2.0 mg L−1 phosphinothricin, transformation of PEMs was verified on the basis of the X-Gluc histochemical reaction for β-glucuronidase activity. All transformed lines (T1–T4) showed intense GUS staining (Fig. 3a). Somatic embryos that developed from T1 and T2 cultures were also positive for GUS expression (Fig. 3b–d). At the time of writing, T2 embryos had germinated (Fig. 4a–d).

X-Gluc histochemical reaction in embryogenic cultures (a) and somatic embryos (b–d) of 'Brewster' ('Chen Tze') litchi.

The GUS (uidA) response in different transgenic leaves (a–d) from T2 line plantlet (e).
The growth of the transformed embryogenic cultures on semi-solid medium with 2.0 mg L−1 phosphinothricin (PPT) was determined after 2, 4, 6, and 8 wk of incubation (Fig. 5). Throughout the measuring period, this growth was lower than the growth of non-transformed cultures in standard maintenance medium. This was probably due to the strong selection for bar gene expression imposed on transformed cells since this gene encodes for PPT-acetyltransferase activity necessary for PPT inactivation. Nevertheless, non-transformed cultures showed no significant growth on selection medium with PPT (Fig. 5).

Growth of transformed proembryogenic cultures compared to non-transformed cultures on semi-solid selection medium with 2.0 mg L−1 phosphinothricin. Results shown are the average of transformed plants T1–T4.
The presence of bar and uidA transgenes, and the PI antisense cDNA in transformed lines was confirmed by standard PCR. As shown in Fig. 6, amplicons with the expected size (Table 1) derived from bar, uidA, and PI sequences were obtained with gDNA templates of the four selected transformed PEMs, whereas PEMs of non-transformed cultures did not show any amplification product.

Detection of the bar (a), uidA (b), and PI (c) genes by PCR analysis of transformed PEMs (lines 1–4; transformed plants T1–T4, respectively). M molecular weight ladder (Promega, Ladder 100 bp, ref. PRG2101), C− control without DNA template, U non-transformed PEM (negative control), P plasmid pCambia3301/B-PISTILLATA (positive control).
Transgene relative copy number.
To determine the relative copy number of the construct in the four transgenic lines, qPCR was performed with three independent gDNA samples from each transformed line. Transgenic line 2 (T2) showed the lowest amplification signal with PI (156) primers (Table 1) and it was used as calibrator, with an assigned value of 1. Gene copy number in T1 was 2-fold higher than in T2, while in T3 and T4 the increment was 1.6-fold relative to the T2 line (Fig. 7). All three differences with respect to T2 were highly significant.

Relative copy number of A. thaliana PI cDNA in four transgenic lines (T1–T4). The LMLY11 gene was used for calculating normalization factors. T2 was used as calibration sample. Error bars indicate confidence intervals (95%) estimated from a randomization assay with n = 3. **Data highly significantly different (p < 0.01) from calibrator.
Expression analysis of transgene and endogenous PISTILLATA homolog.
Initially, the suitability of GAPDH, EF-1α, and RPII housekeeping genes were tested as internal references for normalization of mRNA level measures. Analysis of pairwise variation between two sequential normalization factors with the geNorm software revealed that the EF-1α and RPII genes were sufficient for accurate mRNA quantification. The mean M and CV values for the pair EF-1α/RPII in our set of cDNA samples were M = 0.416 and CV = 0.144. These values of expression stability are inside the ranges M ≤ 0.5 and CV ≤ 0.25, which have been proposed by Hellemans et al. (2007) as acceptable for relatively homogeneous sample panels.
Transgene expression in all the transformed lines was measured. The specificity of this analysis was confirmed by amplicon sequencing. The A. thaliana PI cDNA expression level was significantly lower in line T1 than in the other transgenic lines, whose expression values were estimated as fold changes over T1. Line T3 showed an expression level ca. 9-fold higher than T1. For lines T2 and T4, the average increases were 5-fold and 3-fold, respectively (Fig. 8).

Relative expression level of A. thaliana PI antisense mRNA (PI) in four transgenic lines (T1–T4) and relative expression level of Litchi PISTILLATA (PI-Lch) in the same transgenic lines and the wild type (WT). EF-1α and RPII genes were used for calculating normalization factors. For PI analyses, line T1 was used as the calibration sample, whereas for PI-Lch the calibrator was the WT line. Error bars indicate confidence intervals (95%) estimated from a randomization test with n = 3. *Data significantly different (p < 0.05) and **highly significantly different (p < 0.01) from the respective calibrator.
