
Abstract
Two synthetic cannabinoid analogues were detected using high-performance liquid chromatography (HPLC)–diode array detector, and gas chromatography–time-of-flight-mass spectrometry during the inspection of illegal products in an airmail package. The analogues were separated by semi-preparative HPLC, and their structures were determined by performing liquid chromatography–high-resolution-mass spectrometry, infrared analysis, and nuclear magnetic resonance spectroscopy. Compound 1 was MDMB(N)-Bz-F, which has been reported previously. Compound 2 was elucidated as adamantan-1-yl 1-pentyl-1H-indazole-3-carboxylate (APINAC), in which the amide group of APINACA was replaced with an ester group. Because there has been no chemical or pharmacological data about this compound until now, this is the first report of its detection in illegal products.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The recent rise in the abuse of new synthetic cannabinoids has become a worldwide issue. Synthetic cannabinoids act on cannabinoid CB1 (central type) and/or CB2 (peripheral type) receptors to produce psychoactive effects [1–5]. Synthetic cannabinoids have been classified into several groups based on their structural motif by the National Forensic Services: naphthylindoles, phenylacetylinoles, benzoylindoles, cyclopropylindoles, aminocarbonylindazoles, adamantylindoles, adamantylindazoles, quinolinylindoles, CP-47,497 homologs, and cyclopropylthiazoles [6–9]. Although the use of cannabinoids for restrictive medicinal purposes is permitted, their recreational use is illegal in most countries. Several adverse effects due to abuse of cannabinoids include hallucination, psychosis, hypertension, tachycardia, agitation, vomiting, seizures, and convulsions [10–13].
Illegal herbal products and dietary supplements containing synthetic cannabinoids are available for purchase via the Internet and through international postal services [14–17]. Since unapproved synthetic cannabinoids have not been assessed pharmacologically and toxicologically, the products that contain these ingredients could pose a significant risk to public health [18, 19]. In spite of great efforts and the cooperation of authorities in most countries to protect public health from the toxic effects of synthetic cannabinoids, new synthetic cannabinoids have been detected in a variety type of products [20–24]. Since first synthetic cannabinoids was identified at the end of 2008, more than 130 synthetic cannabinoids have been reported to the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) [25].
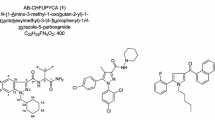
We identified two synthetic cannabinoids in illegal products in an airmail package. These compounds were isolated and structurally elucidated by means of high-performance liquid chromatography (HPLC), gas chromatography–time-of-flight-mass spectrometry (GC–TOF-MS), liquid chromatography–high-resolution-mass spectrometry (LC–HR-MS), infrared (IR) analysis, and nuclear magnetic resonance (NMR) spectroscopy. Compound 1 in herbal-type products was MDMB(N)-Bz-F with an indazole-3-carboxamide group that has been reported recently [26]. Compound 2 in a powder-type product was determined as adamantan-1-yl 1-pentyl-1H-indazole-3-carboxylate (APINAC), an analogue of APINACA that is a type of adamantylindazole synthetic cannabinoid. The APINACA was modified to APINAC by replacement of an amide group with an ester group. In this article, we described two synthetic cannabinoids in illegal products in detail. The structures of the compounds are shown in Fig. 1.

Chemical structures of ADB-FUBINACA, APINACA, compound 1 and compound 2
Materials and methods
Chemicals and reagents
Methanol and acetonitrile were obtained from Merck KGaA (Darmastadt, Germany); sodium 1-hexanesulfonate and formic acid from Sigma-Aldrich (St. Louis, MO, USA); deuterated chloroform and methyl alcohol (99.9 %, isotopic) from Alfa Aesar (Ward Hill, MA, USA); deuterated dimethyl sulfoxide (99.8 %) from AMAR chemicals (Dottingen, Switzerland). Deionized water (18.2 MΩ cm) was generated using a Milli-Q water system (Millipore, Billerica, MA, USA). All solvents and reagents were of HPLC grade.
Sample preparation
About 1 mg of powder-type or 0.2 g of herbal product was dissolved in 10 mL methanol, respectively. The mixture was sonicated for 10 min and filtered through a polytetraflouroethylene filter (0.2 μm). The resulting residue was diluted appropriately with methanol for instrumental analyses.
