
Abstract
Amyloid-beta (Aβ) peptides are the major neuropathological hallmarks related with Alzheimer’s disease (AD). Aβ peptides trigger several biochemical mechanisms of neurotoxicity, including neuroinflammation and glutamatergic neurotransmission impairment. Guanosine is the endogenous guanine-derived nucleoside that modulates the glutamatergic system and the cellular redox status, thus acting as a neuroprotective agent. Here, we investigated the putative neuroprotective effect of guanosine in an AD-like mouse model. Adult mice received a single intracerebroventricular injection of Aβ1–40 (400 pmol/site) or vehicle and then were treated immediately, 3 h later, and once a day during the subsequent 14 days with guanosine (8 mg/kg, intraperitoneally). Aβ1–40 or guanosine did not alter mouse locomotor activity and anxiety-related behaviors. Aβ1–40-treated mice displayed short-term memory deficit in the object location task that was prevented by guanosine. Guanosine prevented the Aβ1–40-induced increase in latency to grooming in the splash test, an indicative of anhedonia. Aβ1–40 increased Na+-independent glutamate uptake in ex vivo hippocampal slices, and guanosine reversed it to control levels. The repeated administration of guanosine increased hippocampal GDP levels, which was not observed in the group treated with Aβ plus guanosine. Aβ1–40 induced an increase in hippocampal ADP levels. Aβ1–40 decreased GFAP expression in the hippocampal CA1 region, an effect not modified by guanosine. No differences were observed concerning synaptophysin and NeuN immunolabeling. Together, these results show that guanosine prevents memory deficit and anhedonic-like behavior induced by Aβ1–40 that seem to be linked to glutamate transport unbalance and alterations on purine and metabolite levels in mouse hippocampus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyloid peptide toxicity is a common feature associated with Alzheimer’s disease (AD), a condition that affects approximately 36 million people worldwide—a number that is expected to raise once life span increases [1–3]. Along with neuronal death, amyloid-β (Aβ) plaques and neurofibrillary tangles are the main neuropathological hallmarks found in the brain of AD patients. The major components of Aβ plaques are amyloid peptides (40–42 amino acids) derived from amyloid precursor protein misprocessing [4, 5]. Aβ peptide-induced neurotoxicity involves different molecular mechanisms, particularly glutamatergic neurotransmission impairment. For instance, Aβ peptides lead to overstimulation of N-methyl-D-aspartate (NMDA) receptors and the massive entry of calcium in the cell, promoting a toxic effect that culminates in synaptic loss and later in neuronal death [6, 7].
Central nervous system (CNS) infusion of Aβ1–40 has been extensively used as a model of AD-like pathology. In mice, intracerebroventricular (i.c.v.) Aβ1–40 infusion causes behavioral disturbances (as cognitive deficits), and biochemical changes in the CNS, which include oxidative stress, neuroinflammation, glutamatergic excitotoxicity associated with a decrease in glutamate transporter activity and expression, and neuronal death [8–12]. A single i.c.v. infusion of Aβ peptides can also induce cognitive and emotional alterations in mice [13, 14], thus modeling AD symptomatology [15, 16]. One of these symptoms may be depression, a psychiatric condition characterized by an unbalance in various neurotransmitter systems including the glutamatergic transmission, and drugs that modulate glutamatergic system present great potential as anti-depressant agents [17, 18].
Guanosine is an endogenous nucleoside known to act as intercellular signaling modulator at the CNS, and it can modulate glutamatergic transmission. Guanosine capacity of counteracting glutamatergic toxicity was shown in vivo, since guanosine reduces seizure incidence induced by quinolinic acid, an NMDA receptor agonist [19–23]. In vitro, guanosine prevents glutamate uptake impairment caused by an ischemia model both in hippocampal slices and in cultured astrocytes [24–26]. Ex vivo guanosine decreased K+-stimulated glutamate release from hippocampal synaptosomal preparation in naive rats [27] and also prevents glutamatergic excitoxicity in hippocampal slices by decreasing glutamate release [28]. Altogether, these results indicate that guanosine neuroprotective effects involve, at least partially, the modulation of glutamate transport. Recently, different research groups have pointed to guanosine as a potential therapeutic agent against depression, considering its modulatory action on glutamatergic transmission (for review, see [29]). For instance, the anti-depressant-like effect exerted by guanosine in mice subjected to the tail suspension test was blocked by the activation of glutamate NMDA receptors [30].
Regarding Aβ peptide toxicity, by inhibiting apoptosis and reactive oxygen (ROS) production, guanosine showed neuroprotective effects against Aβ25–35 and Aβ1–42 toxicities in SH-SY5Y neuroblastoma cells [31, 32]. Besides these previous studies showing the neuroprotective effects of guanosine in in vitro models of Aβ toxicity, there is no study addressing its effect in an in vivo AD-like model. Therefore, by combining behavioral and neurochemical assays, we investigated the putative neuroprotective effect of guanosine in adult Swiss mice submitted to the damage induced by a single i.c.v. injection of Aβ1–40.
Material and Methods
Animals
Three-month-old male Swiss mice (weighing 45–50 g) supplied by the animal facility of the Federal University of Santa Catarina (UFSC, Florianópolis, Brazil) were kept in collective cages (15 animals per cage) and maintained in a room under controlled temperature (23 ± 1 °C) and 12-h light cycle (lights on 7:00 a.m.), with free access to food and water. All tests were carried out during the light phase of the cycle. Efforts were made to minimize the number of animals used and their suffering. The procedures used in the present study complied with the guidelines on animal care of the UFSC Ethics Committee on the Use of Animals, which follows the “Principles of Laboratory Animal Care” from NIH.
Drugs and Treatments
Aβ1–40 (Sigma-Aldrich, Germany) was dissolved in sterile deionized water to 6 mg/mL, and incubated at 37 °C for 4 days to aggregate, according to [33]. The solution was diluted to 1 mg/mL in 0.1 M phosphate-buffered saline (PBS), and aliquots were maintained at −20 °C until use. An aliquot of the peptides used in this study was added to copper formvar grids and contrasted with 2 % phosphotungstic acid for observation in a transmission electronic microscope (Fig. 1). The aggregated form of Aβ1–40 (400 pmol/site) or PBS (vehicle) was administered i.c.v. as previously described [9, 11, 12]. Mice were anesthetized with isoflurane 0.96 % (0.75 CAM; Abbot Laboratórios do Brasil Ltda., RJ, Brazil) until loss of postural reflex and then gently restrained by hand. An incision of approximately 1 cm was performed over mouse skull midline, and the i.c.v. injections were made free-hand with a 27-G needle into a virtual point located 1 mm right and 1 mm posterior to the bregma (coordinates from bregma anteroposterior = −0.1 mm, mediolateral = 1 mm, and dorsoventral = −3 mm). Aβ1–40 or PBS solutions (3 μL) were injected with an infusion pump (Insight) during 1 min. In preliminary experiments, the correct injection site was confirmed by Evans blue injections. Immediately after i.c.v. injections, 3 h later, and daily during 14 consecutive days, animals were treated with saline 0.9 % (control group) or guanosine (8 mg/kg) via intraperitoneal (i.p.). Figure 1 summarizes the protocol used in this study.

