
Abstract
Reverse transcription quantitative real-time PCR (RT-qPCR), a sensitive technique for quantifying gene expression, depends on the stability of the reference gene(s) used for data normalization. Only a few studies on reference genes have been done in fruit trees and none in litchi. In the present study, seven frequently used candidate reference genes, including actin (ACTIN), glyceraldehyde-3-phosphate-dehydrogenase (GADPH), elongation factor 1-alpha (EF-1α), poly ubiquitin enzyme (UBQ), α-tubulin (TUA), β-tubulin (TUB) and RNA polymerase-II transcription factor (RPII), were evaluated for their expression stability in litchi. A total of 78 samples, including different varieties, tissues, organs, developmental stages and treatments, such as NAA, shading and girdling plus defoliation, were addressed in this analysis. Our results showed that GAPDH was the most suitable reference gene among all the tested samples, different organs and NAA treatment. ACTIN was stably expressed in varieties and fruit developmental stages. RPII and UBQ exhibited better expression stability in tissues. EF-1α was the most stable gene in shading and girdling plus defoliation treatments. Moreover, using combination of two genes as reference genes might improve the reliability of gene expression by RT-qPCR in litchi. A better combination was GAPDH + EF-1α or GAPDH + ACTIN for all the examined samples. In addition, the validated reference genes were further relied on to quantify the expression of an interested gene, LcARF13 under different experimental conditions. These results first provide guidelines for reference genes selection under different experimental conditions and also a foundation for more accurate and widespread use of RT-qPCR in litchi.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Litchi (Litchi chinensis Sonn.) is a tropical and subtropical plant native to southern China and one of the major fruit crops in Southeast Asia (Wu 1998; Huang et al. 2005). Litchi fruit is highly attractive in international trade with its high nutritional value and desirable flavor (Yi et al. 2010). In addition, litchi fruits have been used to produce various types of healthy products and foods, e.g., medicinal beverages, drinks or soups (Chyau et al. 2003; Jiang 2000; Jiang and Li 2003). Studies on litchi have become increasingly extensive and important due to the contradictions between its low production and low quality, and between its perishability and high value in the international trade (Munish et al. 2003; Li et al. 2001; Jiang et al. 2004), and gene expression analysis is also being widely employed in these studies (Lu et al. 2006; Wang et al. 2006; Xiang et al. 2007).
Reverse transcription quantitative real-time PCR (RT-qPCR) is verified to be a very powerful and accurate technique used to quantify transcript expression levels for understanding the biological and molecular mechanisms (Bustin 2002; Gachon et al. 2004; Li et al. 2010) and is becoming the first choice for quantitative gene expression (Martin et al. 2008). RT-qPCR has been widely applied in clinical diagnostics (Bustin and Dorudi 1998), tissue-specific gene expression in animals (Terzi et al. 2010) and humans (Zampieri et al. 2010), and gene expression in plants (Lin and Lai 2010). However, discrepancies of RT-qPCR for obtaining reliable results may exist in different samples, such as the quantity of the initial material, the quality of the RNA, the efficiency of cDNA synthesis, primer performance, selection of reference genes and the methods to be used for statistical analysis, are accounted for in obtaining reliable results (Vandesompele et al. 2002; Exposito-Rodriguez et al. 2008; Tong et al. 2009). Therefore, normalization is predicatively required during RT-qPCR analysis to get more accurate results.
The most reliable genes are those constitutively expressed and involved in basic cellular processes, such as protein and sugar metabolism and cell structure (Silveira et al. 2009). Until recently, several housekeeping genes, such as those encoding glyceraldehyde-3-phosphate-dehydrogenase (GAPDH), actins, elongation factor 1-alpha (EF-1α), 18S rRNA and 25S rRNA, had been most frequently evaluated in different species and tissues as they are supposed to be uniform. Nevertheless, many research findings indicate that any so-called uniform control gene is relative. The transcript levels of these genes can vary considerably under different experimental conditions, making them unsuitable for normalization of gene expression (Bustin 2002; Jain et al. 2006; Lovdal and Lillo 2009). Hence, the use of a reference gene as an internal control gene may not be able to identify small discrepancies in gene expression and might even cause a wrong conclusion. It has become obvious that it is necessary to validate the expression stability of a candidate reference gene in each experimental system prior to its use for normalization to obtain accurate and reliable results (Hong et al. 2008).
At the present time, studies on validating the stability of internal reference genes have been performed in some fruit trees, including longan (Lin and Lai 2010), peach (Tong et al. 2009) and grape berry (Reid et al. 2006); however, no such study has been reported for litchi. In the current study, seven commonly used reference genes, including actin (ACTIN), glyceraldehyde-3-phosphate-dehydrogenase (GADPH), elongation factor 1-alpha (EF-1α), poly ubiquitin enzyme (UBQ), α-tubulin (TUA), β-tubulin (TUB) and RNA polymerase-II transcription factor (RPII), were selected to be validated among various samples using two algorithm methods, geNorm (Vandesompele et al. 2002) and NormFinder (Andersen et al. 2004), to find out suitable internal reference gene(s) for litchi RT-qPCR analysis under different experimental conditions.
