
Abstract
Neurodegenerative diseases and brain tumours represent important health challenges due to their severe nature and debilitating consequences that require substantial medical care. Interestingly, these conditions share common physiological characteristics, namely increased glutamate, and adenosine transmission, which are often associated with cellular dysregulation and damage. Guanosine, an endogenous nucleoside, is safe and exerts neuroprotective effects in preclinical models of excitotoxicity, along with cytotoxic effects on tumour cells. However, the lack of well-defined mechanisms of action for guanosine hinders a comprehensive understanding of its physiological effects. In fact, the absence of specific receptors for guanosine impedes the development of structure–activity research programs to develop guanosine derivatives for therapeutic purposes. Alternatively, given its apparent interaction with the adenosinergic system, it is plausible that guanosine exerts its neuroprotective and anti-tumorigenic effects by modulating adenosine transmission through undisclosed mechanisms involving adenosine receptors, transporters, and purinergic metabolism. Here, several potential molecular mechanisms behind the protective actions of guanosine will be discussed. First, we explore its potential interaction with adenosine receptors (A1R and A2AR), including the A1R-A2AR heteromer. In addition, we consider the impact of guanosine on extracellular adenosine levels and the role of guanine-based purine-converting enzymes. Collectively, the diverse cellular functions of guanosine as neuroprotective and antiproliferative agent suggest a multimodal and complementary mechanism of action.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ageing of the population is a worldwide phenomenon that increases the risk factor for neurodegenerative diseases, mental disorders, and tumours that affect the central nervous system (CNS) [1, 2]. These illnesses, including Alzheimer's, Parkinson's, Huntington's disease (i.e., AD, PD and HD, respectively), and gliomas, can be hereditary or sporadic and could be triggered by environmental factors. Additionally, it should be mentioned that the occurrence of mental disorders is markedly higher in people diagnosed with neurodegenerative diseases [3]. The worldwide count of people with AD dementia, prodromal AD, and preclinical AD has been estimated at 32, 69, and 315 million, respectively, which collectively comprises 416 million across the continuum of AD, or 22% of the population aged 50 years and older [4]. On the other hand, approximately 19.3 million new cancer cases (18.1 million excluding non-melanoma skin cancer) and nearly 10 million cancer-related deaths (9.9 million excluding non-melanoma skin cancer) occurred worldwide in 2020 [5]. Therefore, the combination of cancer and neurodegenerative diseases accounts for the highest mortality rates. Intriguingly, some recent observational studies have reported the appearance of an inverse correlation between cancer and neurodegenerative diseases [6, 7]. Consequently, further exploration of the mechanistic basis of this inverse relationship appears to be of vital importance in improving the design and development of innovative therapies for these devastating diseases.
The pathogenesis of neurodegenerative diseases involves several key factors, such as protein aggregation, oxidative stress, neuroinflammation, and marked imbalance in glutamatergic neurotransmission [8]. The amino acid glutamate is the main excitatory neurotransmitter of the mammalian CNS, mediating the processes of intercellular communication, plasticity, cell growth, and differentiation. Thus, glutamate is behind important brain functions such as learning and memory, emotion and motivation, and locomotor activity [9]. Significantly, increased glutamate levels within the synaptic cleft give rise to excitotoxicity, a phenomenon driven by the excessive activation of ionotropic glutamate receptors (iGluR), such as N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, along with G protein-coupled metabotropic glutamate receptors (mGluR). This excessive activation of glutamate receptors leads to ionic influx, increased reactive oxygen species (ROS), mitochondrial dysfunction, and calcium overload. These processes promote alterations in oxidative metabolism and mitochondrial dynamics, ultimately contributing to neurodegenerative [10] and neuropsychiatric [11] disorders. Consequently, understanding the endogenous mechanisms used by the CNS to counteract excitotoxicity is crucial. Here, we focus on the role of guanine-based purines (GBPs), including guanosine, as endogenous neuroprotective and antiproliferative agents.
Adenosine signalling in the brain: Beyond receptors
Adenosine, an adenine-based purine, plays a pivotal role in various physiological processes. Its significant functions include a negative chronotropic impact on the heart, vasodilatation of the coronary arteries, and involvement in ischemia [12]. These initial observations not only shed light on the various effects of adenosine but also laid the foundation for the purinergic signalling field by establishing its connection to specific adenosine receptors. Interestingly, within the brain, adenosine serves as a neuromodulator, actively participating in pivotal processes such as synaptic plasticity (implicated in learning and memory), the budding of nervous processes, apoptosis, regulation of the sleep–wake cycle, and its function as a neuroprotective agent against detrimental stimuli [13]. Importantly, these effects are mediate by adenosine receptors (ARs) on the cell surface. Extracellular levels of adenosine are regulated by ectoenzymes that metabolise nucleotides and nucleosides, and by concentrative and equilibrative nucleoside transporters (CNTs and ENT, respectively), as part of the adenosinergic transmission system [13, 14].
The neuroprotective properties of adenosine are related to its ability to modulate the glutamatergic system [15]. Consequently, impaired adenosinergic transmission has been associated with several pathological conditions such as pain, migraine, epilepsy, stroke, drug addiction, neurodegeneration, and brain cancer [16, 17]. ARs belong to the G protein-coupled receptor (GPCR) superfamily and are divided into four subtypes: A1R, A2AR, A2BR, and A3R [18]. A1Rs, which are ubiquitously expressed within the CNS, are coupled with inhibitory G proteins (Gi/o), leading to a decrease in adenylate cyclase (AC) activity and a reduction in intracellular levels of cyclic AMP (cAMP). Furthermore, A1R can be coupled with the Gq protein, triggering phospholipase C (PLC) activation, and producing inositol-triphosphate (IP3), thus promoting intracellular calcium release from the rough endoplasmic reticulum. A2ARs, predominantly expressed primarily in the striatum, nucleus accumbens, hippocampus, and cerebral cortex, are coupled with the Gs protein, leading to increased levels of cAMP. A2BRs, which are poorly expressed in the brain, are also coupled to the Gs protein. Finally, A3Rs, mainly distributed within the periphery and with moderate expression in the cerebellum and hippocampus, are coupled with the Gi/o and Gq proteins [19,20,21].
The central effects of adenosine are predominantly mediated by A1Rs and A2ARs. [22]. The activation of presynaptic A1Rs leads to a reduction in synaptic transmission, achieved by decreasing the probability of neurotransmitter release. Consequently, A1R activity has been shown to be instrumental in providing neuroprotection against excitotoxic conditions, such as ischemia and epilepsy [23]. In this context, A1Rs play a crucial role in the preconditioning mechanism, actively promoting the defence of the brain against damage [24, 25]. On the contrary, A2AR is known as a synaptic transmission facilitatory receptor. Consequently, blocking A2AR has shown efficacy in promoting neuroprotection in various models of epilepsy, depression, AD, and PD [26]. Overall, the balance between A1R and A2AR activation is responsible for promoting the neuroprotective effect of adenosine in the CNS.
The existence of direct molecular interactions among adenosine receptors, which lead to the formation of macromolecular complexes known as oligomers, constitutes a mechanism that serves as a superior molecular device that allows fine-tuning control of adenosinergic transmission and its associated signalling processes. Precisely, the formation of functional A1R-A2AR heteromers in the brain supports adenosine-mediated control of glutamate release at the striatal glutamatergic terminals [27, 28]. Therefore, activation of A2AR within the A1R-A2AR heteromer reduces A1R affinity for adenosine-based ligands, while interaction of A1R with A2AR decreases the constitutive activity of the latest. This mutually negative allosteric modulation of adenosine receptors defines the function of A1R-A2AR heteromers. Consequently, low levels of adenosine in the striatum prevent glutamate release through the activation of A1Rs, while high levels promote glutamate release by A2AR signalling [29]. Furthermore, additional studies demonstrated the existence of homomers and heteromers of A1R and A2AR modulating other neurotransmitter systems, including dopamine and endocannabinoid signalling [30].
In essence, the existence of GPCR-effector macromolecular membrane assemblies containing GPCR effector (GEMMA) that house GPCR oligomers, G proteins, plasma membrane effector molecules and other associated transmembrane proteins will ground the functional interaction between different neurotransmitter systems [31]. Consequently, these GEMMAs would provide new opportunities to pharmacologically modulate different neurotransmitter systems in a multimodal manner. This contention is reinforced by the idea that the formation of GPCR oligomers within the GEMMA enables both canonical (i.e., G protein-based) and noncanonical (i.e., Allosteric) crosstalk between these receptors. Accordingly, purinergic GPCRs oligomerization has become a valuable source for identifying novel molecular targets and developing more selective ligands for modulating purinergic signalling in health and disease.
Guanine-based purinergic system
Initially known for their metabolic functions as essential components of the nucleic acid structure and key contributors to energy homeostasis, GBPs have also gained recognition for their signalling properties. Neuronal and glial cells release guanine nucleotides (GTP, GDP, and GMP) along with the nucleoside guanosine under physiological and pathological conditions [32]. Interestingly, GTP is co-stored with neurotransmitters in synaptic vesicles [33] through an electrochemical gradient-dependent transport system similar to other neurotransmitters [34]. The kinetics of the uptake of GTP into synaptic vesicles is comparable to that observed for ATP [35]. Thus, a role for GTP as a co-transmitter has been suggested [36], which also involves the neuromodulatory effect of guanosine generated after its hydrolysis by extracellular purinergic enzymes. GBP signalling involves modulation of monomeric and heterotrimeric G-protein activity (GTP binding proteins) [37, 38]. Interestingly, within the CNS, GBPs exhibit extracellular signalling properties, exerting an antagonistic-like effect over iGluRs, kainate (KAR), NMDAR, and AMPAR [39,40,41,42,43]. In addition, guanine derivatives have demonstrated the ability to reduce mGluR signalling by modulating its cAMP accumulation [44,45,46]. Indeed, several studies have shown that GBPs reduce glutamate-induced toxicity and metabolic alterations in vitro [47, 48] as well as in in vivo neurodegeneration models [49,50,51]. Overall, these findings reinforce the GBP-mediated extracellular effects as paracrine signalling molecules, complementing the effects of adenine-based counterparts [43].