Timeline of experimental protocol of drug administration, behavioral tests, and biochemical analysis. In day 1, mice received an intracerebroventricular infusion of PBS or Aβ1–40 (400 pmol/site, 3 μL) and immediately treated intraperitoneally (i.p.) with saline (0.9 % NaCl) or guanosine (8 mg/kg in saline solution). Three hours later, mice received another i.p. injection of saline or guanosine. In the next 14 consecutive days, mice received one i.p. injection per day of saline or guanosine. Mouse behavior was analyzed from day 16 to 20, and then, mouse hippocampi were used for biochemical and immunohistochemical analyses at 21st day. left panel Photomicrographs of Aβ1–40 aggregates obtained in a transmission electronic microscopy after contrast with 2 % phosphotungstic acid (a scale bar 1 μm and b scale bar 100 nm)
Behavioral Tests
At the 16th day after the Aβ1–40 or PBS infusion, mice were tested in the open field. In the next day, short-term spatial memory was assessed in the novel object location and Y-maze tasks at day 18. Anhedonic- and depressive-like behaviors were analyzed in the sucrose-splash test and tail suspension test at days 19 and 20, respectively. All animals performed the behavioral tests described here. Behavioral tests were carried out between 9:00 and 14:00 h, and they were scored by the same rater in an observation sound-attenuated room under low-intensity light (12 lx), where the mice had been habituated for at least 1 h before the beginning of the tests. Behavior was monitored through a video camera positioned above the apparatuses, and the videos were later analyzed with the ANY Maze video tracking system (Stoelting Co., Wood Dale, IL, USA). The apparatus were cleaned with 10 % ethanol between animals to avoid odor clues.
Open Field
Spontaneous locomotor activity of mice in the open field (a square arena with 50 cm wide × 50 cm deep × 40 cm high) was recorded during 15 min (modified from [34]). Traveled distance corresponds to the first 15 min of exploration. Data presented on average speed, total time spend in the center, and number of entries in the center zone of the open field correspond to analysis made in the first 5 min, as exploration decreases time dependently, and mice were more active during this period. The apparatuses were cleaned with a solution of 10 % ethanol between tests in order to avoid odor animal clues.
Novel Object Location
Short-term spatial memory of mice was assessed using the object location task as described elsewhere [35]. Mice were placed in the center of the open field with two identical objects for 5 min, and object exploration was recorded using a stopwatch when mice sniffed, whisked, or looked at the objects from no more than 1 cm away. After 90 min, one object was moved to a new location, and the time spent by the animals exploring the objects in new (novel) and old (familiar) locations was recorded over 5 min. Objects were thoroughly cleaned with 10 % ethanol after each trial to minimize the presence of olfactory trails. To analyze the cognitive performance, a location index was calculated as (T novel × 100)/(T novel + T familiar), where T novel is the time spent exploring the displaced object and T familiar is the time spent exploring the non-displaced object.
Y-Maze
Spontaneous alternation in the Y-maze was used as a measure of spatial working memory. The Y-maze is composed by three equally spaced arms (120°, 41 cm long, 15 cm high) with 5-cm-wide floor. Arms were labeled A, B, or C. Each mouse was place facing one arm end and allowed to move freely by all compartments for 5 min, and one arm entry was counted after the animal entered the arm with all four paws. One alternation is defined as entries in all three arms consecutively, and results are given as the percentage of correct alternations, calculated as [(real alternations/maximum alternations) × 100]. Maximum alternation is the number of total arm entries minus two [36]. The maze was cleaned with 10 % alcohol between sessions.
Sucrose Splash
The splash test was carried out as described by [37], with minor modifications, and consists of squirting a 10 % sucrose solution on the dorsal coat of a mouse placed individually in clear Plexiglas boxes (9 × 7 × 11 cm). After applying sucrose solution, the latency and time spent grooming were recorded for a period of 5 min. The apparatuses were cleaned with a solution of 10 % ethanol between tests in order to hide animal clues.
Tail Suspension
Immobility time during the tail suspension test was measured according to the method described by [38]. Briefly, mice both acoustically and visually isolated were suspended 50 cm above the floor by adhesive tape placed approximately 2 cm from the tip of the tail. Immobility time was recorded during a 6-min period [39].
Preparation of Hippocampal Slices
Mice were decapitated 21 days after the Aβ1–40 i.c.v. injection. The brain was removed, and hippocampi were rapidly dissected in ice-cold Krebs–Ringer bicarbonate buffer (KRB) of the following composition: 122 mM NaCl, 3 mM KCl, 1.2 mM MgSO4, 1.3 mM CaCl2, 0.4 mM KH2PO4, 25 mM NaHCO3, and 10 mM D-glucose, bubbled with 95 % O2–5 % CO2 up to pH 7.4. Hippocampal slices (400-μm-thick transverse sections) obtained with a McIlwain tissue chopper were incubated for 30 min before performing glutamate transport assays.
Glutamate Uptake and Release Assays
L-[3H]glutamate uptake into hippocampal slices was evaluated as previously described [40]. After incubation, hippocampal slices were washed for 15 min at 37 °C in a Hank’s balanced salt solution (HBSS), composition in mM, 1.29 CaCl2, 136.9 NaCl, 5.36 KCl, 0.65 MgSO4, 0.27 Na2HPO4, 1.1 KH2PO4, and 5 HEPES pH 7.4. Uptake was assessed by adding 0.33 μCi/mL of L-[3H]glutamate with 100 μM of unlabeled glutamate in a final volume of 300 μL for 7 min. Incubation was stopped immediately by discarding the incubation medium followed by two ice-cold washes with 1 mL HBSS. Slices were solubilized with 200 μL 0.1 % NaOH/0.01 % SDS and stored overnight. High-affinity Na+-dependent glutamate uptake was considered as the difference between the measured total uptake and the Na+-independent uptake, assayed in the presence of choline chloride and without sodium ions. Incubations were performed in triplicates. Results are expressed as nanomole of glutamate taken up per minute per milligram of protein.
D-[3H]aspartate release was measured modified from [22]. Previous studies from our laboratory showed similar results by using D-[3H]aspartate or L-[3H]glutamate [41]. Slices were incubated in HBSS (pH 7.4) for 15 min at 37 °C in the presence of 0.33 μCi/mL D-[3H]aspartate plus 100 μM aspartate (final concentration) for the uptake period, thus loading the intracellular pool of D-[3H]aspartate. Slices were then washed four times with 1 mL cold HBSS. To measure the basal D-[3H]aspartate release, slices were incubated in HBSS for 3 min at 37 °C. K+-stimulated D-[3H]aspartate release was assayed as described for basal release, except that the incubation medium contained 40 mM KCl (NaCl decreased accordingly) to induce slice depolarization. Results are expressed as a ratio of K+-stimulated/basal D-[3H]aspartate release. Radioactivity was measured by using a liquid scintillation counter (Wallac 1409).