Materials and methods
Plant materials and treatments
Plant materials were collected in an orchard located at South China Agricultural University, Guangzhou, China in 2009. A total of 78 samples were collected as follows: pericarp samples of different varieties including ‘Hehuadahongli’, ‘Chenzi’, ‘Siliangguo’, ‘Longyanben’, ‘Quanzhouzaohong’, ‘Maquechun’, ‘Guangxigoubei’ and ‘Feizixiao’ were taken at 63 days after anthesis (DAA). Ovaries, fruitlets or pericarp samples at eight different developmental stages of ‘Hehuadahongli’ (female flowers at 7 days before anthesis, fruitlets at 7 and 21 DAA, and pericarp at 35, 49, 63, 77 and 91 DAA) were collected. Young leaf, shoot, fruit and tender root of ‘Hehuadahongli’, and pericarp, aril and seed at 63 DAA, were also sampled.
The 1-naphthylacetic acid (NAA, Sigma-Aldrich Co., USA) treatment was applied to three ‘Heiye’ litchi trees at 30 DAA. Twenty fruit-bearing shoots from each tree were tagged based on the uniform diameter of the shoots. Ten of them were sprayed with 100 mg/L NAA, and the other ten were sprayed with distilled water as control. The pericarp of ten randomly harvested fruit from ten fruit-bearing shoots were sampled 7 days after treatment.
Shading and girdling plus defoliation treatments were carried out to induce carbohydrate starvation stress. For shading treatment, six ‘Nuomici’ trees with uniform vigor and moderate initial crop load were selected. Three of them were randomly selected as untreated controls and the other three were shaded to 15% of available light with neutral density, black-polypropylene shade cloth at 30 DAA. The pericarp of 20 randomly harvested fruit from each tree was sampled 10 days after treatment. The girdling plus defoliation treatment was done on three ‘Kulin’ litchi trees at 25 DAA. Twenty fruit-bearing shoots from each tree were tagged based on the uniform diameter of the shoots. Ten of them were treated by girdling plus defoliation (removing all leaves above girdling), and another ten were untreated controls. The pericarp of ten randomly harvested fruits from ten fruit-bearing shoots was sampled 4 days after treatment.
All samples mentioned above are summarized in Table 1. They were frozen in liquid nitrogen immediately after harvest and stored at −80°C until future use.
RNA isolation, purification and cDNA synthesis
Total RNA was isolated according to the improved hot borate method described by Lu and Jiang (2003). Total RNA from all samples were then treated with DNase I to remove any genomic DNA contamination according to the manufacturer’s instructions (TaKaRa). RNA concentration and purity were evaluated by measuring absorbance at 230, 260 and 280 nm, respectively. Integrity of the RNA was verified by gel electrophoresis on an ethidium bromide-stained 1% agarose-TBE gel and denaturing agarose-MOPS gel. Only the RNA samples with A260/A280 ratios between 1.8 and 2.1 and A260/A230 ratios higher than 2.0, as well as both 28S and 18S ribosomal RNA bands with a density ratio about 2:1 were used for further analysis.
The primary cDNA was synthesized from equal amounts of purified total RNA (2 μg) using the M-MLV first-strand synthesis system according to the manufacturer’s procedure (Promega). All cDNA samples were quantified using absorbance at 260 nm with bio-spectrophotometer to ascertain concentration and diluted to 40 ng/μL as stock solution for further use.
Cloning of part of the selected reference genes
Seven candidate reference genes were selected for investigation to identify the most stably expressed reference gene(s) to be used in RT-qPCR studies; four of them (ACTIN, GAPDH, UBQ and RPII) were cloned by our research group using RT-PCR and RACE-PCR. Degenerate primers were designed using Primer Premier 5.0 software within the conserved region of nucleotide sequences aligned by DNAMAN 6.0 software from numerous plants found on National Center for Biotechnology Information (NCBI) for PCR amplification. Amplicons were sequenced subsequently. To amplify 3′-end fragments, 3′-RACE nested primers were designed according to the sequences of cDNA fragments. 3′-RACE-PCR was performed using 3′-Full RACE Core Set Ver.2.0 Kit (TaKaRa) and then the amplicons were also sequenced. Nucleotide sequences for litchi TUA and TUB were obtained from their PCR products using the quantitative primer pairs of tomato (Exposito-Rodriguez et al. 2008) and sugarcane (Iskandar et al. 2004), respectively. Nucleotide sequences for six reference genes mentioned above were listed in the additional file 1. In addition, nucleotide sequence for litchi EF-1α was obtained from NCBI (DQ471426.1).