Intracellular and extracellular levels of guanine derivatives, such as their adenine derivatives, are regulated by the activity of soluble cytosolic and membrane-bound (ecto) enzymes. These enzymes play a pivotal role in converting nucleotides and nucleosides into their respective nucleobases. Ecto-nucleotidases triphosphatases (i.e., ecto-NTPases) encompass various enzymes, including ecto-ATPase, responsible for the hydrolysis of ATP and GTP to ADP and GDP, respectively. Furthermore, apyrase or ecto-ATP-diphosphohydrolase (i.e., ecto-NTPDase) hydrolyses either ATP/GTP or ADP/GDP in AMP/GMP [52]. Finally, ecto-5'-nucleotidase (i.e., ecto-5’-NT) hydrolyses AMP/GMP to adenosine/guanosine nucleosides [53,54,55]. Interestingly, in cultured astrocytes, inhibition of ecto-5'-NT activity significantly reduced extracellular guanosine accumulation, suggesting that extracellular guanosine, like adenosine, was derived primarily from extracellular hydrolysis of guanine nucleotides, as expected [43]. In particular, these purinergic enzymes have the potential to be released into the cerebrospinal fluid by the choroid plexus, endothelial cells, and even microglial cells, thus exerting an important function in pathophysiological conditions [56,57,58]. Certainly, the enzyme that participates in the nucleotide salvage pathway, purine nucleoside phosphorylase (PNP), could be released by astrocytes into extracellular medium [59]. Thus, the hydrolysis of guanosine to guanine and adenosine to adenine also in the extracellular medium opens the possibility of producing more purinergic mediators in the CNS. Overall, after brain injury, the released nucleotides will undergo hydrolysis, leading to the formation of their respective nucleosides and nucleobases, which in turn will play protective or even restorative roles. It is important to mention that in pathological conditions such as hypoxia/ischemia, there is an elevation in the extracellular concentration of guanine purines compared to adenine derivatives [60, 61]. According to this contention, guanine has been shown to have beneficial effects on learning and memory in animal models [62, 63].
Astrocytes play a crucial role in various processes associated with brain damage, including isolating the affected area and participating in lesion repair [64]. In cultured cortical astrocytes, guanosine and guanine are actively reuptaken, likely through facilitated transport mechanisms [65, 66]. Equilibrative nucleoside transporters (ENTs) operate bidirectionally according to the concentration gradient, while the concentrative nucleoside transporters (CNTs) allow nucleoside influx coupled to the Na+ transmembrane gradient [67]. These nucleoside transporters are crucial components of the purine salvage pathway, particularly significant in the CNS, which has limited capacity for de novo purine synthesis [68].
Neuroprotective and neurotrophic effects of guanosine
Guanosine triggers intercellular signalling on neural cells promoting neuroprotection, neuromodulation and trophic effects [43, 69, 70]. The neuroprotective role of guanosine has been demonstrated in neurotoxicity models and in neurological and neurodegenerative diseases models.
In vitro models of brain diseases are applied to unravel the mechanism of action of guanosine. In in vitro models of brain ischemia guanosine presents neuroprotective effects by preventing glutamate excitotoxicity, oxidative stress, and disruption of mitochondrial polarity, and ATP production [71,72,73,74,75,76,77,78,79,80,81,82]. An important activity that has been attributed to guanosine is its ability to re-establish changes in glutamate transport associated with glutamatergic excitotoxicity. In hippocampal slices, and in astrocytes in culture, guanosine recovered the decrease in glutamate uptake induced by in vitro ischemia and prevented the increase of glutamate release [78, 80, 83, 84]. Since glutamate uptake by astrocytes is the most important mechanism for clearance of this neurotransmitter at synapses, the modulation caused by guanosine can be considered a relevant process for the regulation of glutamatergic transmission, contributing to the protection of neuronal cells against glutamate-induced excitotoxicity [36].
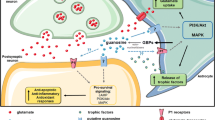
In SH-SY5Y cells submitted to oxidative stress, guanosine promoted a protective effect through the induction of heme-oxygenase-1, an important enzyme in antioxidant cell defence [85]. Heme-oxygenase-1 is also involved in guanosine-induced protection against oxidative stress and increased pro-inflammatory cytokines due to mitochondrial respiratory chain inhibition in C6 astroglial cells [86]. In addition, guanosine reduces inflammation and oxidative damage induced by incubation with bacterial lipopolysaccharides (LPS) in hippocampal astrocyte culture, decreasing the levels of tumour necrosis factor α (TNF-α) and nuclear factor κB (NF-κB) by induction of heme-oxygenase-1 [87], as already demonstrated in in vitro ischemia [78]. The modulation of these antioxidant and anti-inflammatory mechanisms by guanosine involves the participation of the phospho-inositol-3-kinase (PI3K) signalling pathway, a cellular survival pathway [85, 88, 89]. In addition, the effect of guanosine in restoring excess glutamate uptake is also dependent on the intracellular signalling pathway modulated by PI3K/Akt (protein kinase B) [83], and by mitogen-activated protein kinase (MAPK) ERK1/2 (kinase regulated by extracellular signal 1/2), and protein kinase C (PKC) [80], suggesting an intricate activation profile of cell survival pathways (Fig. 1).

Summary of the mechanisms related to the neuroprotective effects of guanosine uncovered through in vitro models of brain diseases. Guanosine (GUO) presents neuroprotective effects through a mechanism involving adenosine A1R and A2AR receptors, with a possible interaction with the A1R/A2AR heteromer. The functional interaction of GUO with the K+-channel (BK) has also been demonstrated. GUO neuroprotective effect increases glutamate (Glu) uptake, preventing oxidative stress and ROS production, and increasing mitochondrial ETS activity, and ATP levels, contributing to neuronal protection against glutamate-induced excitotoxicity. GUO induces an important enzyme in antioxidant cell defence, HO-1, the activity of which prevents inflammatory and NF-κB signalling. The antioxidant and anti-inflammatory actions of GUO depend on PI-3 K signalling pathway, and GUO effect on glutamate uptake is dependent on PI3K/Akt, MAPK/ERK1/2 and PKC pathways. GUO also promotes the covalent conjugation of SUMO protein to target proteins, a post-translational modification related to neuroprotection. A1R/A2AR: adenosine receptors heteromer; BK: high-conductance Ca+2-dependent K+-channel; EAAT: excitatory aminoacid transporters; ETS: mitochondrial electron transport system; GUO: guanosine; Glu: glutamate; HO-1: Heme-oxygenase-1; MAPK/ERK1/2: mitogen-activated protein kinase/kinase regulated by extracellular signal 1/2; NF-κB: Nuclear factor κB; PI3K/Akt: phospho-inositol-3-kinase/protein kinase B; PKC: protein kinase C; ROS: reactive oxygen species; SUMO: Small Ubiquitin-like Modifier. This figure was created using BioRender.com
Moreover, we recently demonstrated that guanosine promotes the covalent conjugation of SUMO (Small Ubiquitin-like MOdifier) protein to lysin residues of target proteins [90], a post-translational modification related to neuroprotection. Guanosine increases global SUMOylation (SUMO2/3 conjugation) in astrocytes and neurons maintained in physiological conditions in culture [91]. Additionally, these effect of guanosine of increasing SUMOylation was also observed in vivo in the hippocampus of adult and aged mice subject to guanosine treatment [92].
Regarding trophic effects, guanosine promotes an increase in the number of granular cerebellar neurons in co-culture with astrocytes [88], by improving the neuronal adhesion in cultures treated with guanosine [93]. Concerning guanosine effects on cultured stem cells in vitro, it promotes the proliferation of hippocampal dentate gyrus stem cells obtained from adult mice, and the differentiation to a neuronal phenotype [94].
In vivo assessment of neurological and neurodegenerative diseases models also confirmed the neuroprotective effects of guanosine. The first studies showed guanosine presents an anticonvulsant effect in mice, preventing seizures and neurotoxicity induced by substances that overstimulate the glutamatergic system [49, 51, 95, 96]. The neuroprotective effect was also evidenced in animal models of brain hypoxia or ischemia [97,98,99,100], and in animal models of traumatic brain injury [101, 102]. Still, the protective effect has been observed in animal models of Parkinson's disease, where guanosine reverses parkinsonian motor impairments and reduces dyskinesia induced by treatment for Parkinon´s [103,104,105,106,107]. In Alzheimer's disease rodent models, guanosine prevents behavioural and neurochemical alterations, as anhedonic-like behaviour and spatial memory impairment, and glutamate transport unbalance, oxidative stress and hippocampal damage caused by amyloid-beta peptide [108, 109]. Furthermore, in vivo guanosine treatment induces neurogenesis in the hippocampal dentate gyrus of adult animals [94].
Behavioural effects of guanosine in healthy animals have also been reported: guanosine has antinociceptive effect [110,111,112], anxiolytic [113, 114] and antidepressant-like effects in mice [89, 115, 116]. Additionally, the antidepressant effect is accompanied by neuroprotection against glutamate neurotoxicity in the hippocampus and cerebral cortex of murine [117].
Despite all this evidence of neuroprotection and trophic effects, and modulation of intracellular signalling pathways, the molecular targets of these effects are not fully elucidated, and guanosine is still considered an orphan ligand with no specific receptor [70, 118].
Guanosine effects in tumour proliferation
Purinergic signalling is often altered in human cancer [119, 120] and the key mediators of the purinergic enzymatic cascade have been extensively investigated to identify potential "conductive biomarkers" for diagnostic, prognostic, and therapeutic monitoring of tumour progression.
Guanosine binding to GPCRs or uptake via nucleoside transporters followed by interaction with signalling pathways involved in cell proliferation, differentiation, or apoptosis, was also reported in cancer settings (Fig. 2a). Recent findings highlighted the importance of GBPs and its converting enzymes in tumour proliferation and in radio- and chemo-therapy resistance. The formation of GTP through reactions catalysed by enzymes of the salvage pathway, namely PNP and hypoxanthine phosphoribosyltransferase (HGPRT), correlates with the arrest of cancer growth, while de novo GTP biosynthesis dependent on phosphoribosyl pyrophosphate (PRPP) is responsible for increased cancer cell viability (Fig. 2b). It is noteworthy that tumour cells compared with non-proliferating cells exhibit a metabolic reprogramming with greater de novo nucleotide biosynthesis ensuring rapid DNA and RNA synthesis to fuel their growth [121].