Purine-Level Measurement
Hippocampi were dissected and homogenized in 300 μL of PBS and TCA 10 % and centrifuged at 10.000×g at 4 °C for 10 min. The supernatant was collected and neutralized with KHCO3 1.5 M (×2 volume). Aliquots of 100 μL were filtered in 0.22-μm membrane and frozen at −70 °C until use.
High-performance liquid chromatography (HPLC) was performed in tissue homogenate aliquots to quantify adenine and guanine derivatives [42, 43]. Briefly, sample separation was carried out with a reverse-phase column (LC-18, 250 × 4.6 mm, Supelco) in a Shimadzu Instruments liquid chromatograph (1-mL loop valve injection). The mobile phase flowed at a rate of 1.2 mL/min, and column temperature was 24 °C. Buffer composition is A 150 mM potassium chloride in 150 mM phosphate buffer, pH 5.5, and B 15 % acetonitrile in buffer A. The gradient profile was modified according to the content of buffer B in the mobile phase, 0 % at 0.00 min, 7 % at 2.45 min, 50 % at 10 min, 100 % at 12.30 min, and 0 % at 12.40 min. Absorbance was read at 254 nm in a Shimadzu UV detector and compared with relative standards.
Immunohistochemistry
For the immunohistochemical analyses, animals (four mice per group) were anesthetized and perfused with iced saline solution, then 4 % paraformaldehyde [44] in 0.1 M PBS, pH 7.4. Brains were removed and maintained in PFA 4 % solution overnight and then cryoprotected in a 30 % sucrose solution in PBS at 4 °C [45]. Coronal sections (40 μm) obtained in a cryostat (Leica, Germany) were collected for immunohistochemistry in a 1-in-6 random series. Sections were incubated for 30 h at 4 °C with rabbit GFAP antibody (Sigma-Aldrich, 1:500), mouse NeuN antibody (Sigma-Aldrich, 1:500), or mouse synaptophysin antibody (Sigma-Aldrich, 1:500) in phosphate-buffered saline Tris-buffered saline/Tween-20 (TBS-T) containing 5 % bovine serum albumin (BSA). After several washes with PBS, sections were incubated with Alexa 594-conjugated donkey anti-mouse and Alexa 488-conjugated goat anti-rabbit antibodies for 2 h at room temperature. To determine fluorescence intensity, images were obtained in a fluorescent microscope (Olympus, Japan) and analyzed using NIH ImageJ 1.36b imaging software (NIH, Bethesda, MD, USA).
Immunoblotting Analysis
Expression of synaptophysin immunocontent was determined by Western blot analysis as described in [41], with minor modifications. Mouse hippocampi solubilized with SDS-stopping solution (4 % SDS, 2 mM EDTA, 8 % β-mercaptoethanol, and 50 mM Tris, pH 6.8) were separated (50 μg of total protein/track) in 10 % SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5 % BSA (1 h) in TBS (Tris 10 mM, NaCl 150 mM, pH 7.5), followed by three-time washing with TBS-T (0.05 %, pH 7.5) and incubation with primary antibody anti-synaptophysin (dilution 1:1000, Sigma-Aldrich) or β-actin (dilution 1:1000, Santa Cruz) overnight, at 4 °C. Then, membranes were incubated with a secondary antibody (anti-mouse IgG, dilution 1:2500, Santa Cruz) for 1 h at room temperature. Immunocomplexes visualized with the enhancing chemiluminescence (ECL) detection system (GE Healthcare) at ChemiDoc (Bio-Rad) were quantified using the Scion Image software (Scion Corporation).
Protein Measurement
Protein content was evaluated by the method of [46] for glutamate uptake and by the method of Peterson [47] for Western blotting analysis. Bovine serum albumin (Sigma) was used as standard.
Statistical Analysis
Data were tested for normality by using the Kolmogorov-Smirnov normality test. For data obtained in the novel object location, one-sample t test was used to determine whether the discrimination index was different from chance performance (50 %). For the rest of data, comparisons among groups were performed by two-way analysis of variance [48], followed by Newman-Keuls test if necessary, with P < 0.05 considered to be statistically significant.
Results
Guanosine Reduces the Cognitive Deficit Induced by Aβ1–40
Figure 1 presents the timeline of experimental protocol for drug administration, behavioral tests, and biochemical analysis performed in the present study. Sixteen days after Aβ1–40 i.c.v. infusion, an initial open field analysis of mice treated with Aβ1–40 and/or guanosine showed no differences among groups on spontaneous locomotion or time spent in the central zone of the apparatus (Fig. 2). Furthermore, results collected in minute-by-minute intervals indicated a habituation response of all groups, as mouse exploratory behavior decreased over time (Fig. 2a).

Evaluation of mouse locomotor activity in the open field test following Aβ1–40 i.c.v. infusion and guanosine treatment. Mice received guanosine (8 mg/kg, i.p.) or saline (10 μL/mg, i.p.) for 14 consecutive days after the i.c.v. infusion of Aβ1–40 (400 pmol/site) or PBS (control group). Mice were evaluated at the 16th day in the open field test for the following parameters: total distance traveled (a), average speed (b), total time spent at the center zone (c), and entry number at the center zone of the open field apparatus (d). b–d represent the data from the first 5 min of exposition. Data represent means + SEM (N = 9–10). Statistical analysis shows no difference among groups (two-way ANOVA)
Additionally, mouse spatial memory was analyzed in the object location test and in the Y-maze task. Aβ1–40-treated mice displayed impaired short-term spatial memory in the object location test (a decrease in the index of recognition of the moved object in the object location test; Fig. 3a). Two-way ANOVA revealed a significant difference between control group and Aβ1–40 (F(1,29) 4.9329, P < 0.05). Guanosine treatment reduced partially the decreased discrimination index induced by Aβ1–40, as statistical analysis showed no difference of Aβ1–40 plus guanosine group from control or from Aβ1–40 plus saline group.

Evaluation of mouse short-term spatial memory in the object location task (a) and in the Y-maze (b) after Aβ1–40 i.c.v. infusion and guanosine treatment. Mice received guanosine (8 mg/kg, i.p.) or saline (10 μL/mg, i.p.) for 14 consecutive days after the i.c.v. infusion of Aβ1–40 (400 pmol/site) or PBS (control group). Mice were evaluated at the 17th and 18th days in the novel object location (a) and Y-maze (b), respectively. Data represent means + SEM (N = 9–10). *P < 0.05 compared to the hypothetical 50 % (random investigation). Statistical analysis shows a significant difference compared to PBS + saline group (# P < 0.05; two-way ANOVA followed by post hoc test Newman-Keuls)
Mouse spatial memory assessed in the Y-maze revealed no alterations in correct alternations among all groups in this test (Fig. 3b).