Primer design and RT-qPCR analysis
To facilitate the RT-qPCR analysis of all the investigated genes under the same reaction conditions, primer pairs of five genes (EF-1α, ACTIN, GAPDH, UBQ and RPII) were designed using Primer Premier 5.0 software under default parameters, while primers for TUA and TUB were referred to the quantitative primer pairs obtained from tomato (Exposito-Rodriguez et al. 2008) and sugarcane (Iskandar et al. 2004), respectively. All the primer pairs were custom ordered from a commercial supplier (Sangon, Guangzhou). PCR was performed on cDNA to check the specificity of all primers. To confirm the sequences of amplification, PCR products of desired size were analyzed on 1.2% agarose gel and then sequenced. The primers were further confirmed with a melting curve analysis after amplification for the seven reference genes. No further analysis was performed on any primer that showed non-single product or product in no template control (NTC). Standard curve using a dilution series of the mixed cDNA (spanning six orders of magnitude) was made to calculate the gene-specific PCR efficiency and regression coefficient (R 2) for each gene.
RT-qPCR was performed on ABI 7500 Real-Time PCR System and 7500 System Software (Applied Biosystems, USA) using a SYBR Green based PCR assay. Each reaction mixture containing 6 μL of diluted cDNA (10 ng of starting amount for ACTIN, EF-1α, GAPDH and UBQ, and 80 ng for TUA, TUB and RPII), 10 μL of SYBR Green PCR Master Mix (Invitrogen), 250 nM of each primer and 0.1 μL ROX to a final volume of 20 μL was subjected to the following conditions: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 55°C for 30 s and 72°C for 30 s in 96-well optical reaction plates (Bio-Rad, USA). The melting curves were analyzed at 60–95°C after 40 cycles. Each RT-qPCR analysis was performed in triplicate. The genes amplicon characteristics and their primer sequences are listed in Table 2.
Data analysis
Expression levels of the tested reference genes were determined by CT values (Bustin et al. 2009), the number of amplification threshold cycles needed to reach a specific threshold level of detection. For each candidate gene, where the highest relative quantity (the minimum CT value) was set to 1, the other average CT value of each duplicate reaction of sample was converted to relative quantity data. Log-transformed data were then exported into geNorm (version 3.5) (Vandesompele et al. 2002) and NormFinder (Exposito-Rodriguez et al. 2008) to analyze gene expression stability. GeNorm calculates a gene stability M value, defined as the mean pairwise variation for a given gene compared to the remaining tested genes, which relies on the principle that the expression ratio of two ideal reference genes should be identical in all samples, regardless of the experimental conditions and cell types (Huggett et al. 2005; Tong et al. 2009; Zampieri et al. 2010; Lin and Lai 2010). Hence, the lower the M value, the more stably is the gene expressed. GeNorm can also enable the exclusion of the most unstable gene, so as to allow recalculating the M value by setting a cutoff value (Silveira et al. 2009). We chose 1.5 as the cutoff in this study according to Vandesompele et al. (2002). NormFinder, another algorithm for identifying the optimal normalization gene(s) among a set of candidates, uses a solid statistical framework to estimate both intra- and inter-group variation and a separate analysis of the sample subgroups in expression levels are included into the calculation of a gene expression stability value. The lowest stability value will be top ranked according to the analysis (Andersen et al. 2004).
Normalization of an interest gene, LcARF13
ARF gene is a member of the auxin response factors (ARFs) family, which are unique to plants and bind with specificity to auxin response elements (AuxREs) in promoters of primary or early auxin response genes (Guilfoyle et al. 1998). LcARF13 was cloned from litchi pericarp in our laboratory (data not shown) and used as a target gene to demonstrate the usefulness of the validated candidate reference genes in RT-qPCR. Gene expression levels of LcARF13 were quantified under different experimental conditions using the one or two most stable reference genes and the most unstable gene with the lowest or highest M values, respectively, as determined by geNorm in the same RT-qPCR conditions mentioned above. Primer pairs (forward: 5′-AGG GGT TGA ATC CTT AAA TCC AAG-3′ and reverse: 5′-ATT TTC GTG GGA TTA TGT TAT GTC-3′) of LcARF13 were also verified by melting curve analysis and sequencing as described for reference genes.
Results
Verification of quantitative amplicons, primer specificity and gene-specific amplification efficiency
Agarose gel electrophoresis (Fig. 1a) revealed that all primer pairs amplified a single PCR product with the expected size. Specific amplifications were also confirmed by the presence of a single peak in the melting curve obtained after 40 cycles of amplification, while no amplicon was observed in NTC for each selected reference gene (Fig. 1b). Furthermore, sequence analysis of cloned amplicons revealed that all sequenced amplified fragments were identical or nearly identical to the sequences used for primer design (data not shown). The gene-specific PCR amplification efficiency (E) was calculated by the regression coefficient (R 2) of the slope of the standard curve. The PCR amplification efficiency for the seven reference genes varied from 89.426% for EF-1α to 94.347% for TUA, and regression coefficients ranged between 0.983 for TUA and 0.998 for ACTIN, respectively (Table 2).