a. Schematic representation of the proliferative and anti-proliferative effects of guanosine in cancer: Guanosine can enter cells via NTs or bind to GPCRs (A1R, A2AR). The proliferative effect is attributed to radiotherapy resistance following DSBs repair in DNA (left panel), while the cytotoxic effect is associated with cell cycle arrest in S-phase, induction of cancer cell apoptosis and differentiation of cancer stem cells leading to cell senescence and tumour arrest (right panel). b. The de novo and salvage pathways for nucleotide biosynthesis are shown. Cancer cells rely on IMPDH-dependent GTP formation to meet higher energy demand and boost their growth (de novo pathway) whereas non-proliferating cells prefer to generate GTP by recycling GUA and HYPO (salvage pathways). DSBs: double-strand breaks; GPCRs: G protein-coupled receptors; GUA: guanine; GUO: guanosine; HYPO: hypoxanthine; IMPDH: inosine monophosphate dehydrogenase; INO: inosine; NTs: nucleoside transporters; PNP: purine nucleoside phosphorylase; PRPP: phosphoribosyl pyrophosphate; RT: radiotherapy. This figure was created using BioRender.com
In cancer, nucleotide biosynthesis is controlled by several upstream master transcriptional regulators, such as myelocytomatosis (Myc) signalling, which activates GTP synthesis through inosine monophosphate dehydrogenase (IMPDH) [122,123,124], or PI3K-E2F1 that promotes E2F1-dependent retinoblastoma protein phosphorylation, up-regulation of MAPK/RAS signalling and cAMP/cGMP production [125]. IMPDH is the rate-limiting enzyme that catalyses the conversion of inosine monophosphate (IMP) to xanthosine monophosphate (XMP) and is responsible for de novo purine biosynthesis and the maintenance of the guanine nucleotide pool [54]. IMPDH increases in several malignancies including glioblastoma [126], renal carcinoma [127] and small cell lung cancer (SCLC) [128]. Inhibitors of IMPDH have already been investigated in numerous clinical trials in tumours [129, 130]. Combination therapy encompassing IMPDH inhibitor mycophenolate mofetil (MMF, 120 mg/kg twice a day) and PI3K inhibitor (PI-103, 10 mg/kg daily) strongly reduced purine biosynthesis and tumour growth in patients with hepatocellular carcinoma (HCC) characterized by elevated expression of IMPDH and high guanosine levels. Knockdown of IMPDH in HCC and treatment with MMF reduced cancer cell proliferation in vitro and mitigated tumour burden in vivo [125]. In a subset of SCLC cell lines, Myc-stimulated IMPDH promoted de novo GTP biosynthesis. Cell treatment with IMPDH inhibitor, mycophenolic acid (MPA), for 24 h depleted GTP levels and suppressed ribosome biogenesis, a fundamental pathway for cancer cell progression. Guanosine supplementation for 48 h overcame inhibition of de novo biosynthesis, restored the GTP pool through the purine salvage pathway, and recovered ribosome synthesis [131]. According to these findings, MPA inhibited in a concentration and time- dependent manner the proliferation of normal MCF-12A and tumour MCF-7 breast cell lines, with an IC50 of 0.38 ± 0.01 µM and 1.43 ± 0.13 µM, respectively [132]. The authors observed a time- and dose-dependent cytotoxic effect of guanosine in MCF-7 breast cancer cells but not in normal MCF12-A cells, thus exploiting the different metabolic signature of MCF7 over normal MCF12A cells. However, the molecular mechanisms underlying the antiproliferative effect of guanosine have yet to be elucidated, although the authors hypothesized an excessive generation of guanosine-derived GTP affecting cell proliferation [132].
However, GTP may also have an antiproliferative effect after exogenous administration and rapid conversion to the metabolite guanosine. As an example, GTP has been proposed as an antiproliferative and differentiation promoting agent in acute myeloid leukemia, given its ability to induce terminal differentiation of cancer stem cells causing senescence and tumour arrest [133, 134]. Differentiating agents such as retinoic acid, bryostatin and all-trans-retinoic acid, have been investigated in myeloid leukemia or myelodysplastic syndromes as a therapeutic strategy to generate populations of non-tumorigenic cancer cell progeny (135, 136). In the human chronic myelogenous leukemia cell line (K562), GTP treatment (50–200 µM) up to 6 days suppressed tumour growth and DNA synthesis by inducing S-phase cell cycle arrest and erythroid differentiation, revealed by increased expression of glycophorine A, an erythroid marker. It is noteworthy that GTP did not affect the proliferation rate of human normal peripheral lymphocytes. The concentration- and time-dependent inhibitory effect of GTP involved guanosine formation and uptake, as the use of heat-inactivated medium inhibiting purine-converting enzymes or the addition of adenosine competing for guanosine uptake would reduce the GTP effect. Similar results were obtained after cell exposure to GDP, GMP, or guanosine 100 µM [137]. Accordingly, in a model of acute myeloid leukemia (AML), the growth suppression induced by supplementation with GTP occurred following its conversion to the corresponding nucleoside guanosine. Specifically, guanosine treatment caused a time- and dose-dependent inhibition of cell growth (IC50 30–50 µM) and increased the expression of myeloid differentiation markers such as CD11b and CD14. In the U937 orthotopic xenograft model, guanosine delayed leukemia progression through the formation of GTP dependent on PNP and HPRT. Finally, direct delivery of GTP by using a fluorescently labelled GTP analogue, MANT-GTP, also induced differentiation. Interestingly, the effect of exogenous guanosine was weaker compared to that of GTP-derived guanosine [138]. Nevertheless, increased levels of purine guanylates are also one of the metabolic features of glioblastoma (GBM) cells resistant to radiotherapy, as they correlated with the ability of repairing DNA double-stranded breaks (DSBs). Supplementation with exogenous guanosine and other nucleosides (80–240 µM) was able to confer radioprotection to GBM cells by decreasing RT-induced DSBs. Treatment with MPA 10 μM and the prodrug MMF impaired DNA repair and radiosensitized RT-resistant GBM cells and primary patient-derived GBM neurospheres. In the same study, using an orthotopic GBM PDX model, combination therapy with RT and MMF prolonged mouse survival and revealed high intracranial drug permeability and efficacy [139].
However, the notion of exogenous guanine derivatives as cytotoxic agents is reported in CNS tumours as well. In gliomas, it was shown that the nucleoside guanosine increased apoptosis and reduced tumour cell migration, the cytotoxic effect being independent of glutamate transport while relying on interaction with the adenosine A1R and A2AR receptors [140]. GBMs are characterised by a decrease in glutamate uptake from the extracellular space and an increase in glutamate release in the peritumoral spaces, causing glutamatergic excitotoxicity and promoting a path of invasion and tumour growth [141,142,143]. Furthermore, in the U87 glioblastoma cell line, the nucleobase guanine revealed the highest potency compared to guanosine and GMP, and the cytotoxic effect depended on the up-regulation of HGPRT expression which would provoke an imbalance of nucleotide pool thus causing an S-phase cell cycle arrest [144]. In a more recent study, the authors observed that guanine elicited a growth inhibition in U87 cells, this effect being mediated by activation of the GPR23 receptor or lysophosphatidic acid (LPA) 4 receptor. In line with the previous study, HGPRT-negative melanoma cell lines expressed low levels of GPR23 and were less sensitive to the antiproliferative effect of guanine [145].
Aligning with previous work, cell exposure to guanine, guanosine, deoxyguanosine, and GMP reduced the viability of embryonic kidney cells (HEK293) (IC50 = 40 − 90 μM) and acute T lymphoblastic leukemia cells (Jurkat E6-1) (IC50 = 25 μM). The molecular mechanisms underpinning this effect were the alteration of nucleotide metabolism, the induction of DNA damage response, and the downregulation of RNA transport-related proteins, all resulting in cell apoptosis and death, caused by cell cycle arrest in the S phase. Supplementation with adenosine and cytidine partially re-established a balance of nucleotides/nucleosides and recovered cell viability, thus depicting adenine and guanine nucleosides competing for the same enzymes [146]. In another study, Jurkat cells exposed to guanosine showed a reduction in ATP levels and a dramatic accumulation of GTP derived from salvage pathway activation. The de novo synthesis of ATP was reduced by guanosine-derived GMP with a feedback regulation mechanism. Cell death by necrosis or apoptosis was provoked by ATP depletion and accumulation of GTP responsible for inhibition of deoxyribonucleotide synthesis. Once again, healthy peripheral blood mononuclear cells (PBMC) viability was not affected by guanosine treatment thus conveying selectivity towards tumour cells [147].
An additional molecular mechanism attributed to GBPs to stop cancer growth is the activation of NO-cGMP pathway, which is involved in chemotaxis, cell-survival/apoptosis, as well as oxidative stress and angiogenesis [148,149,150].
In summary, the antiproliferative effect of guanosine was mediated by the nucleoside uptake and intracellular metabolism achieved by a series of enzymes of the purine salvage pathway. The principal molecular mechanisms include: (1) unbalance of the nucleotide/nucleoside pool through competition with other nucleosides for the same enzymes that determine adenine nucleotides and deoxynucleotides depletion, which would arrest DNA synthesis. This hypothesis was corroborated by the addition of adenosine or other nucleosides thwarting guanosine effects; (2) differentiation of cancer stem cells leading to tumour arrest and (3) crosstalk with NO-cGMP-mediated anti-proliferative and pro-apoptotic signalling pathways. The dichotomous nature of guanine derivatives that have pro- and anti-tumour effects may depend on factors such as the experimental setting (i.e. compound concentration, time of exposure, cancer genotype) and the different tissue expression of purinergic enzymes rapidly converting one metabolite to another. Additionally, the lack of a specific receptor for GBPs poses limits regarding the pharmacological potential of these compounds and their clinical application. Nonetheless, it is intriguing to speculate that guanosine effects in cancer may involve the activation of adenosine receptors whose role in cancer is widely documented [17, 151].
Guanosine’s mechanism of action: Therapeutic opportunities
GBPs show efficacy in counteracting glutamate excitotoxicity, underscoring a significant interplay between GBPs and glutamatergic transmission. GUO nucleotides (i.e., GTP, GDP, and GMP) have shown the ability to reduce NMDA-induced neurotoxicity in cultured hippocampal and cortical neurons [152], and GMP exhibits neuroprotective effects against NMDA-induced apoptosis in hippocampal slices [48, 153, 154]. Neuroprotection of GBPs has been suggested to be associated with increased astrocytic glutamate uptake, thus avoiding excitotoxic glutamate accumulation [155]. Although the effect of GUO on glutamate transport has been described [78, 156], no evidence of direct interaction with glutamate transporters was demonstrated. Instead, GUO has been shown to increase glutamate transporter-1 (GLT-1) cell surface targeting in astrocytes, after ischemic damage, through a mechanism involving adenosine A1R and A2AR [80]. In general, since this neuroprotective mechanism is largely accepted, the development of GBP derivatives for the treatment of disorders linked to glutamate excitotoxicity has been postulated [157], despite the lack of a well-defined mechanism of action. Interestingly, a functional connection has been established between GBP-mediated neuroprotection and potassium (K+) channel activity, adding an interesting layer to the understanding of the neuroprotective effects of GBPs and suggesting a potential link to K+ channel modulation in the therapeutic action of GBPs.