Guanosine Displays Anti-Anhedonic Effect in Mice After Aβ1–40 Infusion
Mouse anhedonic- and depressive-like behaviors were also investigated after Aβ1–40 infusion. No significant differences were observed among groups in the immobility time in the tail suspension test (Fig. 4a). On the other hand, Aβ1–40-treated mice presented an increase in latency for initiating grooming behavior in the splash test (F(1,30) = 7.3079, P < 0.05; Fig. 4b). Guanosine treatment in mice that received Aβ1–40 evoked similar grooming latency as compared to control group. No significant differences were observed among groups regarding the total time of grooming (Fig. 4c).

Evaluation of mouse immobility in the tail suspension test (a) and self-cleanness behavior (b, c) after Aβ1–40 i.c.v. infusion and guanosine treatment. Mice received guanosine (8 mg/kg, i.p.) or saline (10 μL/mg, i.p.) for 14 consecutive days after the i.c.v. infusion of Aβ1–40 (400 pmol/site) or PBS (control group). a Total immobility time of mice submitted to the tail suspension test. b Latency for initiating the self-cleanness behavior in the sucrose splash test and c total self-cleanness time in the sucrose splash test. Data represent means + SEM (N = 9–10). Statistical analysis shows a significant difference compared to PBS + saline group (*P < 0.05) and to Aβ1–40 + saline group (# P < 0.05; two-way ANOVA followed by post hoc test Newman-Keuls)
Guanosine Counteracts Aβ1–40-Increased Na+-Independent Glutamate Uptake
After behavioral analyses, we performed ex vivo glutamate uptake and release assays in mouse hippocampal slices. Aβ1–40 or guanosine did not alter total glutamate uptake (Fig. 5a). However, by discriminating the Na+-dependent and Na+-independent components of glutamate transport, it was observed that Aβ1–40 induced a tendency of decreasing high-affinity Na+-dependent glutamate uptake, although there are no statistical differences among groups (Fig. 5b). Additionally, analysis of the low-affinity Na+-independent glutamate uptake showed that Aβ1–40 induced a statistical significant increase in Na+-independent transport (F(1,42) = 0.6692, P < 0.05 compared to PBS + saline group), and guanosine treatment counteracted this effect induced by Aβ1–40 infusion (F(1,42) = 4.10, P < 0.05; Fig. 5b). Aβ1–40 also induced a non-significant tendency of increasing glutamate release (Fig. 5c).

Evaluation of glutamate uptake (a, b) and release (c) after Aβ1–40 i.c.v. infusion and guanosine treatment. Mice received guanosine (8 mg/kg, i.p.) or saline (10 μL/mg, i.p.) for 14 consecutive days after the i.c.v. infusion of Aβ1–40 (400 pmol/site) or PBS (control group). After behavioral analysis, mice were killed and their hippocampi were dissected for preparing slices. a Total glutamate uptake obtained values. b High-affinity Na+-dependent and low-affinity Na+-independent glutamate uptake values. c Glutamate release values. Data represent means ± SEM (N = 6–8). Statistical analysis shows a significant difference compared to PBS + saline group (*P < 0.05) and to Aβ1–40 + saline group (# P < 0.05; two-way ANOVA followed by post hoc test Newman-Keuls)
Measurement of Guanine and Adenine Derivative Levels in the Hippocampus of Mice After Aβ1–40 Infusion and Guanosine Treatment
Guanosine distribution in CNS structures and influence on other purine derivative levels were shown elsewhere, but no studies so far investigated adenine and guanosine derivative levels after a chronic treatment protocol in mice (for review, see [49]). Here, we analyzed guanine and adenine derivative levels after guanosine chronic treatment. Data presented in Table 1 show HPLC analysis of purine concentration in mouse hippocampus 21 days after Aβ1–40 infusion. Aβ1–40 induced a significant increase in ADP levels compared to control group (PBS; F(1,17) 8.28, P = 0.01) and a slight increase in ATP levels, effects that were not modified by guanosine treatment. Guanosine treatment increased GDP levels compared to PBS group (F(1,17) 11.06, P < 0.05), and this effect was not observed in the Aβ1–40 plus guanosine group.
Guanosine Effects on GFAP, NeuN, and Synaptophysin Immunocontents
We performed GFAP immunohistochemistry in order to investigate the occurrence of reactive astrogliosis. Immunohistochemistry was also used to investigate neuronal and synaptic losses, respectively, by analyzing NeuN and synaptophysin expressions. Our results suggest that 21 days following i.c.v. Aβ1–40 infusion decreased GFAP immunocontent at hippocampal CA1 region (F(1,7) = 22.715, P < 0.05), and guanosine did not affect this alteration. No statistical differences were observed at regions CA3 and dentate gyrus (DG) for GFAP expression (Fig. 6).

GFAP immunoreactivity evaluation 21 days after Aβ1–40 i.c.v. infusion and guanosine treatment at hippocampal CA3, CA1, and dentate gyrus (DG) regions. Mice received guanosine (8 mg/kg, i.p.) or saline (10 μL/mg, i.p.) for 14 consecutive days after the i.c.v. infusion of Aβ1–40 (400 pmol/site) or PBS (control group). a Representative photomicrographs of GFAP immunohistochemistry in hippocampal slices performed 21 days after Aβ1–40 i.c.v. infusion. Scale bar 25 μm. b Graphs represent means ± SEM (N = 3–4) of optic density obtained with ImageJ analysis software. Statistical analysis suggests a significant difference compared to PBS + saline group (*P < 0.05; two-way ANOVA followed by post hoc test Newman-Keuls)
Immunohistochemistry analysis regarding NeuN expression 21 days after Aβ1–40 i.c.v. infusion revealed no differences among treated groups, although we observed a slight decrease in the hippocampal CA1 region in the Aβ1–40 group (Fig. 7). Immunohistochemistry and Western blotting performed to analyze synaptophysin expression also showed no alterations in synaptic density at mouse hippocampus after Aβ1–40 i.c.v. infusion (Fig. 8).