Analytical conditions
High-performance liquid chromatography–diode array detector (DAD) analysis was performed using an Agilent 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA) with a DAD. A ZORBAX Eclipse XDB-C18 (250 mm × 4.6 mm i.d., particle size 5 μm) column (Agilent Technologies) was used with the column chamber set at 40 °C. The injection volume was 10 μL, and the flow rate was 1.0 mL/min. Gradient elution was applied using 0.5 mM aqueous sodium 1-hexanesulfonate containing 0.1 % phosphoric acid at pH 2.3 (A) and 95 % acetonitrile (B), and was programmed as follows: 0–2 min (A: 80 %, B: 20 %), 2–10 min (A: 80–20 %, B: 20–80 %), 10–15 min (A: 20–0 %, B: 80–100 %), 15–28 min (A: 0 %, B: 100 %), 28–28.1 min (A: 0–80 %, B: 100–20 %), 28.1–30 min (A: 80 %, B: 20). The ultraviolet (UV) spectra were recorded from 210 to 400 nm, while the chromatograms were acquired at 210 nm.
GC–TOF-MS analysis was carried out using an Agilent 7890A system (Agilent Technologies) with a LECO Pegasus HT TOF-MS (LECO Corp., St. Joseph, MI, USA). The column was Agilent HP 5MS (30 m × 0.25 mm i.d., 0. 25 μm; Agilent Technologies) and the extracts were injected in split mode (10:1). Helium at a constant flow rate of 1.0 mL/min was used as the carrier gas and the injection volume was 1 μL. The initial column temperature (100 °C) was increased to 200 °C at a rate 15 °C/min, ramped up to 300 °C at 10 °C/min, and held at 300 °C for 8 min. The GC injector was set at 250 °C and the transfer line was maintained at 280 °C. The mass conditions were as follows: ionization mode, electron ionization; electron energy 70 eV; source temperature, 230 °C; scan range, 50–500 amu; selected ion monitoring dwell time, 100 ms.
The LC–HR-MS experiments were conducted on an Agilent 1200 series HPLC system connected to an Agilent 6530 Accurate-Mass Quadrupole (QTOF-MS) spectrometer equipped with an electrospray ionization Jet Stream Technology source (Agilent Technologies). The chromatographic separation was performed on an X-Bridge C18 column (150 mm × 2.1 mm i.d, particle size 3.5 μm) at 35 °C. The mobile phase was composed of 0.1 % formic acid in both distilled water (A) and acetonitrile (B). The elution profile was as follows: 0–3 min (A: 80 %, B: 20 %), 3–13 min (A: 80–40 %, B: 20–60 %), 13–16 min (A: 40 %, B: 60 %), 16–18 min (A: 40–0 %, B: 60–100 %), 18–21 min (A: 0 %, B: 100 %), 21–22 min (A: 0–80 %, B: 100–20 %), 22–25 min (A: 80 %, B: 20 %). The injected sample volume was 3 μL, and the flow rate was 0.3 mL/min. The MS conditions were as follows: positive ion mode; gas temperature, 350 °C; drying gas flow, 8 L/min; nebulizer pressure, 35 psig; sheath gas temperature, 350 °C; sheath gas flow, 11 mL/min; capillary voltage, 3.5 kV; nozzle voltage, 1.0 kV; fragment voltage, 0.12 kV. After acquiring a full scan MS spectrum, tandem mass spectrometry (MS/MS) experiment was performed.
An Agilent 1200 series semi-preparative HPLC system with a DAD was used for isolation of analogues. A ZORBAX Eclipse XDB C18 (250 mm × 9.4. mm i.d, particle size 5 μm) column was used, and the column chamber was maintained at 40 °C. The flow rate was 3.0 mL/min, and the injection volume was 100 μL, with detection at 210 nm. Gradient elution was applied using distilled water (A) and methanol (B). The mixed isocratic and gradient elution profile was as follows: 0–2 min (A: 80 %, B: 20 %), 2–20 min (A: 70–0 %, B: 30–100 %), 20–23 min (A: 0 %, B: 100 %), 23–25 min (A: 0–80 %, B: 100–20 %), 25–28 min (A: 80 %, B: 20 %). The fractions were collected and concentrated in vacuo.
The analogues isolated from powder- and herbal-type samples were dissolved in CDCl3 and (CD3)2CO, respectively. All NMR experiments [1H, 13C, distortionless enhancement by polarization transfer (DEPT), heteronuclear single quantum coherence (HSQC), 1H-1H correlation spectroscopy (COSY), heteronuclear multiple bond correlation (HMBC), and nuclear Overhauser effect spectroscopy (NOESY)] were performed using a Bruker Avance II 600 MHz FT-NMR spectrometer (Bruker, Ettlingen, Germany). IR spectra were recorded over the spectral range 4000–400 cm−1 using a Perkin Elmer Frontier Fourier transform (FT)-IR spectrometer (Perkin Elmer, Waltham, MA, USA).