Astrocytes treated with GUO showed increased activity and expression of voltage-rectifying K+ channels [158]. Using different selective pharmacological blockers of K+ channels, a functional interaction of GUO with the high-conductance Ca+2-dependent K+-channel (maxiK or BK) has been demonstrated. Therefore, blocking BK channels with charybdotoxin precluded the beneficial effects of GUO on cell viability in an in vitro ischemia model [73, 83]. Importantly, this functional interaction of GUO is selective for BK channel, since inhibition of the small conductance Ca2+-activated K+ channels with apamine and the ATP-sensitive K + channels with glibenclamide does not interfere with the neuroprotective effect of GUO [83]. BK channels are activated by increased intracellular calcium levels and belong to a large family of channels that have various physiological functions, such as neurotransmitter release, cell excitability, vascular reactivity, and smooth muscle tone [159]. In the CNS, BK channels are considered an "emergency brake," limiting the frequency of neuronal firing and contributing to the repolarization of the action potential [160]. BK channels expressed presynaptically where they regulate the termination of neurotransmitter release upon the activation by the influx of calcium into the synaptic terminal [147]. Therefore, presynaptic BK channel activators play a promising neuroprotective role in situations of cerebral ischemia [161]. Blocking BK channels in SH-SY5Y neuroblastoma cells prevented the neuroprotective effects of GUO (i.e., reduction of oxidative stress and cell viability), an effect also observed upon blockade of adenosine A1R or A2AR [85]. Interestingly, we recently reported that GUO and ADO evoked K+-outward currents in SH-SY5Y cells. Both nucleosides were observed to influence repolarization time by reducing its duration, indicating a neuroprotective profile [162]. However, this effect was specific to repolarization and did not affect depolarization time. In particular, when GUO and ADO were administered simultaneously, there was a synergistic potentiation of their effects on delayed rectifying K+-outward currents, supporting the hypothesis of a functional interaction between these nucleosides [162]. In the absence of a direct interaction with the BK channel, it is suggested that GUO probably modulates channel function through an interaction with a GPCR. Thus, the existence of putative, yet-to-be discovered, GUO GPCR has been suggested. Alternatively, GUO can operate through the on-demand recruitment of ARs, either directly or by modulating the availability of these receptors to endogenous ADO.
Initial investigations that provided evidence of a potential interaction site for GUO in the cell membrane were conducted using isolated membranes from rat brain. These studies suggested the presence of a selective binding site for GUO in the rat brain [163, 164]. Thus, a single binding site for [3H]-GUO was demonstrated (KD = 95.4 ± 11.9 nM; Bmax = 0.57 0.03 pmol/mg of 0.57 ± 0.03 pmol/mg protein). This binding site was selective for guanosine as other purines (GMP, GDP, adenosine, ATP, hypoxanthine, and xanthine) and naturally occurring purines (i.e., caffeine, theophylline) were unable to significantly displace 50 nM [3H]-GUO. However, a partial, but not significant, displacement was observed for xhantosine (50%), GTP (30%), ATP (20%) and nitrobenzylthioinosine (NBTI; 30%), a blocker of the equilibrative nucleoside transport (ENT) system [163, 164]. In general, this GUO binding site is different from the well-characterised receptors for adenosine and other purines. Importantly, when the brain membranes were incubated with pertussis toxin (PTX), shown to ADP-ribosylate Gi or Go proteins (inhibitory Gi/o protein family), the specific binding of guanosine was reduced by 45% [163]. However, in slices from the rat brain cortex, GUO induced a dose-dependent increase in intracellular cAMP accumulation, suggesting the activation of a stimulatory Gs-protein [163]. Nevertheless, using a DELFIA Eu-GTP binding assay for G-protein binding, the guanosine-mediated activation of putative GPCRs in rat brain membranes was observed only under specific experimental assay conditions (i.e., 1 mM MgCl2, 10 mM GDP, and 20 mM NaCl in 50 mm HEPES). However, under these experimental conditions adenosine receptor agonists (i.e., CPA, CGS21680, NECA, Cl-IBMECA) had no effect on guanosine binding, suggesting the identification of a GUO GPCR [165]. Hence, the specific G-protein activation profile of GUO remains an intriguing aspect. Interestingly, the GPR23 or LPA4 receptor, for lysophosphatidic acid, has been suggested as a potential GUO receptor. The silencing of the GPR23 gene in U87 glioma cultures reduced GUO-mediated inhibition of cell proliferation, while its overexpression promoted the GUO-induced antiproliferative effect [166]. Remarkably, the highest binding of [3H]-GUO within the brain has been shown to be found in the cerebral cortex, precisely where GPR23 also has its highest expression [167]. However, the direct interaction of guanosine with GPR23 has not yet been demonstrated, thus not ruling out the possibility that GUO may interact with other GPCRs. Overall, despite some evidence of the existence of putative GUO receptors in the brain, this nucleoside is currently considered an orphan neuromodulator, with no assigned receptors.
GUO effects are eventually abolished by selective ligands of adenosine receptors. For instance, the trophic effect of GUO that promotes cell adhesion and survival of cultured cerebellar neurones was blocked by the adenosine A2AR antagonist ZM241385 [93]. Likewise, A1R blockade and A2AR activation precluded GUO-mediated recovery of glutamate uptake homeostasis in cultured astrocytes after ischemia [80]. Also, and as previously mentioned, the protective effect of GUO on SH-SY5Y cells was abolished by A1R and A2AR antagonists and by BK channel blockers [77]. In ex vivo experiments, A1R blockade prevented GUO-mediated reduction in ROS production and preserved mitochondrial membrane potential in hippocampal slices subjected to ischemia [78], while it had no effect on GUO-induced recovery of glutamate uptake. Finally, numerous in vivo findings have consistently demonstrated the dependence on A1R and A2AR receptors for the protective effects of GUO in models of cerebral ischemia [77, 78] and in Parkinson's disease rodent models [104, 105, 107, 168]. Many of GUO's protective effects appear to depend on adenosine receptor signalling, often being hindered by selective A1R and A2AR antagonists. Given the minimal or negligible affinity of A1R and A2AR for GUO, alternative adenosine-based mechanisms of action for GUO have been considered. We have identified that GUO can modulate A2AR ligand binding and intracellular signalling only when co-expressed with A1R in HEK293 cells [169]. Our research has revealed that GUO can modulate A2AR ligand binding and intracellular signalling only in the presence of co-expressed A1R in HEK293 cells. On the contrary, GUO had no effect on A1R signalling, regardless of the presence or absence of A2AR [169]. Interestingly, the protective effect of GUO is absent in hippocampal slices but remains unaffected in striatal slices obtained from A2AR−/− mice [106]. These findings suggest a region-specific role for A2AR, which is instrumental in GUO-mediated neuroprotection in the hippocampus, while its involvement in the striatum is limited, where only A1R appears to be necessary. In general, we have provided evidence that some GUO fine-tuning effects may be based on the presence of an oligomeric organisation of adenosine receptors, specifically the A1R-A2AR heteromer [169] (Fig. 1).
The GUO-ADO interaction mechanism is based on the fact that extracellular GUO can regulate extracellular ADO levels in vivo [170], which in turn will modulate indirectly the adenosinergic signalling. Therefore, GUO signalling will rely on an ADO-dependent mechanism of action by which extracellular GUO, by stimulating ADO release, facilitates the indirect activation of adenosine receptors, particularly those with the highest affinity for ADO (A1R > A2AR). Indeed, previous findings supported the GUO-ADO interaction mechanism [171, 172]. In fact, at least in terms of the mitogenic effect of GUO, GUO-mediated release of ADO is partially responsible for this feat, since the addition of adenosine deaminase (ADA, the enzyme that metabolises adenosine to inosine) in the medium partially precludes the effect of GUO [140, 173]. However, some trophic effects of GUO have also been observed in the presence of dipyridamole, a nucleoside transport inhibitor, and in this situation ADO release is unlikely to occur [88, 140], although in vivo ADO release has not been monitored. However, adenosine receptor antagonists blocked the trophic effect of GUO, suggesting an interaction of GUO with these receptors. Overall, the GUO-ADO interaction signalling mechanism will be independent and compatible with the existence of putative cell surface GUO GPCRs, thus providing a genuine multimodal mechanism of action of GUO and allowing us to consider whether manipulating this GUO-ADO interaction could be of interest for therapeutic purposes.
Perspectives
GBPs are crucial players in neurogenesis and cancer cell metabolism, but their precise functions under physiological and pathological conditions are not fully understood. Therefore, further research will be necessary to gain deeper insights into the role of GBPs in modulating purinergic signalling, which could lead to novel treatments for neurodegenerative diseases and cancer. It is noteworthy that both guanine- and adenine-based purinergic systems play essential roles in healthy cells. Therefore, any therapeutic strategies that aim to modulate these systems in disease must carefully consider the potential risks of severe adverse effects. Guanosine stands out as a differentiating agent with potential clinical relevance in leukaemia and other diseases where differentiating stem cells could be therapeutically beneficial in impeding cancer progression. However, the development of guanosine as a potential therapeutic agent is challenging due to the lack of a specific target receptor, which precludes structure–activity relationship studies needed for the implementation of drug development programs. However, the emergence of the GUO-ADO interaction signalling mechanism, coupled with the longer half-life of GUO compared to ADO, offers new opportunities to indirectly manipulate the adenosinergic system to achieve long lasting and tuneable ADO-mediated therapeutic effects. Overall, further exploration in these directions is promising for uncovering innovative GBP-based approaches to disease treatment.
Data availability
No datasets were generated or analysed during the current study.