NeuN immunoreactivity evaluation 21 days after Aβ1–40 i.c.v. infusion and guanosine treatment at hippocampal CA3, CA1, and dentate gyrus (DG) regions. Mice received guanosine (8 mg/kg, i.p.) or saline (10 μL/mg, i.p.) for 14 consecutive days after the i.c.v. infusion of Aβ1–40 (400 pmol/site) or PBS (control group). a Representative photomicrographs of NeuN immunohistochemistry in hippocampal slices performed 21 days after Aβ1–40 i.c.v. infusion. Scale bar 25 μm. b Graphs represent means ± SEM (N = 3–4) of optic density obtained with ImageJ analysis software. Statistical analysis suggests no difference among groups (two-way ANOVA)

Synaptic density evaluation 21 days after Aβ1–40 i.c.v. infusion. Mice received guanosine (8 mg/kg, i.p.) or saline (10 μL/mg, i.p.) for 14 consecutive days after the i.c.v. infusion of Aβ1–40 (400 pmol/site) or PBS (control group). a Representative photomicrographs of synaptophysin immunohistochemistry in CA3, CA1, and dentate gyrus (DG) hippocampal regions, performed 21 days after Aβ1–40 i.c.v. infusion. Scale bar 25 μm. b Fluorescence quantification of photomicrographs from CA3, CA1, and DG. Graphs represent means ± SEM (N = 3–4) of optic density obtained with ImageJ analysis software. c Representative immunoblotting of synaptophysin (Syn) and β-actin content in whole hippocampal extracts. Graph represents the quantitative analysis expressed as the ratio of synaptophysin/β-actin. Statistical analysis suggests no difference among groups (two-way ANOVA)
Discussion
Amyloid-β peptide toxicity is tightly involved with AD occurrence. Several models of AD-like pathology use Aβ peptides in order to mimic some features of AD, aiming to further understand AD pathophysiology and investigate promising neuroprotective agents. A study performed in SH-5YSY neuroblastoma cells showed that guanosine, an endogenous guanine-derived nucleoside, has neuroprotective effect against ROS production and apoptotic cell death induced by Aβ (Tarozzi et al. 2011). No further studies have investigated guanosine neuroprotection in rodent models of AD. Therefore, in the present study, we investigated guanosine putative neuroprotective effects against Aβ1–40 i.c.v. infusion in mice.
Mouse exploratory behavior and locomotor activity were analyzed in the open field task after Aβ1–40 i.c.v. infusion and guanosine chronic treatment, and we reported no differences among treatments used in this study. Mouse spatial memory was analyzed in the object location and in the Y-maze task. Aβ1–40-treated mice displayed impaired short-term spatial memory in the object location test, a task dependent on CA1 region of the hippocampus [35]. We also reported here, for the first time, guanosine effect on reducing the cognitive deficit induced by Aβ1–40 i.c.v. infusion in mice. Guanosine effect on preventing cognitive deficits was shown before in a hepatic encephalopathy model in rats [50]. We observed no differences among groups in the Y-maze task. Although AD transgenic models present an alteration in this task [51], no previous studies using Aβ1–40 i.c.v. infusion in mice showed cognitive deficits in the Y maze. The lack of cognitive deficit in this test could be related to the fact that this task evaluates working memory more related to the prefrontal cortex function [52, 53]. Taken together, these results suggest that Aβ1–40 i.c.v. infusion protocol causes impairment in hippocampal-dependent memories, although additional behavioral analysis must be performed to confirm this hypothesis.
Because of its viscosity, the sucrose solution dirties the mouse fur and animals initiate grooming behavior as an index of self-cleanness and motivational behavior. The absence or delay in the onset of this behavior is considered to parallel with some symptoms of depression such as anhedonia [54]. Bettio and colleagues [30, 55] have already demonstrated guanosine anti-depressant-like effects in mice, and guanosine has been proposed acts as an anti-depressant agent via the modulation of glutamatergic transmission [29]. For instance, guanosine anti-depressant-like effect occurs through interaction with the glutamatergic system, as pretreatment with NMDA (the selective NMDA receptor agonist) inhibited guanosine effect in the tail suspension test [30]. In the present study, we did not observe a significant effect of guanosine in the tail suspension test. This observation might be due to the treatment schedule used in our study. Bettio and colleagues [30] exposed mice to the test 60 min after guanosine administration, while in the present study, there was a gap of 5 days between the last treatment with guanosine and the behavioral evaluation of mice in this task. However, even in this protocol where an anti-depressant-like effect of guanosine per se was not observed, it was effective in reducing the anhedonic-like behavior caused by Aβ1–40.
After analyzing mouse behavioral alterations, we evaluated biochemical changes in the hippocampus following Aβ1–40 and guanosine treatment. Regarding glutamatergic transmission, the Na+-independent glutamate transport, carried out mainly by cystine–glutamate exchanger (Xc−) system, has call attention due to its effect of increasing intracellular cysteine levels for the anti-oxidant glutathione synthesis [56]. However, in excitotoxic conditions, a reversal activity of these transporters can be observed, thus increasing intracellular glutamate levels and decreasing cystine and consequently cysteine, the limiting substrate to glutathione synthesis [57]. Our group has previously reported a decrease in the hippocampal immunoreactivity of GLAST and GLT-1 (astrocytic Na+-dependent glutamate transporters responsible for the high affinity and major capacity of glutamate uptake activity) [58] and reduced glutathione levels 16 days after Aβ1–40 i.c.v. infusion in mice [11]. More recently, we also demonstrated that there is an increase in Na+-independent glutamate uptake in the prefrontal cortex, 18 days after Aβ1–40 infusion [9]. Herein, we observed an increase in hippocampal Na+-independent glutamate uptake 21 days after Aβ1–40 infusion, and guanosine treatment prevented this glutamate uptake alteration. Interestingly, Aβ1–40 injection at mouse hippocampus induced an increase in microglial xCT expression, the functional component of Xc− system [59], and Albrecht and colleagues [60] showed that guanosine increased normal Xc− activity in HT22 neuroblastoma cells exposed to glutamatergic excitotoxicity. Identification on whether Aβ toxicity relates to Xc− reversed activity and if guanosine prevents this effect is under investigation in our laboratory.
Postmortem analysis of AD patient brains revealed that energetic deficiency is related to mitochondrial dysfunction [61], and this alteration could be induced by Aβ accumulation at mitochondrial membrane. Figueiredo and colleagues [8] reported the increase in mitochondrial complexes I, II, and IV activities caused by Aβ, although there was a decrease in the energetic metabolism. Here, we observed that Aβ1–40 infusion induced a significant increase in ADP and a slight increase in ATP levels in the hippocampus. Taken together, these data suggest a compensatory mechanism against Aβ1–40 damage, aiming the maintenance of brain energetic metabolism.
We also observed that guanosine treatment for 15 days induced an increase in hippocampal GDP levels. Previous studies demonstrated that after systemic guanosine treatment, guanosine and guanine levels raise soon after 15 min in rat brains, accompanied by an increase in the levels of the purine nucleoside phosphorilase (PNP), an enzyme of the salvage pathway that is responsible for guanosine metabolism [62]. In this regard, considering that we did not observe any differences in guanosine metabolite levels (as uric acid or xanthine), we can suggest that guanosine might be used directly in the salvage pathway for GDP synthesis, as GTP levels also slightly increase, although it did not reach significance. It is important to notice that Aβ plus guanosine-treated group did not show the increase in GDP levels, so an effect of Aβ1–40 in the purine salvage pathway or involved enzymes can be hypothesized, although more studies are needed to clarify the Aβ influence over the purinergic system.
Several studies showed that i.c.v. Aβ1–40 infusion induces reactive astrogliosis [11, 63], but no study reported GFAP immunoreactivity 21 days after Aβ1–40. Corroborating our results, studies performed in 3Tg-DA mice showed a decrease in GFAP expression in 3-month-old animals [64, 65]. A decrease in the GFAP immunoreactivity was reported in depressive patients and in rodent models of depression [66, 67], and it could be related to the depressive-like behavior (increase in the latency for grooming) observed in this study. Besides guanosine anti-depressant-like effect, no alterations on GFAP immunoreaction were observed after guanosine treatment.