Specificity of primer pairs for RT-qPCR amplification. a A 1.2% non-denaturing agarose gel electrophoresis showed amplification of a single product of the expected size for each reference gene. b Melt curve showed a single peak for each selected reference gene and no amplicon was observed in no template control (NTC) indicated by arrows
Expression profiling of candidate reference genes
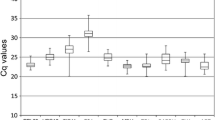
CT values analyzed using the raw expression data across all samples varied within a range from 20.96 to 32.10 among the seven reference genes across all tested samples (Fig. 2). ACTIN, GAPDH, UBQ and EF-1α were highly expressed compared to the other three genes, TUA, TUB and RPII. The least transcriptional level was TUA, with CT values from 23.69 to 32.12. The individual reference gene had different CT value ranges across all studied samples. Variation was lower in ACTIN, GAPDH, UBQ, EF-1α and RPII (below seven cycles), but much higher in TUA and TUB (above eight cycles). No single reference gene had a constant expression level in different litchi samples, suggesting that it was extremely important to choose suitable reference gene(s) to normalize gene expression under given conditions in litchi.

RT-qPCR CT values for seven candidate reference genes. The raw expression data CT values of all tested litchi samples for each reference gene were analyzed by Origin 6.11. The box represents the 25th and 75th percentiles of data. A line across the box is depicted as the median. Whiskers represent the maximum and minimum values
Expression stability of reference genes
GeNorm analysis
Average expression stability (M) value of the seven reference genes in all tested samples calculated by geNorm are presented in Table 3. Among the seven genes used for analysis, not all of the most stable reference genes were identical in different sample series. For instance, the ACTIN gene was most stably expressed (the lowest M values) throughout the different developmental stages as well as in different varieties, while the RPII gene had the highest expression stability in different tissue samples with an M value of 0.972. For the girdling plus defoliation and shading samples, the most stable gene was EF-1α with an M value of 0.822 and 0.236, respectively. GAPDH had the minimum M value in NAA-treated samples and was also the most stable gene in organ samples. When all samples were considered, GAPDH showed most stable expression and might be widely used as a single reference gene for multiple samples, while TUA and TUB showed the largest variations, indicating that these two genes were not suitable for normalization. In addition, it is noteworthy that ACTIN, TUA and TUB in different tissues and TUA in different organs showed an M value higher than the cutoff value of 1.5, indicating less stable expression that was not suitable for the expression study in these samples.
NormFinder analysis
NormFinder (Andersen et al. 2004) is another tool that ranks all reference gene candidates based on intra- and inter-group variations and combines both results into a stability value for each reference gene candidate. Table 4 shows the results of the NormFinder analysis. It was found that there were two common features in the NormFinder output between different sample subgroups and no subgroups: (1) GAPDH, ACTIN, EF-1a and RPII were more stably expressed, indicating lower average expression stability values, and were always classified as the top four stable genes with GAPDH and ACTIN ranked as the top two orders. (2) UBQ, TUB and TUA exhibited unstable expression profiles and had the same rank order. When the seven reference genes were analyzed in different sample series, GAPDH was calculated to be the most stable single gene in fruit development, organs and NAA-treated samples. ACTIN ranked the first in varieties and girdled samples with lower average expression stability values of 0.008 and 0.001, respectively. EF-1a was always located above average except in organs and NAA-treated sample series. TUA, TUB, RPII and UBQ presented in unstable positions more frequently with some exceptions. For example, RPII and UBQ showed more stable profiles in tissue series with the same value of 0.001, and TUB was identified as the more reliable gene in organs and shading series.
Determination of the optimal number and most reliable combination of reference genes
GeNorm analysis
Although some authors agree on using only one single gene as an internal control for normalization, it has been suggested that the use of two or more reference genes for normalization might produce more reliable results (Tricarico et al. 2002; Lin and Lai 2010). GeNorm performs a stepwise calculation of the pairwise variation between sequential normalization factors to determine the optimal number of reference genes required for accurate normalization. V n values were calculated by stepwise inclusion of more reference genes until the (n + 1) gene had no significant contribution to the newly calculated normalization factor. Vandesompele et al. (2002) proposed 0.15 as a cutoff value for the variation below which the inclusion of an additional reference gene is not required. As shown in Fig. 3, the V2/3 values in most experimental sample series except for all samples (A) and organs (D) were lower than the cutoff value (0.15) recommended in the geNorm manual. The V values for all the samples and different organs were only slightly higher than the threshold. Moreover, the inclusion of a third gene did not result in noticeable decrease of their V3/4 from the V2/3 value (from 0.157 to 0.159, and from 0.18 to 0.125, respectively). Therefore, the inclusion of an additional reference gene would not be required, implicating that it was unnecessary to use more than two reference genes in quantitative analysis in these sample series on litchi.