Results and discussion
Identification of compound 1
The UV spectrum of compound 1 at 15.5 min was maximally absorbed at 210 and 302 nm (Fig. 2). For GC–TOF-MS, this compound at 17.1 min produced ions at m/z 109 and 253 (Fig. 3). We predicted that compound 1 was ADB-FUBINACA analogue in comparison to the previous report [27]. The accurate mass spectrum of compound 1 was measured by LC–QTOF-MS (Fig. 4). The ion peak was detected at m/z 398.1864 and suggested as the protonated molecular formula of C22H25FN3O3 + (calcd. 398.1874) (Table 1). The product ions appeared at m/z 109.0449 (C7H6F+), 253.0772 (C15H10FN2O+), and 338.1667 (C20H21FN3O+) indicated the presence of indazole-3-carboxamide moiety and fluorotropylium ion like ADB-FUBINACA.
By NMR analyses in CD3OD, we confirmed that compound 1 is MDMB-Bz-F reported as an ADB-FUBINCA analogue [26]. Most of 1H- and 13C spectral data were similar to those of ADB-FUBINACA (Fig. 5). For example, the 1H NMR spectra were identified the presence of t-butyl group (δ H 1.08), benzylic proton (δ H 5.71) and eight protons of the aryl group. The two carbonyl groups were proved by the peaks at δ C 173.1 and 164.1 while the doublet peak at δ C 163.9, (d, J C−F = 243 Hz) was assigned as a peak for C-5b due to coupling between aryl carbon and fluoride. The oxygenated methyl group was indicated by the peaks at δ H 3.76 and δ C 52.4, which suggested that amide group of ADB-FUBINACA was substituted by methyl ester. When compound 1 was dissolved in DMSO-d 6, its NMR spectral data were in good agreement with the reported data (Table 2) [26].

a High-performance liquid chromatography chromatograms at 210 nm and ultraviolet spectra (190–400 nm) for compound 1 and b compound 2

a Gas chromatography–time-of-flight- mass spectrometry chromatograms of compounds 1 and 2 in the sample solution, and b mass spectra of the compounds

a Liquid chromatography–quadrupole time-of-flight-mass spectrometry (LC–QTOF-MS) spectra in the single-stage mode for compounds 1 and 2 with parent ions and LC–QTOF-MS/MS spectra of b protonated compound 1 with a parent ion at m/z 398.1864, of c protonated compound 2 with a parent ion at m/z 367.2375, and of d sodium adducted compound 2 with a parent ion at m/z 389.2226

a 1H nuclear magnetic resonance (NMR) and b 13C NMR (600 MHz, CD3OD) spectra of the compound 1
Identification of compound 2
Compound 2 was detected on the HPLC at 22.5 min and GC–TOF-MS at 18.5 min, as depicted in Figs. 2 and 3. Not only the UV spectrum had two absorption maxima at 210 and 303 nm but also its GC–TOF-MS spectra showed distinguishable two peaks at m/z 135 and 215, which were quite similar to those reported for APINACA [8]. To identify the quasi-molecular ion(s) of the compound, LC–QTOF-MS experiments were carried out under several sets of conditions (Fig. 4). Ions at m/z 367.2375 and 389.2226 were identified as quasi-molecular ions corresponding to the molecular formula C23H30N2O2 + (calcd. 367.2380) and C23H30N2NaO2 + (calcd. 389.2199), respectively. Further fragmentation of the quasi-molecular ions by LC–QTOF-MS/MS produced product ions at m/z 135.1169 and 215.1167 that were also detected in the MS2 spectra of APINACA [9]. Given that the compound 2 is an APINACA analogue, it possessed both an adamantine group and an indazole moiety, as shown in Fig. 6. The ions at m/z 135.1169 corresponded to an adamantine cation and the ion at m/z 215.1167 was predicted as an indazole carbonium ion. The ion at m/z 255.1087 was considered a sodium salt of an indazolecarboxylic acid. The mass spectral data were consistent with an ester analogue of APINACA. To complete the structural determination of the compound, we conducted NMR and IR analyses. As summarized in Table 3, 30 proton and 17 carbon signals were recorded in the NMR spectra in CDCl3. The 1H and 13C NMR spectral data of the compound strongly resembled those of APINACA [24]. The chemical shift values of all the proton and carbon signals, except for two, were slightly different from those of APINACA when the spectra of both compounds were compared. However, the NH signal (δ H 6.81, APINACA) was not observed, and one carbon signal (C-1‴, δ C 52.2, APINACA) was shifted downfield largely (δ C 81.8) in the 1H and 13C NMR spectra of the compound (Fig. 7). These observations further confirmed that the amide group of APINACA was replaced with an ester group. Interpretation of 1H-1H COSY, HSQC, DEPT, and HMBC spectra also indicated that the compound was structurally identical to APINACA except for an “NH” group (supplementary material figure). The existence of an ester group in the compound was further confirmed by the strong IR absorption band at 1718 cm−1 (Fig. 8). Based on all the above data and careful analysis, the structure of the compound 2 was elucidated as “adamantan-1-yl 1-pentyl-1H-indazole-3-carboxylate (APINAC)”.