Synaptic loss is also considered a characteristic of DA at initial stages [68, 69], and it was previously reported 14 days after Aβ1–40 infusion [8, 63]. In the present study, we did not found any differences regarding synaptophysin expression using both immunohistochemistry and Western blotting techniques performed 21 days after Aβ1–40 infusion. Regarding neuronal loss, we did found a slight decrease in NeuN expression in the CA1 region of the hippocampus following Aβ1–40 infusion, although statistical analysis showed no differences among groups. Therefore, additional studies are necessary to clarify the time-course alterations evoked by Aβ1–40 infusion. In fact, it is not possible to delineate a simple linear function between pure amyloidopathy and synaptic dysfunction associated to neuronal depletion. An interesting example relies on hAPP transgenic models presenting extremely high levels of cerebral Aβ but little or no neuronal depletion [70].
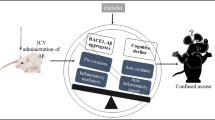
In conclusion, guanosine treatment prevents the cognitive deficit and the anhedonic-like behavior induced by Aβ1–40 in mice, what might be related to guanosine modulation of the hippocampal glutamatergic transmission altered by Aβ1–40. We also showed here for the first time the quantification of guanine and adenine derivative levels after Aβ1–40 infusion, suggesting an effect of Aβ over the purine salvage pathway, although Aβ1–40 effect over purinergic system must be further investigated. A hypothetical and schematic proposal encompassing general effects observed in our study is depicted in Fig. 9.

Schematic view of effects promoted by chronic guanosine treatment following Aβ1–40 i.c.v. infusion in mice. Guanosine treatment (8 mg/kg, i.p., for 15 days) prevented the cognitive deficit and anhedonic-like behavior induced in mice by Aβ1–40 (400 pmol, i.c.v.). right panel In mouse hippocampus, Aβ1–40 increased ATP and ADP levels. Aβ1–40 altered glutamate transport, mainly by increasing Na+-independent glutamate uptake. Guanosine treatment recovered glutamatergic transmission unbalance induced by Aβ1–40. This figure was produced using Servier Medical Art (http://www.servier.com) under creative common license
References
Mazurek AA (2000) Treatment of Alzheimer’s disease. N Engl J Med 342(11):821 author reply 821-822
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10(9):698–712. doi:10.1038/nrd3505
Masters CL, Selkoe DJ (2012) Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harbor Perspect Med 2(6):a006262. doi:10.1101/cshperspect.a006262
Selkoe DJ (2001) Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis: JAD 3(1):75–80
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256(5054):184–185
Parameshwaran K, Dhanasekaran M, Suppiramaniam V (2008) Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol 210(1):7–13. doi:10.1016/j.expneurol.2007.10.008
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27(11):2866–2875. doi:10.1523/JNEUROSCI.4970-06.2007
Figueiredo CP, Bicca MA, Latini A, Prediger RD, Medeiros R, Calixto JB (2011) Folic acid plus alpha-tocopherol mitigates amyloid-beta-induced neurotoxicity through modulation of mitochondrial complexes activity. J Alzheimers Dis: JAD 24(1):61–75. doi:10.3233/JAD-2010-101320
Martins WC, dos Santos VV, dos Santos AA, Vandresen-Filho S, Dal-Cim TA, de Oliveira KA, Mendes-de-Aguiar CB, Farina M, Prediger RD, Viola GG, Tasca CI (2015) Atorvastatin prevents cognitive deficits induced by intracerebroventricular amyloid-beta1–40 administration in mice: involvement of glutamatergic and antioxidant systems. Neurotox Res 28(1):32–42. doi:10.1007/s12640-015-9527-y
Medeiros R, Prediger RD, Passos GF, Pandolfo P, Duarte FS, Franco JL, Dafre AL, Di Giunta G, Figueiredo CP, Takahashi RN, Campos MM, Calixto JB (2007) Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J Neurosci 27(20):5394–5404. doi:10.1523/JNEUROSCI.5047-06.2007
Piermartiri TCB, Figueiredo CP, Rial D, Duarte FS, Bezerra SC, Mancini G, de Bem AF, Prediger RDS, Tasca CI (2010) Atorvastatin prevents hippocampal cell death, neuroinflammation and oxidative stress following amyloid-β1–40 administration in mice: evidence for dissociation between cognitive deficits and neuronal damage. Exp Neurol 226:274–284. doi:10.1016/j.expneurol.2010.08.030
Prediger RD, Franco JL, Pandolfo P, Medeiros R, Duarte FS, Di Giunta G, Figueiredo CP, Farina M, Calixto JB, Takahashi RN, Dafre AL (2007) Differential susceptibility following beta-amyloid peptide-(1-40) administration in C57BL/6 and Swiss albino mice: evidence for a dissociation between cognitive deficits and the glutathione system response. Behav Brain Res 177(2):205–213. doi:10.1016/j.bbr.2006.11.032
Pamplona FA, Pandolfo P, Duarte FS, Takahashi RN, Prediger RDS (2010) Altered emotionality leads to increased pain tolerance in amyloid beta (a beta 1-40) peptide-treated mice. Behav Brain Res 212(1):96–102. doi:10.1016/j.bbr.2010.03.052
Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, De Felice FG, Ferreira ST (2013) Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol Psychiatry 18(10):1053–1054. doi:10.1038/mp.2012.168
Modrego PJ, Ferrandez J (2004) Depression in patients with mild cognitive impairment increases the risk of developing dementia of Alzheimer type: a prospective cohort study. Arch Neurol 61(8):1290–1293. doi:10.1001/archneur.61.8.1290
Geerlings MI, den Heijer T, Koudstaal PJ, Hofman A, Breteler MM (2008) History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology 70(15):1258–1264. doi:10.1212/01.wnl.0000308937.30473.d1
Sanacora G, Treccani G, Popoli M (2012) Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62(1):63–77. doi:10.1016/j.neuropharm.2011.07.036
Tokita K, Yamaji T, Hashimoto K (2012) Roles of glutamate signaling in preclinical and/or mechanistic models of depression. Pharmacol Biochem Behav 100(4):688–704. doi:10.1016/j.pbb.2011.04.016
de Oliveira J, Moreira EL, Mancini G, Hort MA, Latini A, Ribeiro-do-Valle RM, Farina M, da Rocha JB, de Bem AF (2013) Diphenyl diselenide prevents cortico-cerebral mitochondrial dysfunction and oxidative stress induced by hypercholesterolemia in LDL receptor knockout mice. Neurochem Res 38(10):2028–2036. doi:10.1007/s11064-013-1110-4