References
Camandola S, Mattson MP (2017) Brain metabolism in health, aging, and neurodegeneration, (in eng). EMBO J 36(11):1474–1492. https://doi.org/10.15252/embj.201695810
Mattson MP, Arumugam TV (2018) Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States, (in eng). Cell Metab 27(6):1176–1199. https://doi.org/10.1016/j.cmet.2018.05.011
Galts CPC et al (2019) Depression in neurodegenerative diseases: Common mechanisms and current treatment options, (in eng). Neurosci Biobehav Rev 102:56–84. https://doi.org/10.1016/j.neubiorev.2019.04.002
Gustavsson A et al (2023) Global estimates on the number of persons across the Alzheimer’s disease continuum, (in eng). Alzheimers Dement 19(2):658–670. https://doi.org/10.1002/alz.12694
Sung CY, Huang CC, Chen YS, Hsu KF, Lee GB (2021) Isolation and quantification of extracellular vesicle-encapsulated microRNA on an integrated microfluidic platform, (in eng). Lab Chip 21(23):4660–4671. https://doi.org/10.1039/d1lc00663k
Hayes-Larson E et al (2020) The competing risk of death and selective survival cannot fully explain the inverse cancer-dementia association, (in eng). Alzheimers Dement 16(12):1696–1703. https://doi.org/10.1002/alz.12168
Ospina-Romero M et al (2020) Association Between Alzheimer Disease and Cancer With Evaluation of Study Biases: A Systematic Review and Meta-analysis, (in eng). JAMA Netw Open 3(11):e2025515. https://doi.org/10.1001/jamanetworkopen.2020.25515
Segovia G, Porras A, Del Arco A, Mora F (2001) Glutamatergic neurotransmission in aging: a critical perspective, (in eng). Mech Ageing Dev 122(1):1–29. https://doi.org/10.1016/s0047-6374(00)00225-6
Nedergaard M, Takano T, Hansen AJ (2002) Beyond the role of glutamate as a neurotransmitter, (in eng). Nat Rev Neurosci Research Support, U.S. Gov't, P.H.S. Review 3(9):748–755. https://doi.org/10.1038/nrn916
Rueda CB et al (2016) Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs), (in eng). Biochim Biophys Acta 1857(8):1158–1166. https://doi.org/10.1016/j.bbabio.2016.04.003
Gerhard DM, Wohleb ES, Duman RS (2016) Emerging treatment mechanisms for depression: focus on glutamate and synaptic plasticity, (in eng). Drug Disc Today Rev 21(3):454–464. https://doi.org/10.1016/j.drudis.2016.01.016
Drury AN, Szent-Gyorgyi A (1929) The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart, (in eng). J Physiol 68(3):213–237
Burnstock G (2020) Introduction to Purinergic Signaling, (in eng). Methods Mol Biol 2041:1–15. https://doi.org/10.1007/978-1-4939-9717-6_1
Burnstock G (2018) Purine and purinergic receptors, (in eng). Brain Neurosci Adv 2:2398212818817494. https://doi.org/10.1177/2398212818817494
Boison D, Aronica E (2015) Comorbidities in Neurology: Is adenosine the common link?, (in eng). Neuropharmacology 97:18–34. https://doi.org/10.1016/j.neuropharm.2015.04.031
Chen JF, Eltzschig HK, Fredholm BB (2013) Adenosine receptors as drug targets–what are the challenges?, (in eng). Nat Rev Drug Discov 12(4):265–286. https://doi.org/10.1038/nrd3955
Yegutkin GG, Boison D (2022) ATP and Adenosine Metabolism in Cancer: Exploitation for Therapeutic Gain. Pharmacol Rev 74(3):797–822. https://doi.org/10.1124/pharmrev.121.000528
Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM (2005) Adenosine and brain function, (in eng). Int Rev Neurobiol Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S. Review 63:191–270. https://doi.org/10.1016/S0074-7742(05)63007-3
Palmer TM, Stiles GL (1995) Adenosine receptors, (in eng). Neuropharmacol Rev 34(7):683–694
Burnstock G (2007) Purine and pyrimidine receptors, (in eng). Cell Mol Life Sci: CMLS Rev 64(12):1471–1483. https://doi.org/10.1007/s00018-007-6497-0
Ciruela F, Sotelo E (2017) Special Issue: adenosine receptors, (in eng). Mol Editorial 22(7). https://doi.org/10.3390/molecules22071220
Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system, (in eng). Annu Rev Neurosci, Review 24:31–55. https://doi.org/10.1146/annurev.neuro.24.1.31
Boison D (2016) The Biochemistry and Epigenetics of Epilepsy: Focus on Adenosine and Glycine, (in eng). Front Mol Neurosci Rev 9:26. https://doi.org/10.3389/fnmol.2016.00026
Constantino LC et al (2015) Adenosine A1 receptor activation modulates N-methyl-d-aspartate (NMDA) preconditioning phenotype in the brain, (in eng). Behav Brain Res Research Support, Non-U.S. Gov't 282:103–110. https://doi.org/10.1016/j.bbr.2014.12.056
Constantino LC, Vandresen-Filho S, Tasca CI (2015) Neuroprotection induced by NMDA preconditioning as a strategy to understand brain tolerance mechanism, (in eng). Neural Regen Res 10(4):542–543. https://doi.org/10.4103/1673-5374.155415
Cunha RA (2016) How does adenosine control neuronal dysfunction and neurodegeneration?, (in eng). J Neurochem Review Research Support, Non-U.S. Gov't 139(6):1019–1055. https://doi.org/10.1111/jnc.13724
Ciruela F et al (2006) Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers, (in eng). J Neurosci 26(7):2080–2087. https://doi.org/10.1523/JNEUROSCI.3574-05.2006
Ciruela F et al (2012) G protein-coupled receptor oligomerization and brain integration: focus on adenosinergic transmission, (in eng). Brain Res 1476:86–95. https://doi.org/10.1016/j.brainres.2012.04.056
Ciruela F et al (2006) Heterodimeric adenosine receptors: a device to regulate neurotransmitter release, (in eng). Cell Mol Life Sci 63(21):2427–2431. https://doi.org/10.1007/s00018-006-6216-2
Köfalvi A et al (2020) Control of glutamate release by complexes of adenosine and cannabinoid receptors, (in eng). BMC Biol 18(1):9. https://doi.org/10.1186/s12915-020-0739-0
Ferré S et al (2022) G protein-coupled receptor-effector macromolecular membrane assemblies (GEMMAs), (in eng). Pharmacol Ther 231:107977. https://doi.org/10.1016/j.pharmthera.2021.107977
Di Liberto V et al (2016) The Guanine-Based Purinergic System: The Tale of An Orphan Neuromodulation, (in eng). Front Pharmacol 7:158. https://doi.org/10.3389/fphar.2016.00158
Wagner JA, Carlson SS, Kelly RB (1978) Chemical and physical characterization of cholinergic synaptic vesicles, (in eng). Biochem Research Support, U.S. Gov't, P.H.S. 17(7):1199–1206
Santos TG, Souza DO, Tasca CI (2006) GTP uptake into rat brain synaptic vesicles, (in eng). Brain Res Research Support, Non-U.S. Gov't 1070(1):71–76. https://doi.org/10.1016/j.brainres.2005.10.099
Gualix J, Pintor J, Miras-Portugal MT (1999) Characterization of nucleotide transport into rat brain synaptic vesicles, (in eng). J Neurochem Research Support, Non-U.S. Gov't 73(3):1098–1104
Lanznaster D, Dal-Cim T, Piermartiri TC, Tasca CI (2016) Guanosine: a Neuromodulator with Therapeutic Potential in Brain Disorders, (in eng). Aging Dis Rev 7(5):657–679. https://doi.org/10.14336/AD.2016.0208
Rodbell M, Birnbaumer L, Pohl SL, Krans HM (1971) The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action, (in eng). J Biological Chem 246(6):1877–82
Hepler JR, Gilman AG (1992) G proteins, (in eng). Trends Biochem Sci Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S. Review 17(10):383–387
Paz MM, Ramos M, Ramirez G, Souza D (1994) Differential effects of guanine nucleotides on kainic acid binding and on adenylate cyclase activity in chick optic tectum, (in eng). FEBS lett Comparative Study Research Support, Non-U.S. Gov't 355(2):205–208
Souza DO, Ramirez G (1991) Effects of guanine nucleotides on kainic acid binding and on adenylate cyclase in chick optic tectum and cerebellum, (in eng). J Mol Neurosci MN Research Support, Non-U.S. Gov't 3(1):39–45
Paas Y, Devillers-Thiery A, Changeux JP, Medevielle F, Teichberg VI (1996) Identification of an extracellular motif involved in the binding of guanine nucleotides by a glutamate receptor, (in eng). EMBO J Research Support, Non-U.S. Gov't 15(7):1548–1556
Porciuncula LO, Vinade L, Wofchuk S, Souza DO (2002) Guanine based purines inhibit [(3)H]glutamate and [(3)H]AMPA binding at postsynaptic densities from cerebral cortex of rats," (in eng), Brain Res Research Support, Non-U.S. Gov't 928(1–2):106–112
Schmidt A, Lara DR, Souza DO (2007) Proposal of a guanine-based purinergic system in the mammalian central nervous system, (in eng). Pharmacol Therapeutics Research Support, Non-U.S. Gov't Review 116(3):401–416. https://doi.org/10.1016/j.pharmthera.2007.07.004
Tasca CI, Cardoso LF, Martini LH, Ramirez G, Souza DO (1998) Guanine nucleotides inhibit cAMP accumulation induced by metabotropic glutamate receptor activation, (in eng). Neurochem Res Research Support, Non-U.S. Gov't 23(2):183–188
Tasca CI, Cardoso LF, Souza DO (1999) Effects of guanine nucleotides on adenosine and glutamate modulation of cAMP levels in optic tectum slices from chicks, (in eng). Neurochem Int Research Support, Non-U.S. Gov't 34(3):213–220
Tasca CI, Wofchuk ST, Souza DO, Ramirez G, Rodnight R (1995) Guanine nucleotides inhibit the stimulation of GFAP phosphorylation by glutamate, (in eng). Neuroreport Research Support, Non-U.S. Gov't 6(2):249–252
Frizzo ME et al (2002) Guanosine enhances glutamate uptake in brain cortical slices at normal and excitotoxic conditions. Cell Mol Neurobiol 22(3):353–363
Molz S, Decker H, Oliveira IJ, Souza DO, Tasca CI (2005) Neurotoxicity induced by glutamate in glucose-deprived rat hippocampal slices is prevented by GMP, (in eng). Neurochem Res Comparative Study Research Support, Non-U.S. Gov't 30(1):83–89
Schmidt AP, Lara DR, de Faria Maraschin J, da Silveira Perla A, Onofre Souza D (2000) Guanosine and GMP prevent seizures induced by quinolinic acid in mice. Brain Res 864:40–43. https://doi.org/10.1016/S0006-8993(00)02106-5