Determination of the optimal number of reference genes. Pairwise variation calculated by geNorm to determine the minimum number of reference genes for accurate normalization in all tested samples (A), different fruit developmental stages (B), different varieties (C), different organs (D), different tissues (E), girdling plus defoliation (F), shading (G) and NAA (H). Arrows indicated the V2/3 value higher than 0.15
GeNorm also establishes a rank order of gene stability via stepwise exclusion of the least stable gene, which allows identifying the remaining couple of genes having the lowest M value (Fig. 4). ACTIN and EF-1α showed the minimum combined M value among the different fruit development stages (B) and varieties (C), as well as shading (G) treatment samples. UBQ and RPII were the most stable reference genes in sample series of different tissues (E) and girdling plus defoliation (F). For all samples (A), different organs (D) and NAA treatment (H) sample series, the best combinations were GAPDH + EF-1α, GAPDH + TUB and TUB + RPII, respectively. It was obvious that all combination of multiple reference genes in each sample series showed a lower M value compared to the single most stable one calculated by genNorm, implying that using two reference genes for normalization would improve the accuracy of the RT-qPCR analysis.

Gene expression stability and ranking of the seven reference genes as calculated by geNorm in all tested samples (a), different fruit developmental stages (b), different varieties (c), different organs (d), different tissues (e), girdling plus defoliation (f), shading (g) and NAA (h). A lower average expression stability M value indicates more stable expression
NormFinder analysis
NormFinder analysis can also estimate the variation between sample groups based on an algorithm of requiring a minimum of two samples per group. In our seven data sets, there were not sufficient samples to make subgroup sets to fully utilize this function. Therefore, only one analysis was carried out for all samples divided into seven subgroups to determine the best combination of two reference genes. For all samples, the best two combination reference genes were GAPDH and ACTIN (Table 4), with a stability value of 0.192 lower than the best single one GAPDH (0.233). These results also indicated that using the two reference genes in combination would be more accurate than using only the most stable gene, which was the same as that indicated by geNorm analysis.
Reference genes validation under different experimental conditions
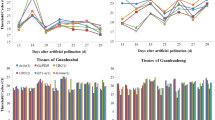
It has been documented that the use of inappropriate references can dramatically change the interpretation of the expression pattern of a given target gene (Gutierrez et al. 2008; Lin and Lai 2010). To demonstrate the usefulness of the validated candidate reference genes in RT-qPCR, the expression patterns of litchi LcARF13 under different experimental conditions, including different fruit development stages, and different organs, tissues and varieties, were analyzed using the one or two most stable reference gene(s), and the most unstable gene for normalization, which had been validated above by geNorm as shown in Table 3 and Fig. 4. The expression level of LcARF13 decreased progressively during litchi fruit development and showed similar change in patterns with slight difference when using ACTIN alone and the combination of ACTIN and EF-1α as reference genes for normalization (Fig. 5a). However, normalization using the least stable reference gene (TUB) led to over-estimation of the LcARF13 expression level during 42–77 DAA (Fig. 5a). Similar results were also observed in that normalization using the least stable reference gene led to over-estimation of the LcARF13 expression level in different organs (Fig. 5b), different varieties (Fig. 5c) and tissues of ‘Hehuadahongli’ (Fig. 5d), while the expression level of LcARF13 showed slight difference when one or two most stable reference gene(s) were used for normalization among these sample series. These results further confirmed the importance of validating reference gene stability to ensure that low precision or misleading results did not occur.

Relative quantification of LcARF13 expression using validated reference genes for normalization under different experimental conditions. a The validated reference gene(s) used as normalization factors were one (ACTIN) or two (ACTIN + EF-1α) most stable reference genes, and the most unstable one (TUB) during fruit development of “Hehuadahongli”. b The validated reference gene(s) used as normalization factors were one (GAPDH) or two (GAPDH + TUB) most stable reference genes, and the most unstable one (TUA) in different organs. c The validated reference gene(s) used as normalization factors were one (ACTIN) or two (ACTIN + EF-1α) most stable reference genes, and the most unstable one (TUA) in different varieties. d The validated reference gene(s) used as normalization factors were one (RPII) or two (RPII + UBQ) most stable reference genes, and the most unstable one (TUB) in different tissues. Each value represent the means of three replicates, and error bars indicate the SE
Discussion
RT-qPCR is widely applied for accurate and sensitive quantification of gene transcript levels, even for those genes for which transcript levels are low. Quantification of gene expression is affected by several factors, such as the quantity of the initial material, the quality of the RNA, the efficiency of cDNA synthesis, primer performance and the methods used for statistical analysis. Thus, the reliability of RT-qPCR data will be greatly improved by inclusion of reference gene (s), the transcription level of which should be invariable in different experimental conditions. The validity of the conclusion depends highly on the reference gene applied. Therefore, it is necessary to validate the expression stability of the reference genes under specific experimental conditions prior to their use for normalization. The present study is the first detailed investigation on the stability of several housekeeping genes, including ACTIN, GAPDH, EF-1α, RPII, TUA, TUB and UBQ, used as internal controls for RT-qPCR studies in litchi. The specificity of the RT-qPCR primer pairs was confirmed by agarose gel electrophoresis (Fig. 1a), melt curve analysis (Fig. 1b) and amplification sequencing (data not shown). The PCR amplification efficiency was estimated by a standard curve. GeNorm has been recently described as one of the best algorithms to validate the most stable reference genes for RT-qPCR analysis (Jain et al. 2006; Gutierrez et al. 2008) and more sensitive to co-regulation as compared to Normfinder. Compared with geNorm, NormFinder can be applied to evaluate the suitability of any normalization gene candidate in any kind of experimental design and should allow more reliable normalization of RT-qPCR data (Exposito-Rodriguez et al. 2008; Hibbeler et al. 2008; Cruz et al. 2009). In addition, NormFinder can also estimate both intra- and inter-group variation of the test samples. Thus, the stability of the seven selected genes was determined using these two methods in the present study. By calculating the stability value and pairwise variation of all selected candidate reference genes, the stability rank and the optional numbers among all kinds of test samples were obtained. Our results indicated that different suitable reference genes should be selected according to the different litchi sample types.