Schmidt AP, Avila TT, Souza DO (2005) Intracerebroventricular guanine-based purines protect against seizures induced by quinolinic acid in mice. Neurochem Res 30(1):69–73
Schmidt AP, Lara DR, de Faria MJ, da Silveira PA, Onofre Souza D (2000) Guanosine and GMP prevent seizures induced by quinolinic acid in mice. Brain Res 864:40–43. doi:10.1016/S0006-8993(00)02106-5
Tavares RG, Schmidt AP, Abud J, Tasca CI, Souza DO (2005) In vivo quinolinic acid increases synaptosomal glutamate release in rats: reversal by guanosine. Neurochem Res 30:439–444
Tavares RG, Schmidt AP, Tasca CI, Souza DO (2008) Quinolinic acid-induced seizures stimulate glutamate uptake into synaptic vesicles from rat brain: effects prevented by guanine-based purines. Neurochem Res 33(1):97–102. doi:10.1007/s11064-007-9421-y
Dal-Cim T, Martins WC, Santos AR, Tasca CI (2011) Guanosine is neuroprotective against oxygen/glucose deprivation in hippocampal slices via large conductance Ca(2)+-activated K+ channels, phosphatidilinositol-3 kinase/protein kinase B pathway activation and glutamate uptake. Neuroscience 183:212–220. doi:10.1016/j.neuroscience.2011.03.022
Dal-Cim T, Martins WC, Thomaz DT, Coelho V, Poluceno GG, Lanznaster D, Vandresen-Filho S, Tasca CI (2016) Neuroprotection promoted by guanosine depends on glutamine synthetase and glutamate transporters activity in hippocampal slices subjected to oxygen/glucose deprivation. Neurotox Res. doi:10.1007/s12640-015-9595-z
Quincozes-Santos A, Bobermin LD, de Souza DG, Bellaver B, Gonçalves CA, Souza DO (2013) Gliopreventive effects of guanosine against glucose deprivation in vitro. Purinergic Signal 9:643–654. doi:10.1007/s11302-013-9377-0
Almeida RF, Comasseto DD, Ramos DB, Hansel G, Zimmer ER, Loureiro SO, Ganzella M, Souza DO (2016) Guanosine anxiolytic-like effect involves adenosinergic and glutamatergic neurotransmitter systems. Mol Neurobiol. doi:10.1007/s12035-015-9660-x
Molz S, Dal-Cim T, Budni J, Martin-de-Saavedra MD, Egea J, Romero A, del Barrio L, Rodrigues AL, Lopez MG, Tasca CI (2011) Neuroprotective effect of guanosine against glutamate-induced cell death in rat hippocampal slices is mediated by the phosphatidylinositol-3 kinase/Akt/ glycogen synthase kinase 3beta pathway activation and inducible nitric oxide synthase inhibition. J Neurosci Res 89(9):1400–1408. doi:10.1002/jnr.22681
Deutschenbaur L, Beck J, Kiyhankhadiv A, Muhlhauser M, Borgwardt S, Walter M, Hasler G, Sollberger D, Lang UE (2016) Role of calcium, glutamate and NMDA in major depression and therapeutic application. Prog Neuro-Psychopharmacol Biol Psychiatry 64:325–333. doi:10.1016/j.pnpbp.2015.02.015
Bettio LE, Cunha MP, Budni J, Pazini FL, Oliveira A, Colla AR, Rodrigues AL (2012) Guanosine produces an antidepressant-like effect through the modulation of NMDA receptors, nitric oxide-cGMP and PI3K/mTOR pathways. Behav Brain Res 234(2):137–148. doi:10.1016/j.bbr.2012.06.021
Pettifer KM, Kleywegt S, Bau CJ, Ramsbottom JD, Vertes E, Ciccarelli R, Caciagli F, Werstiuk ES, Rathbone MP (2004) Guanosine protects SH-SY5Y cells against beta-amyloid-induced apoptosis. Neuroreport 15(5):833–836
Tarozzi A, Merlicco A, Morroni F, Bolondi C, Di Iorio P, Ciccarelli R, Romano S, Giuliani P, Hrelia P (2010) Guanosine protects human neuroblastoma cells from oxidative stress and toxicity induced by amyloid-beta peptide oligomers. J Biol Regul Homeost Agents 24(3):297–306
ElKhoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD (1996) Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 382(6593):716–719
Rial D, Castro AA, Machado N, Garcao P, Goncalves FQ, Silva HB, Tome AR, Kofalvi A, Corti O, Raisman-Vozari R, Cunha RA, Prediger RD (2014) Behavioral phenotyping of Parkin-deficient mice: looking for early preclinical features of Parkinson’s disease. PLoS One 9(12):e114216. doi:10.1371/journal.pone.0114216
Assini FL, Duzzioni M, Takahashi RN (2009) Object location memory in mice: pharmacological validation and further evidence of hippocampal CA1 participation. Behav Brain Res 204(1):206–211. doi:10.1016/j.bbr.2009.06.005
Onaolapo OJ, Onaolapo AY, Mosaku TJ, Akanji OO, Abiodun OR (2012) Elevated plus maze and Y-maze behavioral effects of subchronic, oral low dose monosodium glutamate in Swiss albino mice. J Pharm Biol Sci 3(4):21–27
Isingrini E, Camus V, Le Guisquet AM, Pingaud M, Devers S, Belzung C (2010) Association between repeated unpredictable chronic mild stress (UCMS) procedures with a high fat diet: a model of fluoxetine resistance in mice. PLoS One 5(4):e10404. doi:10.1371/journal.pone.0010404
Steru L, Chermat R, Thierry B, Simon P (1985) The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85(3):367–370
Ludka FK, Zomkowski AD, Cunha MP, Dal-Cim T, Zeni AL, Rodrigues AL, Tasca CI (2013) Acute atorvastatin treatment exerts antidepressant-like effect in mice via the L-arginine-nitric oxide-cyclic guanosine monophosphate pathway and increases BDNF levels. Eur Neuropsychopharmacol 23(5):400–412. doi:10.1016/j.euroneuro.2012.05.005
Molz S, Decker H, Oliveira IJ, Souza DO, Tasca CI (2005) Neurotoxicity induced by glutamate in glucose-deprived rat hippocampal slices is prevented by GMP. Neurochem Res 30(1):83–89
Molz S, Tharine DC, Decker H, Tasca CI (2008) GMP prevents excitotoxicity mediated by NMDA receptor activation but not by reversal activity of glutamate transporters in rat hippocampal slices. Brain Res 1231:113–120. doi:10.1016/j.brainres.2008.07.009
Schmidt AP, Bohmer AE, Leke R, Schallenberger C, Antunes C, Pereira MS, Wofchuk ST, Elisabetsky E, Souza DO (2008) Antinociceptive effects of intracerebroventricular administration of guanine-based purines in mice: evidences for the mechanism of action. Brain Res 1234:50–58. doi:10.1016/j.brainres.2008.07.091
Schmidt AP, Tort AB, Silveira PP, Bohmer AE, Hansel G, Knorr L, Schallenberger C, Dalmaz C, Elisabetsky E, Crestana RH, Lara DR, Souza DO (2009) The NMDA antagonist MK-801 induces hyperalgesia and increases CSF excitatory amino acids in rats: reversal by guanosine. Pharmacol Biochem Behav 91(4):549–553. doi:10.1016/j.pbb.2008.09.009