de Oliveira DL et al (2004) Quinolinic acid promotes seizures and decreases glutamate uptake in young rats: reversal by orally administered guanosine, (in Eng). Brain Res Comparative Study Research Support, Non-U.S. Gov't 1018(1):48–54. https://doi.org/10.1016/j.brainres.2004.05.033
Schmidt AP, Avila TT, Souza DO (2005) Intracerebroventricular guanine-based purines protect against seizures induced by quinolinic acid in mice, (in eng). Neurochem Res Comparative Study Research Support, Non-U.S. Gov't 30(1):69–73
Schadeck RJ, Sarkis JJ, Dias RD, Araujo HM, Souza DO (1989) Synaptosomal apyrase in the hypothalamus of adult rats, (in eng). Braz J Med Biol Res = Revista brasileira de pesquisas medicas e biologicas / Sociedade Brasileira de Biofisica ... [et al.], Research Support, Non-U.S. Gov't 22(3):303–314
Zimmermann H (2001) Ectonucleotidases: some recent developments and a note on nomenclature, (in English). Drug Develop Res 52(1–2):44–56. https://doi.org/10.1002/Ddr.1097
Zimmermann H (2006) Ectonucleotidases in the nervous system, (in eng). Novartis Found Symp Review 276:113–128, discussion 128–130, 233–237, 275–281
Zimmermann H (2006) Nucleotide signaling in nervous system development, (in eng). Pflugers Archiv : Eur J Physiol Research Support, Non-U.S. Gov't Review 452(5):573–588. https://doi.org/10.1007/s00424-006-0067-4
Castro-Gago M, Camina F, Lojo S, Rodriguez-Segade S, Rodriguez-Nunez A (1992) Concentrations of purine nucleotides and purine and pyrimidine bases in cerebrospinal fluid of neurologically healthy children, (in eng). Eur J Clin Chem Clin Bio : j Forum Eur Clin Chem Soc 30(11):761–765
Regner A, Crestana RE, Silveira FJ, Friedman G, Chemale I, Souza D (1997) Guanine nucleotides are present in human CSF, (in eng). Neuroreport Research Support, Non-U.S. Gov't 8(17):3771–3774
Portela LVC et al (2002) Guanine and adenine nucleotidase activities in rat cerebrospinal fluid, (in English). Brain Res 950(1–2):74–78, Pii S0006–8993(02):02987–6 https://doi.org/10.1016/S0006-8993(02)02987-6
Giuliani P et al (2017) Evidence for purine nucleoside phosphorylase (PNP) release from rat C6 glioma cells, (in eng). J Neurochem 141(2):208–221. https://doi.org/10.1111/jnc.14004
Ciccarelli R et al (1999) Rat cultured astrocytes release guanine-based purines in basal conditions and after hypoxia/hypoglycemia, (in eng). Glia 25(1):93–98
Ciccarelli R et al (2001) Involvement of astrocytes in purine-mediated reparative processes in the brain, (in eng). Int J Dev Neurosci: Official J Int Soc Dev NeuroscI Research Support, Non-U.S. Gov't Review 19(4):395–414
Zuccarini M et al (2018) Uncovering the Signaling Pathway behind Extracellular Guanine-Induced Activation of NO System: New Perspectives in Memory-Related Disorders, (in eng). Front Pharmacol 9:110. https://doi.org/10.3389/fphar.2018.00110
Giuliani P et al (2012) Guanine-based purines modulate the effect of L-NAME on learning and memory in rats, (in eng). Panminerva Med Research Support, Non-U.S. Gov't 54(1 Suppl 4):53–58
Chen Y, Swanson RA (2003) Astrocytes and brain injury, (in eng). J Cereb Blood Flow Metab: Official J Int Soc Cerebral Blood Flow Metabolism, Rev 23(2):137–149
Giuliani P et al (2012) Protective activity of guanosine in an in vitro model of Parkinson's disease, (in eng). Panminerva Med Research Support, Non-U.S. Gov't 54(1 Suppl 4):43–51
Nagasawa K, Kawasaki F, Tanaka A, Nagai K, Fujimoto S (2007) Characterization of guanine and guanosine transport in primary cultured rat cortical astrocytes and neurons, (in eng). Glia Comparative Study Research Support, Non-U.S. Gov't 55(14):1397–1404. https://doi.org/10.1002/glia.20550
Peng L, Huang R, Yu AC, Fung KY, Rathbone MP, Hertz L (2005) Nucleoside transporter expression and function in cultured mouse astrocytes, (in eng). Glia 52(1):25–35. https://doi.org/10.1002/glia.20216
Spector R, Johanson CE (2007) The origin of deoxynucleosides in brain: implications for the study of neurogenesis and stem cell therapy, (in eng). Pharmaceutical Res, Rev 24(5):859–867. https://doi.org/10.1007/s11095-006-9221-0
Lanznaster D, Dal-Cim T, Piermartiri TC, Tasca C (2016) Guanosine: a neuromodulator with therapeutic potential in brain disorders. A&D. https://doi.org/10.14336/AD.2016.0208
Tasca CI, Lanznaster D, Oliveira KA, Fernández-Dueñas V, Ciruela F (2018) Neuromodulatory Effects of Guanine-Based Purines in Health and Disease, (in eng). Front Cell Neurosci 12:376. https://doi.org/10.3389/fncel.2018.00376
Oliveira IJ, Molz S, Souza DO, Tasca CI (2002) Neuroprotective effect of GMP in hippocampal slices submitted to an in vitro model of ischemia, (in eng). Cellular Mol Neurobiol Research Support, Non-U.S. Gov't 22(3):335–344
Tasca CI, Santos TG, Tavares RG, Battastini AM, Rocha JB, Souza DO (2004) Guanine derivatives modulate L-glutamate uptake into rat brain synaptic vesicles, (in eng). Neurochem Int Research Support, Non-U.S. Gov't 44(6):423–431
Oleskovicz SP, Martins WC, Leal RB, Tasca CI (2008) Mechanism of guanosine-induced neuroprotection in rat hippocampal slices submitted to oxygen-glucose deprivation, (in eng). Neurochem Int Research Support, Non-U.S. Gov't 52(3):411–418. https://doi.org/10.1016/j.neuint.2007.07.017
Molz S, Dal-Cim T, Tasca CI (2009) Guanosine-5'-monophosphate induces cell death in rat hippocampal slices via ionotropic glutamate receptors activation and glutamate uptake inhibition, (in eng). Neurochem Int Research Support, Non-U.S. Gov't 55(7):703–709. https://doi.org/10.1016/j.neuint.2009.06.015
Molz S et al (2011) Neuroprotective effect of guanosine against glutamate-induced cell death in rat hippocampal slices is mediated by the phosphatidylinositol-3 kinase/Akt/ glycogen synthase kinase 3beta pathway activation and inducible nitric oxide synthase inhibition, (in eng). J Neurosc Res Research Support, Non-U.S. Gov't 89(9):1400–1408. https://doi.org/10.1002/jnr.22681
Ciruela F et al (2012) G protein-coupled receptor oligomerization and brain integration: focus on adenosinergic transmission, (in eng). Brain Res Research Support, Non-U.S. Gov't Review 1476:86–95. https://doi.org/10.1016/j.brainres.2012.04.056
Dal-Cim T et al (2012) Guanosine protects human neuroblastoma SH-SY5Y cells against mitochondrial oxidative stress by inducing heme oxigenase-1 via PI3K/Akt/GSK-3beta pathway, (in eng). Neurochem Int Research Support, Non-U.S. Gov't 61(3):397–404. https://doi.org/10.1016/j.neuint.2012.05.021
Dal-Cim T et al (2013) Guanosine controls inflammatory pathways to afford neuroprotection of hippocampal slices under oxygen and glucose deprivation conditions, (in eng). J Neurochem Research Support, Non-U.S. Gov't 126(4):437–450. https://doi.org/10.1111/jnc.12324
Dal-Cim T et al (2016) Neuroprotection promoted by guanosine depends on glutamine synthetase and glutamate transporters activity in hippocampal slices subjected to oxygen/glucose deprivation, (in Eng). Neurotoxicity Res. https://doi.org/10.1007/s12640-015-9595-z
Dal-Cim T, Poluceno GG, Lanznaster D, de Oliveira KA, Nedel CB, Tasca CI (2019) Guanosine prevents oxidative damage and glutamate uptake impairment induced by oxygen/glucose deprivation in cortical astrocyte cultures: involvement of A, (in eng). Purinergic Signal 15(4):465–476. https://doi.org/10.1007/s11302-019-09679-w
Thomaz DT et al (2016) Guanosine prevents nitroxidative stress and recovers mitochondrial membrane potential disruption in hippocampal slices subjected to oxygen/glucose deprivation, (in eng). Purinergic Signal12(4):707–718. https://doi.org/10.1007/s11302-016-9534-3
Thomaz DT et al (2020) Guanosine Neuroprotective Action in Hippocampal Slices Subjected to Oxygen and Glucose Deprivation Restores ATP Levels, Lactate Release and Glutamate Uptake Impairment: Involvement of Nitric Oxide, (in eng). Neurochem Res 45(9):2217–2229. https://doi.org/10.1007/s11064-020-03083-2
Dal-Cim T, Martins WC, Santos AR, Tasca CI (2011) Guanosine is neuroprotective against oxygen/glucose deprivation in hippocampal slices via large conductance Ca(2)+-activated K+ channels, phosphatidilinositol-3 kinase/protein kinase B pathway activation and glutamate uptake. Neuroscience 183:212–220. https://doi.org/10.1016/j.neuroscience.2011.03.022
Dal-Cim T et al (2016) Glutamate Transporters activity in hippocampal slices subjected to oxygen/glucose deprivation. Neurotoxicity Res
Dal-Cim T et al (2012) Guanosine protects human neuroblastoma SH-SY5Y cells against mitochondrial oxidative stress by inducing heme oxigenase-1 via PI3K/Akt/GSK-3beta pathway, (in Eng). Neurochem Int. https://doi.org/10.1016/j.neuint.2012.05.021
Quincozes-Santos A, Bobermin LD, Souza DG, Bellaver B, Goncalves CA, Souza DO (2014) Guanosine protects C6 astroglial cells against azide-induced oxidative damage: a putative role of heme oxygenase 1, (in Eng). J Neurochem Research Support, Non-U.S. Gov't 130(1):61–74. https://doi.org/10.1111/jnc.12694
Bellaver B, Souza DG, Bobermin LD, Goncalves CA, Souza DO, Quincozes-Santos A (2015) Guanosine inhibits LPS-induced pro-inflammatory response and oxidative stress in hippocampal astrocytes through the heme oxygenase-1 pathway, (in Eng). Purinergic Signal Research Support, Non-U.S. Gov't 11(4):571–580. https://doi.org/10.1007/s11302-015-9475-2
Decker H et al (2007) Guanine derivatives modulate extracellular matrix proteins organization and improve neuron-astrocyte co-culture, (in Eng). J Neurosci Res 85(9):1943–1951. https://doi.org/10.1002/jnr.21332
Bettio LE et al (2012) Guanosine produces an antidepressant-like effect through the modulation of NMDA receptors, nitric oxide-cGMP and PI3K/mTOR pathways, (in eng). Behav Brain Res Research Support, Non-U.S. Gov't 234(2):137–148. https://doi.org/10.1016/j.bbr.2012.06.021