GeNorm
Most of the candidate genes with M value lower than 1.5 in a given sample series were appropriate for normalization under different conditions. ACTIN was the most stable internal control gene in samples of different fruit developmental stages and varieties, which was different from other plant species (Iskandar et al. 2004; Gutierrez et al. 2008; Hong et al. 2008; Li et al. 2010; Artico et al. 2010). RPII and UBQ showed better expression stability in different tissues in litchi, unlike tomato (Exposito-Rodriguez et al. 2008), Brachypodium distachyon (Hong et al. 2008), Arabidopsis (Gutierrez et al. 2008), sugarcane (Iskandar et al. 2004) and cucumber (Wan et al. 2010). For different litchi organs, GAPDH ranked the top and TUB showed the least stable profiles, which was opposite to cotton (Artico et al. 2010). EF-1α and GAPDH were the most stable genes under carbohydrate stress and NAA treatment, respectively. Similar results were also obtained in Brachypodium distachyon (Hong et al. 2008), while contradictory results were observed in cucumber (Wan et al. 2010). All of these results mentioned above indicated that there was no universal reference gene for all sample types.
Increasing evidences have shown that a single reference gene cannot be used to accurately normalize RT-qPCR data and that a combination of multiple reference genes is preferred. Although increasing the number of reference genes for normalization will improve the accuracy of the analysis, as mentioned above, it is expensive due to sample wastage and being time consuming. Therefore, the number of internal controls must be taken into account if the amount of RNA is limited or a large number of samples need to be analyzed (Lin and Lai 2010). In general, the use of two stable reference genes is an effective normalization tactic in most experimental conditions. We also obtained more accurate and reliable normalization compared to the use of a single reference gene (Table 3; Fig. 4). Considering the M value at the same time, it is obvious that all combination of multiple reference genes in each sample series showed a lower M value compared to the single most stable one, implying that using two reference genes for normalization will improve the accuracy of the analysis. Two reference genes (combination of ACTIN + EF-1α for fruit developmental stages, varieties and shading treatment; UBQ + RPII for tissues and girdling plus defoliation; TUB + RPII for NAA treatment; GAPDH + TUB for organs; and GAPDH + EF-1α for examining all tested samples) would be recommended for use in RT-qPCR analysis of litchi according to our study. Different results were also found in many other studies. For instance, UBQ + EF-1α normalized well in longan fruit (Lin and Lai 2010) and rice seed development (Jain et al. 2006). Tong et al. (2009) found that TEF and UBQ was the most suitable combination for all analyzed samples in peach. ACTIN and EF-1α was the best combination for normalizing in Gossypium hirsutum organs (Artico et al. 2010). Yang et al. (2010) suggested that ACTIN + UBQ normalized well in Salvia miltiorrhiza tissues. These results further emphasized that it was important to select appropriate reference gene(s) for accurate gene expression, because reference genes were regulated differently in different plant species and different treatment conditions. Thus, it is suggested that the number of reference genes needed to be employed depends on the considerations of a researcher’s purpose. That is, if one requires to show a rough expression mode of genes, one reference gene may be sufficient if it was confirmed as a stably expressed gene. However, if the researcher hopes to compare the expression among different samples or to obtain the accurate expression level, more reference genes (depends on the geNorm threshold of 0.15) must be taken.
NormFinder
NormFinder algorithm ranked GAPDH as the most stably expressed gene in all tested litchi samples when the samples were assembled into one main group or divided into seven subgroups, unlike in Brachypodium distachyon (Hong et al. 2008), in which the top ranked reference gene was ACTIN. In addition, GAPDH and ACTIN were always ranked at the top two orders in other single sample series. However, in peach (Tong et al. 2009), the top two most reliable genes were TUB and RPII. The rank order of reference genes for different litchi fruit developmental stages was also out of accord with that of logan (Lin and Lai 2010) and tomato (Exposito-Rodriguez et al. 2008). In plant organs, TUB ranked at the top in our study, but at the bottom in Gossypium hirsutum (Artico et al. 2010). UBQ was the most stable gene in tissue samples, but the most variable one in NAA treatment, which was in uniformity partly with peach (Tong et al. 2009) and Brachypodium distachyon (Hong et al. 2008), and totally different from cucumber (Wan et al. 2010) and chicory (Maroufi et al. 2010). Lin and Lai (2010) found that EF-1α was the most reliable gene and ACTIN was the variable one in development samples in longan, while in our study ACTIN revealed a lower value than EF-1α. In summary, most of our results in litchi show great differences from the published results in other plants, implying the importance of validating the reference gene prior to target gene expression. NormFinder could determine the best combination of reference gene only when subgroups were set within a group. In this study, a combination of GAPDH and ACTIN was the best as suggested by NormFinder, while geNorm analysis indicated that GAPDH and EF-1α was the best combination.