Proposed fragmentation process for the quasi-molecular ions [M + H]+ at m/z 367.24, and [M+Na]+ at m/z 389.22 of compound 2

a 1H NMR (600 MHz, CDCl3) and b 13C NMR (150 MHz, CDCl3) spectra of the compound 2

Infrared spectrum of compound 2
Conclusions
A new synthetic cannabinoid was detected by HPLC–DAD and GC–TOF-MS in illegal products, which was seized during the custom inspection of an airmail parcel. Compound 1 was confirmed as MDMB(N)-Bz-F and compound 2 was determined as an analogue of APINACA by combination of LC–HR-MS, IR, and NMR analyses. The amide group of APINACA was modified to an ester group in this analogue, which was thus named APINAC. Synthetic cannabinoids are abused worldwide, and new ones continue to be generated by modification of existing compounds to avoid being detected by authorities’ inspection. Since unsupervised and addictive use of synthetic cannabinoids may cause social and health problems, this new analogue should be included on the list for the inspection of illegal products.
References
Huffman JW (2009) Cannabimimetic indoles, pyrroles, and indenes: structure-activity relationships and receptor interactions. In: Reggio PH (ed) The cannabinoid receptors. Humana Press, New York, pp 49–94
Benford DM, Caplan JP (2011) Psychiatric sequelae of spice, K2 and synthetic cannabinoid receptor agonists. Psychosomatics 52:295
Every-Palmer S (2010) Warning: legal synthetic cannabinoid-receptor agonists such as JWH-018 may precipitate psychosis in vulnerable individuals. Addiction 105:1859–1860
Gandhi AS, Zhu M, Pang S, Wohlfarth A, Scheidweiler KB, Liu HF, Huestis MA (2013) First characterization of AKB-48 metabolism, a novel synthetic cannabinoid, using human hepatocytes and high-resolution mass spectrometry. J AAPS 15:1091–1098
Spaderna M, Addy PH, D’Souza DC (2013) Spicing things up: synthetic cannabinoids. Psychopharmacology 228:525–540
Uchiyama N, Kawamura M, Kikura-Hanajiri R, Goda Y (2013) URB-754: a new class of designer drug and 12 synthetic cannabinoids detected in illegal products. Forensic Sci Int 227:21–32
Chung H, Choi H, Heo S, Kim E, Lee J (2014) Synthetic cannabinoids abused in South Korea: drug identifications by the 123 National Forensic Service from 2009 to June 2013. Forensic Toxicol 32:82–88
Uchiyama N, Kawamura M, Kikura-Hanajiri R, Goda Y (2012) Identification of two new-type synthetic cannabinoids, N-(1-adamantyl)-1-pentyl-1H-indole-3-carboxamide (APICA) and N-(1-adamantyl)-1-pentyl-1H-indazole-3-carboxamide (APINACA), and detection of five synthetic cannabinoids, AM-1220, AM-2233, AM-1241, CB-13 (CRA-13), and AM-1248, as designer drugs in illegal products. Forensic Toxicol 30:114–125
Vikingsson S, Josefsson M, Gre´en H (2015) Identification of AKB-48 and 5F-AKB-48 metabolites in authentic human urine samples using human liver microsomes and time of flight mass spectrometry. J Anal Toxicol 39:426–435
Thornton SL, Wood C, Friesen MW, Gerona RR (2013) Synthetic cannabinoid use associated with acute kidney injury. Clin Toxicol 51:189–190
Seely KA, Lapoint J, Moran JH, Fattore L (2012) Spice drugs are more than harmless herbal blends: a review of the pharmacology and toxicology of synthetic cannabinoids. Prog Neuropsychopharmacol Biol Psychiatry 29:234–243
Ashton AC (1999) Adverse effects of cannabis and cannabinoids. Br J Anaesth 83:637–649
Kronstrand R, Roman M, Andersson M, Eklund A (2013) Toxicological findings of synthetic cannabinoids in recreational users. J Anal Toxicol 37:534–541
Vardakou I, Pistos C, Spiliopoulou C (2010) Spice drugs as a new trend: mode of action, identification and legislation. Toxicol Lett 197:157–162