PI-Lch mRNA in PEMs was detected as revealed by amplicon sequencing; however, the expression levels of the PI-Lch gene in these tissues were very low (C q values around 35–38) and consequently expression measures suffered from a lack of precision. In order to increase accuracy, amplification reactions were performed on six replicates during expression analysis of this endogenous gene. Despite this technical improvement, comparisons involving the T1 transformed line were precluded by the fact that only one out of three independent cDNA preparations from the same T1 PEM gave an amplification signal. The non-transformed line (WT) was used as calibrator for the analysis of PI-Lch expression levels showed in Fig. 8. Compared to WT, PI-Lch mRNA levels in T2 and T4 decreased 7-fold, whereas in T3, the reduction was ca. 1.5-fold. For line T1, PI-Lch expression level was so low that it could not be determined with a minimal accuracy. Expression levels were significantly lower in the transgenic lines T2 and T4 compared to WT, and highly significant for T4. These results suggest that, to a variable extent, post-transcriptional silencing of the PI-Lch gene by the A. thaliana PI antisense mRNA was successful. Interestingly, the amount of antisense mRNA did not correlate with the level of PI-Lch silencing (Fig. 8).
Discussion
The most important tropical fruit crops, including banana (Ganapathi et al. 2001), papaya (Cheng et al. 1996; Ying et al. 1999), pineapple (Firoozabady and Gutterson 1998; Firoozabady et al. 2006), avocado (Cruz-Hernández et al. 1998), and mango (Mathews et al. 1992, 1993), have been transformed using Agrobacterium-mediated protocols based on embryogenic cultures and the recovery of somatic embryos. Das and Rahman (2012) reported Agrobacterium-mediated transformation of litchi zygotic embryos with the regeneration of transgenic plantlets. In the present study, transgenic somatic embryos and plants from elite 'Brewster' ('Chen Tze') litchi have been obtained.
A. tumefaciens strain EHA105 has been successfully used for the transformation of many different fruit tree crops, while transformation with pCambia as the binary plasmid vector has only been reported for kiwifruit (Actinidia deliciosa 'Hayward'; Kim et al. 2008). Under our conditions, transformation efficiency based upon recovery of plants was low (<5%), although recovery of transformed PEMs and somatic embryos was much higher (>80%). Raharjo and Litz (2007) also observed a very high rate of germination/conversion for (non-transformed) 'Brewster' ('Chen Tze') litchi somatic embryos. Similar results were observed with respect to plant recovery following transformation of avocado embryogenic cultures (Raharjo et al. 2008). Somatic embryo development has occurred with two transgenic lines, T1 and T2, and plant recovery only with line T2.
Agrobacterium-mediated transformation usually results in a few copies of the transgenes integrated at a single locus as a tandem array (Jorgensen et al. 1987). Most frequently, transgene copy number in transformed plants has been estimated by Southern blot analysis. Determination of transgene copy number by real-time qPCR has become an effective alternative to Southern blot (Shepherd et al. 2009), and in recent years several studies have demonstrated its effectiveness in comparison with Southern blot hybridization, particularly with woody perennial species (Omar et al. 2008; Casu et al. 2012; Gadaleta et al. 2012). Using qRT-PCR in this study, it has been inferred that transgene copies in T1 was double that of T2, while T3 and T4 had about 1.6 more insertions than T2 (Fig. 7). These results suggest that the most probable absolute number of insertions is two for T2, three for T3 and T4, and four for T1.
The expression of the PI antisense cDNA did not correlate with the estimated number of copies in the obtained transformed lines (Figs. 7 and 8), as has been previously reported (Hobbs et al. 1993; Gelvin 2003). For example, T1 showed the highest relative number of copies among transformed lines even though it had the lowest PI expression level. In the same way, the level of PI antisense mRNA in T3 was roughly 3-fold greater than in T4 (p < 0.05), although their transgene copy numbers were not significantly different. Variable expression of plant transgenes has been explained by position and epigenetic effects, as well as by post-transcriptional silencing (Matzke and Matzke 1998; Meins 2000). Post-transcriptional silencing is frequently associated with multiple transgene copies within a cell, especially when they are inserted in the genome as inverted repeats (Stam et al. 1997; Gelvin 2003).