Comparison of geNorm and NormFinder
The results obtained by geNorm for four sample series (for all samples, organs, varieties and NAA treatment) were identical with those obtained by NormFinder (Tables 3, 4). When seven subgroups were set, the rank order of the top four genes (GAPDH, ACTIN, EF-1α and RPII) was changed. For the other sample series, there were no changes in the top four more reliable genes and the three unreliable genes except in shading treatment samples, although their rank orders showed some differences. It suggested that normalization using the genes defined by geNorm or NormFinder can produce almost the same result in litchi plant. In chicory tissue (Maroufi et al. 2010) and cucumber for hormone treatment (Wan et al. 2010) analysis, the stability order of candidate genes ranked by geNorm and NormFinder was absolutely the same. But there were totally different results using these two software for longan (Lin and Lai 2010) and cucumber in low- and high temperature treatment (Wan et al. 2010). However, no matter how the order changed, the most unstable gene almost always remained the same (Exposito-Rodriguez et al. 2008; Lin and Lai 2010; Artico et al. 2010; Wan et al. 2010). These differences in the two methods are expected because the programs are based on distinct statistical algorithms. Therefore, both methods could provide a stability value for each gene and select the best reference gene for normalization.
Conclusions
To the best of our knowledge, this is the first attempt to select a set of commonly used candidate reference genes in litchi for the normalization of gene expression analysis using RT-qPCR. Our results suggested that different suitable reference genes should be selected according to the different litchi sample types. ACTIN, GAPDH and EF-1α would be more reliable for normalizing the expression levels in most sample series. Using a combination of two genes as reference genes might improve the reliability of gene expression by RT-qPCR in litchi. The best combination used to normalize the expression profiles was ACTIN + EF-1α for development stages, varieties and shading treatment; UBQ + RPII for tissues and girdling plus defoliation; TUB + RPII for NAA treatment; and GAPDH + EF-1α for all tested samples. The expression analysis of LcARF13 under different experimental conditions emphasized the importance of validating reference genes to achieve accurate RT-qPCR analysis. These results provide a foundation for the more accurate and widespread use of RT-qPCR in the analysis of gene expression in litchi.
References
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Artico S, Nardeli SM, Brilhante O, Grossi-de-Sa MF, Alves-Ferreira M (2010) Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol 10:49
Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. Mol Endocrinol 29:23–29
Bustin SA, Dorudi S (1998) Molecular assessment of tumour stage and disease recurrence using PCR-based assays. Mol Med Today 4:389–396
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Chyau CC, Ko PT, Chang CH, Mau JL (2003) Free and glycosidically bound aroma compounds in lychee (Litchi chinensis Sonn.). Food Chem 80:387–392
Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros LMG, Romano E, Grossi-de-Sa M, Vaslin M, Alves-Ferreira M (2009) Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT-PCR. Mol Breed 23:607–616
Exposito-Rodriguez M, Borges AA, Borges-Perez A, Perez JA (2008) Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol 8:131
Gachon C, Mingam A, Charrier B (2004) Real-time PCR: what relevance to plant studies? J Exp Bot 55:1445–1454
Guilfoyle TJ, Ulmasov T, Hagen G (1998) The ARF family of transcription factors and their role in plant hormone-responsive transcription. Cell Mol Life Sci 54:619–627
Gutierrez L, Mauriat M, Guénin S, Pelloux J, Lefebvre JF, Louvet R, Rusterucci C, Moritz T, Guerineau F, Bellini C, Van Wuytswinkel O (2008) The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol 6:609–618
Hibbeler S, Scharsack JP, Becker S (2008) Housekeeping genes for quantitative expression studies in the three-spined stickleback Gasterosteus aculeatus. BMC Mol Biol 9:18
Hong SY, Seo PJ, Yang MS, Xiang F, Park CM (2008) Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol 8:112
Huang XM, Suranant S, Mitra SK, Ben-Arie Stern R (2005) Origin, history, production and processing. In: Menzel C, Waite GK (eds) Litchi and Longan: botany, production and uses. CABI Publishing, Oxford
Huggett J, Dheda K, Bustin S, Zumla A (2005) Real-time RT-PCR normalization strategies and considerations. Genes Immun 6:279–284
Iskandar HM, Simpson RS, Casu RE, Bonnett GD, Maclean DJ, Manners JM (2004) Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Mol Biol Rep 22:325–337
Jain M, Nijhawan A, Tyagi AK, Khurana JP (2006) Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem Biophy Res Commun 345:646–651