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622. doi:10.1373/clinchem.2008.112797
Muller AP, Gnoatto J, Moreira JD, Zimmer ER, Haas CB, Lulhier F, Perry ML, Souza DO, Torres-Aleman I, Portela LV (2011) Exercise increases insulin signaling in the hippocampus: physiological effects and pharmacological impact of intracerebroventricular insulin administration in mice. Hippocampus 21(10):1082–1092. doi:10.1002/hipo.20822
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Peterson GL (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83(2):346–356
Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, S-i O, Dziewczapolski G, Nakamura T, Cao G, Pratt aE, Kang Y-J, Tu S, Molokanova E, McKercher SR, Hires S, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, Powers ET, Kelly JW, Roberts a, Tong G, Fang-Newmeyer T, Parker J, Holland E, Zhang D, Nakanishi N, Chen H-SV, Wolosker H, Wang Y, Parsons LH, Ambasudhan R, Masliah E, Heinemann SF, Pina-Crespo JC, Lipton S (2013) A induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci 110:E2518–E2527. doi:10.1073/pnas.1306832110
Lanznaster D, Dal-Cim T, Piermartiri TC, Tasca CI (2016) Guanosine: a neuromodulator with therapeutic potential in aging-related disorders. Aging Dis 7(5). doi:10.14336/AD.2016.0208
Paniz LG, Calcagnotto ME, Pandolfo P, Machado DG, Santos GF, Hansel G, Almeida RF, Bruch RS, Brum LM, Torres FV, de Assis AM, Rico EP, Souza DO (2014) Neuroprotective effects of guanosine administration on behavioral, brain activity, neurochemical and redox parameters in a rat model of chronic hepatic encephalopathy. Metab Brain Dis 29(3):645–654. doi:10.1007/s11011-014-9548-x
Gotz J, Ittner LM (2008) Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci 9(7):532–544. doi:10.1038/nrn2420
Matheus FC, Rial D, Real JI, Lemos C, Ben J, Guaita GO, Pita IR, Sequeira AC, Pereira FC, Walz R, Takahashi RN, Bertoglio LJ, Cunha CD, Cunha RA, Prediger RD (2016) Decreased synaptic plasticity in the medial prefrontal cortex underlies short-term memory deficits in 6-OHDA-lesioned rats. Behav Brain Res 301:43–54. doi:10.1016/j.bbr.2015.12.011
Garabadu D, Ahmad A, Krishnamurthy S (2015) Risperidone attenuates modified stress-Re-stress paradigm-induced mitochondrial dysfunction and apoptosis in rats exhibiting post-traumatic stress disorder-like symptoms. J Mol Neurosci: MN 56(2):299–312. doi:10.1007/s12031-015-0532-7
Willner P, Towell A, Sampson D, Sophokleous S, Muscat R (1987) Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology 93(3):358–364
Bettio LE, Freitas AE, Neis VB, Santos DB, Ribeiro CM, Rosa PB, Farina M, Rodrigues AL (2014) Guanosine prevents behavioral alterations in the forced swimming test and hippocampal oxidative damage induced by acute restraint stress. Pharmacol Biochem Behav 127:7–14. doi:10.1016/j.pbb.2014.10.002
Bender AS, Reichelt W, Norenberg MD (2000) Characterization of cystine uptake in cultured astrocytes. Neurochem Int 37(2–3):269–276
Bringmann A, Pannicke T, Biedermann B, Francke M, Iandiev I, Grosche J, Wiedemann P, Albrecht J, Reichenbach A (2009) Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem Int 54(3–4):143–160. doi:10.1016/j.neuint.2008.10.014
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M (2006) System xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci 26(12):3345–3356. doi:10.1523/JNEUROSCI.5186-05.2006
Albrecht P, Henke N, Tien ML, Issberner A, Bouchachia I, Maher P, Lewerenz J, Methner A (2013) Extracellular cyclic GMP and its derivatives GMP and guanosine protect from oxidative glutamate toxicity. Neurochem Int 62(5):610–619. doi:10.1016/j.neuint.2013.01.019
Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58(4):495–505. doi:10.1002/ana.20624
Giuliani P, Ballerini P, Ciccarelli R, Buccella S, Romano S, D’Alimonte I, Poli A, Beraudi A, Pena E, Jiang S, Rathbone MP, Caciagli F, Di Iorio P (2012) Tissue distribution and metabolism of guanosine in rats following intraperitoneal injection. J Biol Regul Homeost Agents 26(1):51–65
Bicca MA, Figueiredo CP, Piermartiri TC, Meotti FC, Bouzon ZL, Tasca CI, Medeiros R, Calixto JB (2011) The selective and competitive N-methyl-D-aspartate receptor antagonist, (−)-6-phosphonomethyl-deca-hydroisoquinoline-3-carboxylic acid, prevents synaptic toxicity induced by amyloid-beta in mice. Neuroscience 192:631–641. doi:10.1016/j.neuroscience.2011.06.038
Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58(7):831–838. doi:10.1002/glia.20967
Pekny M, Wilhelmsson U, Pekna M (2014) The dual role of astrocyte activation and reactive gliosis. Neurosci Lett 565:30–38. doi:10.1016/j.neulet.2013.12.071
Rajkowska G, Stockmeier CA (2013) Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets 14(11):1225–1236
Cobb JA, O’Neill K, Milner J, Mahajan GJ, Lawrence TJ, May WL, Miguel-Hidalgo J, Rajkowska G, Stockmeier CA (2015) Density of GFAP-immunoreactive astrocytes is decreased in left hippocampi in major depressive disorder. Neuroscience. doi:10.1016/j.neuroscience.2015.12.044
Masliah E (1995) Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol 10(2):509–519
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8(8):1051–1058. doi:10.1038/nn1503
Zahs KR, Ashe KH (2010) ‘Too much good news’—are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer’s disease? Trends Neurosci 33(8):381–389. doi:10.1016/j.tins.2010.05.004
Acknowledgments
Research was supported by grants from the Brazilian funding agencies, CAPES (PVE 052/2012), CNPq (INCT for Excitotoxicity and Neuroprotection), and FAPESC (NENASC/PRONEX) to C.I.T. C.I.T. is recipient of CNPq productivity fellowship. D.L. was recipient of CAPES/PVE 052/2012 and CAPES/Procad predoctoral scholarships. D.L. kindly thanks Prof. Dra. Zenilda Bouzon (UFSC) for helping with the acquisition of images presented in Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Lanznaster, D., Mack, J.M., Coelho, V. et al. Guanosine Prevents Anhedonic-Like Behavior and Impairment in Hippocampal Glutamate Transport Following Amyloid-β1–40 Administration in Mice. Mol Neurobiol 54, 5482–5496 (2017). https://doi.org/10.1007/s12035-016-0082-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0082-1