PISTILLATA was originally identified as a floral homeotic gene in A. thaliana (Bowman et al. 1989; Meyerowitz et al. 1989). Although its highest expression occurs during early floral development (Goto and Meyerowitz 1994), PI is also expressed at low levels in young seedlings, embryos, and even embryonic culture tissues (Kinoshita et al. 2001; Lehti-Shiu et al. 2005). This suggests that the gene may play an important regulatory function during embryo development, in addition to a role in floral identity (Lehti-Shiu et al. 2005). In this study, we have also detected the expression of the putative litchi PI homolog at very low levels. Although we demonstrated that PI antisense is able to significantly decrease levels of PI-Lch mRNA, we cannot conclude that silencing will be effective during flower development, when the expression level of endogenous genes is notably up-regulated in comparison to embryonic stages. Mature plants derived from T1 and T2 somatic embryos will be available in 7–8 yr (typical juvenile period for litchi) and will allow us to confirm whether PI-Lch mRNA levels will be sufficiently decreased in floral primordial to generate parthenocarpic fruits.
Since PI and PI-Lch cDNAs show a moderate identity (65%) biased towards the 5′-half of the sequence where the conserved MADS domain is encoded, we cannot totally exclude the possibility of the PI antisense sequence affecting the expression of other members of MADS-box gene family; however, the fact that we were able to obtain plants from germinated transgenic somatic embryos suggests that any essential function for early development was not compromised.
References
Block MD, Botterman J, Vandewiele M, Dockx J, Thoen C, Gossele V, Movva NR, Thompson C, Vanmontagu M, Leemans J (1987) Engineering herbicide resistance in plants by expression of a detoxifying enzyme. EMBO J 6:2513–2518
Bowman JL, Smyth DR, Meyerowitz EM (1989) Genes directing flower development in Arabidopsis. Plant Cell 1:37–52
Bowman JL, Smyth DR, Meyerowitz EM (1991) Genetic interactions among floral homeotic genes of Arabidopsis. Development 112:1–20
Callahan A, Scorza R (2007) Effects of a peach antisense ACC oxidase gene on plum fruit quality. Acta Horticult 738:567–573
Casu RE, Selivanova A, Perroux JM (2012) High-throughput assessment of transgene copy number in sugarcane using real-time quantitative PCR. Plant Cell Rep 31:167–177
Cheng YH, Yang JS, Yeh SD (1996) Efficient transformation of papaya by coat protein gene of papaya ringspot virus mediated by Agrobacterium following liquid-phase wounding of embryogenic tissues with carborundum. Plant Cell Rep 16:127–132
Cruz-Hernández A, Gómez-Lim M, Litz RE (1997) Transformation of mango somatic embryos. Acta Horticult 455:292–298
Cruz-Hernández A, Litz RE, Witjaksono, Gómez-Lim M (1998) Agrobacterium tumefaciens-mediated transformation of embryogenic avocado cultures and regeneration of somatic embryos. Plant Cell Rep 17:497–503
Das DK, Rahman A (2012) Expression of a rice chitinase gene enhances antifungal response in transgenic litchi (cv. Bedana). Plant Cell Tissue Organ Cult 109:315–325
Firoozabady E, Gutterson N (1998) Genetically transformed pineapple plants and methods for their production. International Patent Application No. PCT/US98/03681
Firoozabady E, Heckert M, Gutterson N (2006) Transformation and regeneration of pineapple. Plant Cell Tissue Organ Cult 84:1–16
Gadaleta A, Giancaspro A, Cardone MF, Blanco A (2012) Real-time PCR for the detection of precise transgene copy number in durum wheat. Cell Mol Biol Lett 16:652–668
Gamborg OL, Miller RA, Ojima K (1968) Plant cell cultures. I. Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–158
Ganapathi TR, Higgs NS, Balint-Kurti PJ, Arntzen CJ, May GD, Van Eck JM (2001) Agrobacterium-mediated transformation of embryogenic cell suspensions of the banana cultivar Rasthali (AAB). Plant Cell Rep 20:157–162
Gao M, Matsuta N, Murayama H, Toyomasu T, Mitsuhashi W, Dandekar AM, Tao R, Nishimura K (2007) Gene expression and ethylene production in transgenic pear (Pyruscommunis cv. 'LaFrance') with sense or antisense cDNA encoding ACC oxidase. Plant Sci 173:32–42