Jiang YM (2000) Role of anthocyanins, polyphenol oxidase and phenols in lychee pericarp browning. Sci Food Agric 80:305–310
Jiang YM, Li YB (2003) Effects of low-temperature acclimation on browning of litchi fruit in relation to shelf life. J Hortic Sci Biotech 78:437–440
Jiang YM, Duan XW, Joyce D, Zhang ZQ, Li JR (2004) Advances in understanding of enzymatic browning in harvested litchi fruit. Food Chem 88:443–446
Li JG, Huang HB, Gao FF, Huang XM, Wang HC (2001) An overview of litchi fruit cracking. Acta Hort 558:205–208
Li QF, Sun SSM, Yuan DY, Yu HX, Gu MH, Liu QQ (2010) Validation of candidate reference genes for the accurate normalization of real-time quantitative RT-PCR data in rice during seed development. Plant Mol Biol Rep 28:49–57
Lin YL, Lai ZX (2010) Reference gene selection for qPCR analysis during somatic embryogenesis in longan tree. Plant Sci 178:359–365
Lovdal T, Lillo C (2009) Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold, and light stress. Anal Biochem 387:238–242
Lu WJ, Jiang YM (2003) Clone sequence analysis of two enlarged gene in Litchi fruit. Sci Agr Sin 6:1525–1529 (in Chinese with English abstract)
Lu WJ, Wang Y, Jiang YM, Li JG, Liu H, Duan XW, Song L (2006) Differential expression of litchi XET genes in relation to fruit growth. Plant Physiol Biochem 44:707–713
Maroufi A, Van Bockstaele E, De Loose M (2010) Validation of reference genes for gene expression analysis in chicory (Cichorium intybus) using quantitative real-time PCR. BMC Mol Biol 11:15
Martin RC, Hollenbeck VG, Dombrowski JE (2008) Evaluation of reference genes for quantitative RT-PCR in Lolium perenne. Crop Sci 48:1881–1887
Munish A, Kahlon PS, Mahajan BVC (2003) Effect of exogenous application of growth regulators on fruit drop, cracking and quality of litchi (Litchi chinensis Sonn.) Cv. Dehradun. Agr Sci Dig 23:191–194
Reid KE, Olsson N, Schlosser J, Peng F, Lund ST (2006) An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol 6:27
Silveira ED, Alves-Ferreira M, Guimaraes LA, da Silva FR, Carneiro VTD (2009) Selection of reference genes for quantitative real-time PCR expression studies in the apomictic and sexual grass Brachiaria brizantha. BMC Plant Biol 9:84
Terzi V, Morcia C, Spini M, Tudisco R, Cutrignelli MI, Infascelli F, Stanca AM, Faccioli P (2010) Identification and validation of reference genes for gene expression studies in water buffalo. Animal 4:853–860
Tong ZG, Gao ZH, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol 10:71
Tricarico C, Pinzani P, Bianchi S, Paglierani M, Distante V, Pazzagli M, Bustin SA, Orlando C (2002) Quantitative real-time reverse transcription polymerase chain reaction: normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal Biochem 309:293–300
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, DePaepe A, Speleman F (2002) Accurate normalisation of realtime quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034.1–research0034.11
Wan HJ, Zhao ZG, Qian CT, Sui Y, Malik AA, Chen JF (2010) Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal Biochem 399:257–261
Wang Y, Lu WJ, Li JG, Jiang YM (2006) Differential expression of two expansin genes in developing fruit of cracking-susceptible and -resistant litchi cultivars. J Am Soc Hortic Sci 131:118–121
Wu SX (1998) Encyclopaedia of China fruits: Litchi. China Forestry Press, Beijing
Xiang X, Zhang YS, Huang SZ, Ou Lx, Qiu YP, Li ZQ, Chen JZ (2007) Application of suppressive subtraction hybridization in the study of embryo abortion related to gene expression in litchi (Litchi chinensis Sonn.). Acta Hortic 763:83–89
Yang YF, Hou S, Cui GH, Chen SL, Wei JH, Huang LQ (2010) Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol Biol Rep 7:507–513
Yi C, Jiang YM, Shi J, Qu HX, Xue S, Duan XW, Shi JY, Prasad NK (2010) ATP-regulation of antioxidant properties and phenolics in litchi fruit during browning and pathogen infection process. Food Chem 118:42–47
Zampieri M, Ciccarone F, Guastafierro T, Bacalini MG, Calabrese R, Moreno M, Villanueva A, Reale M, Chevanne A, Bürkle P, Caiafa (2010) Validation of suitable internal control genes for expression studies in aging. Mech Age D 131:89–95
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (30871694), Doctoral Fund of Ministry of Education of China (200805640003) and Modern Agro-industry Technology Research System (grant No. nycytx-32-03).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Lakshmanan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhong, HY., Chen, JW., Li, CQ. et al. Selection of reliable reference genes for expression studies by reverse transcription quantitative real-time PCR in litchi under different experimental conditions. Plant Cell Rep 30, 641–653 (2011). https://doi.org/10.1007/s00299-010-0992-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-010-0992-8