Martins WC, Tasca CI, Cimarosti H (2016) Battling Alzheimer’s Disease: Targeting SUMOylation-Mediated Pathways, (in eng). Neurochem Res 41(3):568–578. https://doi.org/10.1007/s11064-015-1681-3
Zanella CA, Tasca CI, Henley JM, Wilkinson KA, Cimarosti HI (2020) Guanosine modulates SUMO2/3-ylation in neurons and astrocytes via adenosine receptors, (in eng). Purinergic Signal16(3):439–450. https://doi.org/10.1007/s11302-020-09723-0
Zanella CA, Marques N, Junqueira S, Prediger RD, Tasca CI, Cimarosti HI (2023) Guanosine increases global SUMO1-ylation in the hippocampus of young and aged mice and improves the short-term memory of young mice, (in eng). J Neurochem. https://doi.org/10.1111/jnc.15920
Decker H et al (2019) Guanosine and GMP increase the number of granular cerebellar neurons in culture: dependence on adenosine A, (in eng). Purinergic Signal 15(4):439–450. https://doi.org/10.1007/s11302-019-09677-y
Piermartiri TCB, Dos Santos B, Barros-Aragão FGQ, Prediger RD, Tasca CI (2020) Guanosine Promotes Proliferation in Neural Stem Cells from Hippocampus and Neurogenesis in Adult Mice, (in eng). Mol Neurobiol 57(9):3814–3826. https://doi.org/10.1007/s12035-020-01977-4
Lara DR, Schmidt AP, Frizzo ME, Burgos JS, Ramirez G, Souza DO (2001) Effect of orally administered guanosine on seizures and death induced by glutamatergic agents, (in eng). Brain Res Research Support, Non-U.S. Gov't 912(2):176–180
Soares FA et al (2004) Anticonvulsant effect of GMP depends on its conversion to guanosine, (in eng). Brain Res Comparative Study Research Support, Non-U.S. Gov't 1005(1–2):182–186. https://doi.org/10.1016/j.brainres.2004.01.053
Ganzella M et al (2012) Effects of chronic guanosine treatment on hippocampal damage and cognitive impairment of rats submitted to chronic cerebral hypoperfusion, (in eng). Neurol Sci Official J Ital Neurol Soc Ital Soc Clin Neurophysiol Research Support, Non-U.S. Gov't 33(5): 985–997. https://doi.org/10.1007/s10072-011-0872-1
Paniz LG et al (2014) Neuroprotective effects of guanosine administration on behavioral, brain activity, neurochemical and redox parameters in a rat model of chronic hepatic encephalopathy, (in eng). Metab Brain Dis Research Support, Non-U.S. Gov't 29(3): 645–654. https://doi.org/10.1007/s11011-014-9548-x
Hansel G et al (2014) The potential therapeutic effect of guanosine after cortical focal ischemia in rats, (in eng). PloS One Research Support, Non-U.S. Gov't 9(2):e90693. https://doi.org/10.1371/journal.pone.0090693
Hansel G et al (2015) Guanosine protects against cortical focal ischemia. Involvement of inflammatory response, (in eng). Mol Neurobiol Research Support, Non-U.S. Gov't 52(3):1791–1803. https://doi.org/10.1007/s12035-014-8978-0
Gerbatin RDR et al (2017) Guanosine Protects Against Traumatic Brain Injury-Induced Functional Impairments and Neuronal Loss by Modulating Excitotoxicity, Mitochondrial Dysfunction, and Inflammation, (in eng). Mol Neurobiol 54(10):7585–7596. https://doi.org/10.1007/s12035-016-0238-z
Dobrachinski F et al (2018) Guanosine attenuates behavioral deficits after traumatic brain injury by modulation of adenosinergic receptors, (in eng). Mol Neurobiol. https://doi.org/10.1007/s12035-018-1296-1
Su C et al (2009) Guanosine improves motor behavior, reduces apoptosis, and stimulates neurogenesis in rats with parkinsonism, (in eng). J Neurosci Res Research Support, Non-U.S. Gov't 87(3):617–625. https://doi.org/10.1002/jnr.21883
Massari CM, López-Cano M, Núñez F, Fernández-Dueñas V, Tasca CI, Ciruela F (2017) Antiparkinsonian Efficacy of Guanosine in Rodent Models of Movement Disorder, (in eng). Front Pharmacol 8:700. https://doi.org/10.3389/fphar.2017.00700
Marques NF, Massari CM, Tasca CI (2019) Guanosine Protects Striatal Slices Against 6-OHDA-Induced Oxidative Damage, Mitochondrial Dysfunction, and ATP Depletion, (in eng). Neurotox Res 35(2):475–483. https://doi.org/10.1007/s12640-018-9976-1
Massari CM et al (2020) Involvement of adenosine A, (in eng). Purinergic Signal 16(3):379–387. https://doi.org/10.1007/s11302-020-09716-z
Massari CM, Constantino LC, Tasca CI (2021) Adenosine A, (in eng). Purinergic Signal 17(2):247–254. https://doi.org/10.1007/s11302-021-09765-y
Lanznaster D et al (2017) Guanosine Prevents Anhedonic-Like Behavior and Impairment in Hippocampal Glutamate Transport Following Amyloid-beta1-40 Administration in Mice, (in eng). Mol Neurobiol 54(7):5482–5496. https://doi.org/10.1007/s12035-016-0082-1
Coelho V, Binder LB, Marques NF, Constantino LC, Mancini G, Tasca CI (2022) Guanosine prevents spatial memory impairment and hippocampal damage following amyloid-β, (in eng). Metabolites 12(12). https://doi.org/10.3390/metabo12121207
Schmidt AP et al (2009) Anti-nociceptive properties of the xanthine oxidase inhibitor allopurinol in mice: Role of A1 adenosine receptors. Br J Pharmacol 156:163–172. https://doi.org/10.1111/j.1476-5381.2008.00025.x
Schmidt AP et al (2008) Antinociceptive effects of intracerebroventricular administration of guanine-based purines in mice: evidences for the mechanism of action, (in eng). Brain Res Research Support, Non-U.S. Gov't 1234:50–58. https://doi.org/10.1016/j.brainres.2008.07.091
Schmidt AP et al (2010) Mechanisms involved in the antinociception induced by systemic administration of guanosine in mice, (in eng). British J Pharmacol Research Support, Non-U.S. Gov't 159(6):1247–1263. https://doi.org/10.1111/j.1476-5381.2009.00597.x
Vinadé ER, Schmidt AP, Frizzo MES, Izquierdo I, Elisabetsky E, Souza DO (2003) Chronically administered guanosine is anticonvulsant, amnesic and anxiolytic in mice. Brain Res 977:97–102. https://doi.org/10.1016/S0006-8993(03)02769-0
Almeida RF et al (2017) Guanosine Anxiolytic-Like Effect Involves Adenosinergic and Glutamatergic Neurotransmitter Systems, (in eng). Mol Neurobiol Research Support, Non-U.S. Gov't 54(1):423–436. https://doi.org/10.1007/s12035-015-9660-x
Bettio LE et al (2014) Guanosine prevents behavioral alterations in the forced swimming test and hippocampal oxidative damage induced by acute restraint stress, (in eng). Pharmacol Biochem Behav Research Support, Non-U.S. Gov't 127:7–14. https://doi.org/10.1016/j.pbb.2014.10.002
Bettio LE et al (2016) The antidepressant-like effect of chronic guanosine treatment is associated with increased hippocampal neuronal differentiation, (in eng). Eur J Neurosci Research Support, Non-U.S. Gov't 43(8):1006–1015. https://doi.org/10.1111/ejn.13172
Camargo A, Dalmagro AP, Altê GA, Zeni ALB, Tasca CI, Rodrigues ALS (2023) NMDA receptor-mediated modulation on glutamine synthetase and glial glutamate transporter GLT-1 is involved in the antidepressant-like and neuroprotective effects of guanosine, (in eng). Chem Biol Interact 375:110440. https://doi.org/10.1016/j.cbi.2023.110440
Ciruela F (2013) Guanosine behind the scene, (in eng). J Neurochem 126(4):425–427. https://doi.org/10.1111/jnc.12328
Boison D, Yegutkin GG (2019) Adenosine Metabolism: Emerging Concepts for Cancer Therapy, (in eng). Cancer Cell 36(6):582–596. https://doi.org/10.1016/j.ccell.2019.10.007
El-Kharrag R, Owen R, Boison D (2019) Adenosine Kinase Deficiency Increases Susceptibility to a Carcinogen, (in eng). J Caffeine Adenosine Res 9(1):4–11. https://doi.org/10.1089/caff.2018.0019
Mullen NJ, Singh PK (2023) Nucleotide metabolism: a pan-cancer metabolic dependency, (in eng). Nat Rev Cancer 23(5):275–294. https://doi.org/10.1038/s41568-023-00557-7
Barfeld SJ et al (2015) Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer, (in eng). Oncotarget 6(14):12587–602. https://doi.org/10.18632/oncotarget.3494
Pozzo AR, Faria FCC, Carvalho LO, Pinho MB, Maia RC (2017) DNA microarray expression profiling of a new t(8;13) AML case allows identification of possible leukemogenic transformation markers, (in eng). Rev Bras Hematol Hemoter 39(4):368–371. https://doi.org/10.1016/j.bjhh.2017.06.003
Lane AN, Yan J, Fan TW (2015) C Tracer Studies of Metabolism in Mouse Tumor Xenografts, (in eng). Bio Protoc 5(22). https://doi.org/10.21769/bioprotoc.1650
Chong YC et al (2020) Targeted Inhibition of Purine Metabolism Is Effective in Suppressing Hepatocellular Carcinoma Progression, (in eng). Hepatol Commun 4(9):1362–1381. https://doi.org/10.1002/hep4.1559
Kofuji S et al (2019) IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma, (in eng). Nat Cell Biol 21(8):1003–1014. https://doi.org/10.1038/s41556-019-0363-9
Ruan B et al (2020) Identification of a Set of Genes Improving Survival Prediction in Kidney Renal Clear Cell Carcinoma through Integrative Reanalysis of Transcriptomic Data, (in eng). Dis Markers 2020:8824717. https://doi.org/10.1155/2020/8824717
Huang F et al (2018) Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers, (in eng). Cell Metab 28(3):369-382.e5. https://doi.org/10.1016/j.cmet.2018.06.005
Garcia-Manero G, Chien KS, Montalban-Bravo G (2020) Myelodysplastic syndromes: 2021 update on diagnosis, risk stratification and management, (in eng). Am J Hematol 95(11):1399–1420. https://doi.org/10.1002/ajh.25950
Liu X et al (2023) IMPDH inhibition activates TLR-VCAM1 pathway and suppresses the development of MLL-fusion leukemia, (in eng). EMBO Mol Med 15(1):e15631. https://doi.org/10.15252/emmm.202115631
Huang F et al (2021) Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer, (in eng). J Clin Invest 131(1). https://doi.org/10.1172/JCI139929