Gelvin SB (2003) Agrobacterium-mediated plant transformation: the biology behind the “gene-jockeying” tool. Microbiol Mol Biol Rev 67:16–37
Gomez-Lim MA, Litz RE (2004) Genetic transformation of perennial tropical fruit crops. In Vitro Cell Dev Biol Plant 40:442–449
Goto K, Meyerowitz EM (1994) Function and regulation of the Arabidopsis floral homeotic gene PISTILLATA. Genes Dev 8:1548–1560
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19
Hobbs SLA, Warkentin TD, DeLong CMO (1993) Transgene copy number can be positively or negatively associated with transgene expression. Plant Mol Biol 21:17–26
Huang H, Stern AS (2005) Fruit set, development and maturation. In: Menzel CM, Waite GK (eds) Litchi and longan: botany, production and uses. CABI, Wallingford, pp 115–140
Huang HB (2001) Towards a better insight into the development of the arilate fruit of litchi and longan. Acta Horticult 558:185–192
Huang X (2005) Fruit disorders. In: Menzel CM, Waite GK (eds) Litchi and longan: botany, production and uses. CABI, Wallingford, pp 141–151
Huang XM, Li JG, Wang HC, Huang HB, Gao FF (2001) The relationship between fruit cracking and calcium in litchi pericarp. Acta Horticult 558:209–216
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS fusion system. Plant Mol Biol Report 5:387–405
Jiménez-Bermúdez S, Redondo-Nevado J, Munoz-Blanco J, Caballero JL, Lopez-Aranda JM, Valpuesta V, Pliego-Alfaro F, Quesada MA, Mercado JA (2002) Manipulation of strawberry fruit softening by antisense expression of a pectate lyase gene. Plant Physiol 128:751–759
Jorgensen R, Snyder C, Jones JDG (1987) T-DNA is organized predominantly in inverted repeat structures in plants transformed with Agrobacterium tumefaciens C58 derivatives. Mol Gen Genet 207:471–477
Kim M, Kim SC, Jang KC, Song EY, Kang SJ, Song KJ (2008) Agrobacterium-mediated transformation of carotenoid synthesis gene in kiwifruit (Actinidia deliciosa 'Hayward'). Acta Horticult 773:41–145
Kinoshita T, Harada JJ, Goldberg RB, Fischer RL (2001) Polycomb repression of flowering during early plant development. Proc Natl Acad Sci U S A 98:14156–14161
Kotoda N, Iwanami H, Takahashi S, Abe K (2006) Antisense expression of MdTFL1, a TFL1-likegene, reduces the juvenile phase in apple. J Am Soc Horticult Sci 131:74–81
Lehti-Shiu MD, Adamczyk BJ, Fernandez DE (2005) Expression of MADS-box genes during the embryonic phase in Arabidopsis. Plant Mol Biol 58:89–107
Litz RE, Raharjo SHT (2005) Sapindaceae. In: Litz RE (ed) Biotechnology of fruit and nut crops. CABI, Wallingford, pp 627–636
Magdalita PM, Laurena AC, Yabut-Perez BM, Mendoza EMT, Villegas VN, Botella JR (2002) Progress in the development of transgenic papaya: transformation of Solo papaya using ACC synthase antisense construct. Acta Horticult 575:171–176
Mathews H, Litz RE, Wilde DH, Merkel S, Wetzstein HY (1992) Stable integration and expression of β-glucuronidase and NPT II genes in mango somatic embryos. In Vitro Cell Dev Biol Plant 28:172–178
Mathews H, Litz RE, Wilde HD, Wetzstein HY (1993) Genetic transformation of mango. Acta Horticult 341:93–97
Matsumoto TK (2006) Genes uniquely expressed in vegetative and potassium chlorate induced floral buds of Dimocarpus longan. Plant Sci 170:500–516
Matzke AJM, Matzke MA (1998) Position effects and epigenetic silencing of plant transgenes. Curr Opin Plant Biol 1:142–148
Meins F (2000) RNA degradation and models for post-transcriptional gene silencing. Plant Mol Biol 43:261–273
Menzel CM (2002) The lychee crop in Asia and the Pacific. FAO, Regional Office for Asia and the Pacific. Bangkok, Thailand
Menzel CM, Huang X, Liu C (2005) Cultivars and plant improvement. In: Menzel CM, Waite GK (eds) Litchi and longan: botany, production and uses. CABI, Wallingford, pp 59–86
Meyerowitz EM, Smyth DR, Bowman JL (1989) Abnormal flowers and pattern formation in floral development. Development 106:209–217
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Nolan T, Hands RE, Bustin SA (2006) Quantification of mRNA using real-time RT-PCR. Nat Protoc 1:1559–1582