UNODC (2012) World drug report 2012. United Nations Office on Drugs and Crime, Vienna
Choi JY, Hea S, Yoo GJ, Park SK, Yoon CY, Baek SY (2015) Development and validation of an LC-MS/MS method for the simultaneous analysis of 28 specific narcotic adulterants used in dietary supplements. Food Addit Contam A 32:1029–1039
Penn HJ, Langman LJ, Unold D, Shields J, Nichols JH (2011) Detection of synthetic cannabinoids in herbal incense products. Clin Biochem 44:1163–1165
Harris CR, Brown A (2013) Synthetic cannabinoid intoxication: a case series and review. J Emerg Med 44:360–366
Grotenhermen F (2007) The toxicology of cannabis and cannabis prohibition. Chem Biodivers 4:1744–1769
Seely KA, Patton AL, Moran CL, Womack ML, Prather PL, Fantegrossi WE, Radominska-Pandya A, Endres GW, Channell KB, Smith NH, McCain KR, James LP, Moran JH (2013) Forensic investigation of K2, spice, and ‘‘bath salt’’ commercial preparations: a 3-year study of new designer drug products containing synthetic cannabinoid, stimulant, and hallucinogenic compounds. Forensic Sci Int 233:416–422
Kikura-Hanajiri R, Uchiyama N, Kawamura M, Goda Y (2013) Changes in the prevalence of synthetic cannabinoids and cathinone derivatives in Japan until early 2012. Forensic Toxicol 31:44–53
Namera A, Kawamura M, Nakamoto A, Saito T, Nagao M (2013) Comprehensive review of the detection methods for synthetic cannabinoids and cathinones. Forensic Toxicol 33:175–194
Uchiyama N, Kikura-Hanajiri R, Kawahara N, Haishima Y, Goda Y (2009) Identification of a cannabinoid analog as a new type of designer drug in a herbal product. Chem Pharm Bull 57:439–441
Amato J, Iaccarino N, Pagano B, Compagnone V, Di Rosa F, Peluso G, Novellino E, Randazzo A (2014) NMR assignment of N-(1-adamantyl)-1-pentyl-1H-indazole-3-carboxamide seized as herbal incense for the first time in Italy. J Forensic Sci Criminol 2(1):103. doi:10.15744/2348-9804.1.403
EMCDDA (2015) New psychoactive substances in Europe: an update from the EU early warning system. European Monitoring Centre for Drugs and Drug Addiction. http://www.emcdda.europa.eu/publications/2015/new-psychoactive-substances. Accessed 8 June 2016
Shevyrin V, Melkozerov V, Nevero A, Eltsov O, Shafran Y, Morzherin Y, Lebedv AT (2015) Identification and analytical characteristics of synthetic caanabinoids with an indazole-3-carboxamide structure bearing a N-1-methoxycarbonylalkyl group. Anal Bioanal Chem 407:3601–3615
Uchiyama N, Matsuda S, Kawamura M, Kikura-Hanajiri R, Goda Y (2013) Two new-type cannabimimetic quinolinyl carboxylates, QUPIC and QUCHIC, two new cannabimimetic carboxamide derivatives, ADB-FUBINACA and ADBICA, and five synthetic cannabinoids detected with a thiophene derivative α-PVT and an opioid receptor agonist AH-7921 identified in illegal products. Forensic Toxicol 31:223–240
Acknowledgments
This research was supported by Research Grants (15181MFDS521 and 15182MFDS523) from the Ministry of Food and Drug Safety (MFDS) in Korea. We thank the Central Laboratory of Kangwon National University for providing us with technical assistance on the spectroscopic experiments.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
J. H. Lee and H. N. Park contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, J.H., Park, H.N., Leem, TS. et al. Identification of new synthetic cannabinoid analogue APINAC (adamantan-1-yl 1-pentyl-1H-indazole-3-carboxylate) with other synthetic cannabinoid MDMB(N)-Bz-F in illegal products. Forensic Toxicol 35, 45–55 (2017). https://doi.org/10.1007/s11419-016-0331-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-016-0331-z