Takizawa Y et al (2023) Specific inhibitory effects of guanosine on breast cancer cell proliferation, (in eng). Biochem Biophys Res Commun 673:67–72. https://doi.org/10.1016/j.bbrc.2023.06.069
Wang ZH, Li W, Dong H, Han F (2022) Current state of NK cell-mediated immunotherapy in chronic lymphocytic leukemia, (in eng). Front Oncol 12:1077436. https://doi.org/10.3389/fonc.2022.1077436
Osti F, Corradini FG, Hanau S, Matteuzzi M, Gambari R (1997) Human leukemia K562 cells: induction to erythroid differentiation by guanine, guanosine and guanine nucleotides, (in eng). Haematologica 82(4):395–401
Frank, NY, Schatton T, Frank MH (2010) The therapeutic promise of the cancer stem cell concept. J Clin Invest 120(1):41–50
Clarkson B, Strife A, Wisniewski D, Lambek CL, Liu C (2003) Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia 17(7):1211–1262
Moosavi MA, Yazdanparast R, Lotfi A (2006) GTP induces S-phase cell-cycle arrest and inhibits DNA synthesis in K562 cells but not in normal human peripheral lymphocytes, (in eng). J Biochem Mol Biol 39(5):492–501. https://doi.org/10.5483/bmbrep.2006.39.5.492
Wang H, He X, Li Z, Jin H, Wang X, Li L (2022) Guanosine primes acute myeloid leukemia for differentiation via guanine nucleotide salvage synthesis, (in eng). Am J Cancer Res 12(1):427–444
Zhou W, Wahl DR (2020) Purine metabolism promotes radioresistance and is a therapeutic target in glioblastoma, (in eng). Mol Cell Oncol 7(6):1834902. https://doi.org/10.1080/23723556.2020.1834902
Oliveira KA, Dal-Cim TA, Lopes FG, Nedel CB, Tasca CI (2017) Guanosine promotes cytotoxicity via adenosine receptors and induces apoptosis in temozolomide-treated A172 glioma cells, (in eng). Purinergic Signal 13(3):305–318. https://doi.org/10.1007/s11302-017-9562-7
de Groot J, Sontheimer H (2011) Glutamate and the biology of gliomas, (in eng). Glia 59(8):1181–1189. https://doi.org/10.1002/glia.21113
Sontheimer H (2011) Glutamate and tumor-associated epilepsy, (in eng). Oncotarget 2(11):823–4. https://doi.org/10.18632/oncotarget.350
Robert SM, Sontheimer H (2014) Glutamate transporters in the biology of malignant gliomas, (in eng). Cell Mol Life Sci 71(10):1839–1854. https://doi.org/10.1007/s00018-013-1521-z
Garozzo R, Sortino MA, Vancheri C, Condorelli DF (2010) Antiproliferative effects induced by guanine-based purines require hypoxanthine-guanine phosphoribosyltransferase activity, (in eng). Biol Chem 391(9):1079–1089. https://doi.org/10.1515/BC.2010.106
Garozzo R et al (2022) Guanine inhibits the growth of human glioma and melanoma cell lines by interacting with GPR23, (in eng). Front Pharmacol 13:970891. https://doi.org/10.3389/fphar.2022.970891
Wang J et al (2019) The Mechanism of the Selective Antiproliferation Effect of Guanine-Based Biomolecules and Its Compensation, (in eng). ACS Chem Biol 14(6):1164–1173. https://doi.org/10.1021/acschembio.9b00062
Wang ZW, Saifee O, Nonet ML, Salkoff L (2001) SLO-1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction, (in eng). Neuron, Research Support, U.S. Gov't, P.H.S. 32(5): 867–81
Le X, Wei D, Huang S, Lancaster JR, Xie K (2005) Nitric oxide synthase II suppresses the growth and metastasis of human cancer regardless of its up-regulation of protumor factors, (in eng). Proc Natl Acad Sci U S A 102(24):8758–8763. https://doi.org/10.1073/pnas.0409581102
Di Iorio P, Beggiato S, Ronci M, Nedel CB, Tasca CI, Zuccarini M (2021) Unfolding New Roles for Guanine-Based Purines and Their Metabolizing Enzymes in Cancer and Aging Disorders, (in eng). Front Pharmacol 12:653549. https://doi.org/10.3389/fphar.2021.653549
Di Iorio P et al (2021) Pros and Cons of Pharmacological Manipulation of cGMP-PDEs in the Prevention and Treatment of Breast Cancer, (in eng). Int J Mol Sci 23(1). https://doi.org/10.3390/ijms23010262
Di Virgilio F, Giuliani AL, Vultaggio-Poma V, Falzoni S, Sarti AC (2018) Non-nucleotide Agonists Triggering P2X7 Receptor Activation and Pore Formation, (in eng). Front Pharmacol 9:39. https://doi.org/10.3389/fphar.2018.00039
Morciano M, Ortinau S, Zimmermann H (2004) Guanine nucleotides inhibit NMDA and kainate-induced neurotoxicity in cultured rat hippocampal and neocortical neurons, (in eng). Neurochem Int 45(1):95–101. https://doi.org/10.1016/j.neuint.2003.11.017
Molz S, Tharine DC, Decker H, Tasca CI (2008) GMP prevents excitotoxicity mediated by NMDA receptor activation but not by reversal activity of glutamate transporters in rat hippocampal slices, (in eng). Brain Res 1231:113–120. https://doi.org/10.1016/j.brainres.2008.07.009
Chang R, Algird A, Bau C, Rathbone MP, Jiang S (2008) Neuroprotective effects of guanosine on stroke models in vitro and in vivo, (in eng). Neurosci Lett 431(2):101–105. https://doi.org/10.1016/j.neulet.2007.11.072
Deutsch SI, Rosse RB, Long KD, Gaskins BL, Mastropaolo J (2008) Guanosine possesses specific modulatory effects on NMDA receptor-mediated neurotransmission in intact mice, (in eng). Eur Neuropsychopharmacol 18(4):299–302. https://doi.org/10.1016/j.euroneuro.2007.07.010
Dal-Cim T et al (2016) Neuroprotection Promoted by Guanosine Depends on Glutamine Synthetase and Glutamate Transporters Activity in Hippocampal Slices Subjected to Oxygen/Glucose Deprivation, (in eng). Neurotox Res 29(4):460–468. https://doi.org/10.1007/s12640-015-9595-z
Deutsch SI, Long KD, Rosse RB, Mastropaolo J, Eller J (2005) Hypothesized deficiency of guanine-based purines may contribute to abnormalities of neurodevelopment, neuromodulation, and neurotransmission in Lesch-Nyhan syndrome, (in eng). Clin Neuropharmacol 28(1):28–37. https://doi.org/10.1097/01.wnf.0000152043.36198.25
Benfenati V, Caprini M, Nobile M, Rapisarda C, Ferroni S (2006) Guanosine promotes the up-regulation of inward rectifier potassium current mediated by Kir4.1 in cultured rat cortical astrocytes, (in eng). J Neurochem, Research Support, Non-U.S. Gov't 98(2): 430–45. https://doi.org/10.1111/j.1471-4159.2006.03877.x
Ghatta S, Nimmagadda D, Xu X,O'Rourke ST (2006) Large-conductance, calcium-activated potassium channels: structural and functional implications, (in eng). PharmacolTherapeutics, Research Support, Non-U.S. Gov't Review 110(1): 103–16. https://doi.org/10.1016/j.pharmthera.2005.10.007
Jin W, Sugaya A, Tsuda T, Ohguchi H, Sugaya E (2000) Relationship between large conductance calcium-activated potassium channel and bursting activity, (in eng). Brain Res 860(1–2):21–28
Hewawasam P et al (2002) The synthesis and characterization of BMS-204352 (MaxiPost) and related 3-fluorooxindoles as openers of maxi-K potassium channels, (in eng). Bioorg Med Chem Lett 12(7):1023–1026
Gambino G et al (2022) Guanosine modulates K, (in eng). Pflugers Arch 474(11):1133–1145. https://doi.org/10.1007/s00424-022-02741-4
Traversa U et al (2003) Rat brain guanosine binding site. Biological studies and pseudo-receptor construction. Bioorg Med Chem 11:5417–5425
Traversa U, Bombi G, Di Iorio P, Ciccarelli R, Werstiuk ES, Rathbone MP (2002) Specific [(3)H]-guanosine binding sites in rat brain membranes. Br J Pharmacol 135:969–976. https://doi.org/10.1038/sj.bjp.0704542
Volpini R et al (2011) Evidence for the existence of a specific g protein-coupled receptor activated by guanosine. ChemMedChem 6:1074–1080. https://doi.org/10.1002/cmdc.201100100
Di Liberto V et al (2012) Identification of GPR23/LPA4 as a candidate G protein-coupled receptor for guanosine. Acta Physiol, Abstract 206(Supplement 692)
Grillo M et al (2012) Brain expression and 3H-guanosine binding analysis of novel G protein-coupled receptor for guanosine (GPR23/LPA4). Acta Physiol 206(Supplement 692)
Massari CM, Zuccarini M, Di Iorio P, Tasca CI (2021) Guanosine Mechanisms of Action: Toward Molecular Targets, (in eng). Front Pharmacol 12:653146. https://doi.org/10.3389/fphar.2021.653146
Lanznaster D et al (2019) Adenosine A, (in eng). Cells 8(12). https://doi.org/10.3390/cells8121630
Jackson EK, Mi Z (2014) The guanosine-adenosine interaction exists in vivo, (in eng). J Pharmacol Exp Ther 350(3):719–726. https://doi.org/10.1124/jpet.114.216978
Rathbone MP et al (1998) The trophic effects of purines and purinergic signaling in pathologic reactions of astrocytes, (in eng). Alzheimer Dis Assoc Disord 12(Suppl 2):S36–45
Jackson EK, Cheng D, Jackson TC, Verrier JD, Gillespie DG (2013) Extracellular guanosine regulates extracellular adenosine levels, (in eng). Am J Physiol. Cell physiol, Research Support, N.I.H., Extramural 304(5): C406–21. https://doi.org/10.1152/ajpcell.00212.2012
Ciccarelli R et al (2000) Cultured astrocyte proliferation induced by extracellular guanosine involves endogenous adenosine and is raised by the co-presence of microglia, (in eng). Glia, Research Support, Non-U.S. Gov't 29(3): 202–11
Funding
Supported by: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq -Brasil) and Instituto Nacional de Ciência e Tecnologia (INCT/EN—CNPq) for CIT; Ministerio de Ciencia, Innovación y Universidades-Agencia Estatal de Investigación-FEDER funds/European Regional Development Fund – “A way to build Europe” grant PID2020-118511RB-I00 and Departament de Recerca i Universitats de la Generalitat de Catalunya (2021 SGR 00698) for FC; PRIN-PNRR 2022 to PDI.
Author information
Authors and Affiliations
Contributions
CIT, MZ and FC wrote the main manuscript text and CT and MZ prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tasca, C.I., Zuccarini, M., Di Iorio, P. et al. Lessons from the physiological role of guanosine in neurodegeneration and cancer: Toward a multimodal mechanism of action?. Purinergic Signalling (2024). https://doi.org/10.1007/s11302-024-10033-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11302-024-10033-y