Omar AA, Dekkers MGH, Graham JH, Grosser JW (2008) Estimation of transgene copy number in transformed citrus plants by quantitative multiplex real-time PCR. Biotechnol Prog 24:1241–1248
Peirson SN, Butler JN, Foster RG (2003) Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res 31:e73
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Pfaffl MW, Horgan W, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36
Raharjo SHT, Litz RE (2007) Somatic embryogenesis and plant regeneration of litchi (Litchi chinensis Sonn.) from leaves of mature phase trees. Plant Cell Tissue Organ Cult 89:113–119
Raharjo SHT, Witjaksono, Gomez-Lim MA, Padilla G, Litz RE (2008) Recovery of avocado (Persea americana Mill.) plants transformed with the antifungal plant defensin gene pdf1.2. In Vitro Cell Dev Biol Plant 44:254–262
Ribas AF, Kobayashi AK, Pereira LFP, Vieira LGE (2006) Production of herbicide-resistant coffee plants (Coffea canephora P.) via Agrobacterium tumefaciens-mediated transformation. Braz Arch Biol Technol 49:11–19
Riechmann JL, Meyerowitz EM (1997) MADS domain proteins in plant development. Biol Chem 378:1079–1101
Samach A, Kohalmi SE, Motte P, Datla R, Haughn GW (1997) Divergence of function and regulation of class B floral organ identity genes. Plant Cell 9:559–570
Schwarz-Sommer Z, Hue I, Huijser P, Flor PJ, Hansen R, Tetens F, Lönnig W, Saedler H, Sommer H (1992) Characterization of the Antirrhinum floral homeotic MADS-box gene deficiens: evidence for DNA binding and autoregulation of its persistent expression throughout flower development. EMBO J 11:251–263
Shepherd CT, Moran Lauter AN, Scott MP (2009) Determination of transgene copy number by real-time quantitative PCR. Methods Mol Biol 526:129–134
Stam M, de Bruijn R, Kenter S, van der Hoorn RAL, van Blokland R, Mol JNM, Kooter JM (1997) Post-transcriptional silencing of chalcone synthase in Petunia by inverted transgene repeats. Plant J 12:63–82
Subhadrabandhu S, Stern RA (2005) Taxonomy, botany and plant development. In: Menzel CM, Waite GK (eds) Litchi and longan: botany, production and uses. CABI, Wallingford, pp 25–34
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:R0034
Viruel MA, Hormaza JI (2004) Development, characterization and variability analysis of microsatellites in lychee (Litchi chinensis Sonn., Sapindaceae). Theor Appl Genet 108:896–902
Weigel D, Meyerowitz EM (1994) The ABCs of floral homeotic genes. Cell 78:203–209
Wong WS, Li GG, Ning W, Xu ZF, Hsaio WLW, Zhang LY, Li N (2001) Repression of chilling induced ACC accumulation in transgenic citrus by overproduction of antisense 1-aminocyclopropane-1-carboxylate synthase RNA. Plant Sci 161:969–977
Wu Y, Yi G, Zhou B, Zeng J, Huang Y (2007) The advancement of research on litchi and longan germplasm resources in China. Sci Hortic 114:143–150
Yao J-L, Dong YH, Morris B (2001) Parthenocarpic apple fruit production conferred by transposon insertion mutations in a MADS-box transcription factor. Proc Natl Acad Sci U S A 98:1306–1311
Ying ZT, Yu X, Davis MJ (1999) New method for obtaining transgenic papaya plants by Agrobacterium-mediated transformation of somatic embryos. Proc Fla State Hortic Soc 112:201–205
Zhong H-Y, Chen J-W, Li C-Q, Chen L, Wu J-Y, Chen J-Y, Lu W-J, Li J-G (2011) Selection of reliable reference genes for expression studies by reverse transcription quantitative real-time PCR in litchi under different experimental conditions. Plant Cell Rep 30:641–653
Acknowledgment
G. Padilla acknowledges financial support from the Cabildo Insular de La Palma.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: J. Forster
Rights and permissions
About this article
Cite this article
Padilla, G., Pérez, J.A., Perea-Arango, I. et al. Agrobacterium tumefaciens-mediated transformation of 'Brewster' ('Chen Tze') litchi (Litchi chinensis Sonn.) with the PISTILLATA cDNA in antisense. In Vitro Cell.Dev.Biol.-Plant 49, 510–519 (2013). https://doi.org/10.1007/s11627-013-9533-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-013-9533-9