
Abstract
SUMO (small ubiquitin-like modifier) conjugation is a critically important control process in all eukaryotic cells, because it acts as a biochemical switch and regulates the function of hundreds of proteins in many different pathways. Although the diverse functional consequences and molecular targets of SUMOylation remain largely unknown, SUMOylation is becoming increasingly implicated in the pathophysiology of Alzheimer’s disease (AD). Apart from the central SUMO-modified disease-associated proteins, such as amyloid precursor protein, amyloid β, and tau, SUMOylation also regulates several other processes underlying AD. These are involved in inflammation, mitochondrial dynamics, synaptic transmission and plasticity, as well as in protective responses to cell stress. Herein, we review current reports on the involvement of SUMOylation in AD, and present an overview of potential SUMO targets and pathways underlying AD pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common cause of chronic dementia among the elderly, and in 2010, an estimated 35.6 million patients were diagnosed with this condition worldwide. Even more crucially, prognoses predict this number to almost double every 20 years reaching projected 65.7 million in 2030 and 115.4 million in 2050 [1]. AD is more likely to occur in later stages of life and increased average life expectancies consequently generate increasing economic and social strain. Currently, only symptomatic treatments are available for AD, and therefore, the identification of novel mechanisms and the development of new therapeutic strategies for AD represent urgent research targets. Despite the complexity and multifactorial nature of AD, studies using animal and cell culture models have contributed considerably to the understanding of the pathophysiology of AD [2]. However, an effective treatment of AD relies on the translation of the disease pathways, as well as molecular mechanisms, into specific pharmacological targets, a currently unachieved goal for most neurological disorders.
AD is a biologically complex neurodegenerative form of dementia. Cerebral plaques laden with β-amyloid peptide (Aβ) and prominent neurofibrillary tangles are important pathological features of AD. While the amyloid cascade hypothesis, which supports that pathological accumulations of Aβ are central to the pathogenesis of AD [3], is still predominant, data that are inconsistent with this hypothesis have emerged [4]. Growing evidence suggests that other important processes contribute to the development and progress of the disease. These processes, which are summarized in Fig. 1, include, but are not restricted to, synaptic dysfunction, oxidative stress, inflammation, and loss of calcium regulation (reviewed in [5, 6]).

General overview of AD pathogenesis. Aβ and tau proteins lead to calcium dysregulation, which in turn can disrupt mitochondrial function. Highly reactive oxygen species (ROS) may be generated by this damage to the mitochondria, by inflammation products or by apolipoprotein E4 (ApoE4) alleles that reduce antioxidant activity and may induce excess oxidative damage to the lipids. ROS can also augment the Aβ levels, leading to a cyclic toxicity effect. Moreover, aging by itself is associated with reduced resistance to oxidation and increased Aβ and tau proteins levels. Aβ amyloid-β, APP amyloid precursor protein, iNOS inducible nitric oxide synthase, NMDAR N-methyl-d-aspartate receptor, NO nitric oxide, P-tau phosphorylated tau
It is generally accepted that neurofibrillary tangles, resulting from tau hyperphosphorylation, and amyloid plaques, formed by aggregation of Aβ peptide, accumulate in the brain of AD patients [7, 8]. In AD, abnormal phosphorylation of tau decreases its affinity for microtubules, thus causing neuronal instability. These hyperphosphorylated tau proteins subsequently dissociate from microtubules and aggregate in the neuron cell body to form neurofibrillary tangles that impair axonal transport and lead to synaptic dysfunction [9].
The proteolytic cleavage of a transmembrane glycoprotein known as the amyloid precursor protein (APP) can be mediated by α-, β-, or γ-secretases [8]. APP is normally cleaved by α-secretase followed by γ-secretase cleavage; the resulting proteins contain approximately 654–670 aminoacids and can participate in neuroprotection and neuroplasticity [10]. In pathogenic situations APP is cleaved by β-secretase, followed once again by γ-secretase cleavage, generating the toxic forms of Aβ, i.e. proteins containing mainly 40 or 42 aminoacids (Aβ1–40 or Aβ1–42). These peptides accumulate and form fibrils and/or other aggregates of low molecular weight. The oligomeric form of Aβ has been described as relatively soluble and diffusible, resulting in an accordingly increased level of potential toxicity. Moreover, both fibrillar and soluble forms have been implicated in various intracellular and extracellular perturbations, e.g. increased levels of reactive oxygen species (ROS), loss of intracellular calcium homeostasis, and excitotoxicity [11–14].
The exact mechanism of Aβ-induced neurotoxicity that eventually culminates in neuronal death is not yet fully understood. The deposition of Aβ peptides in brain areas involved in cognitive functions, leading to its aggregation into oligomeric species and activation of glial cells, might initiate the cascade that results in synaptic dysfunction and loss [15].
SUMOylation
SUMOylation is a post-translational modification, in which SUMO (small ubiquitin-like modifier; a 97-residue protein) covalently binds to specific lysine residues on target proteins. Mammals usually contain three SUMO paralogues (SUMO-1-3). Except for three residues, SUMO-2 and SUMO-3 are identical, but both share only ~50 % sequence identity with SUMO-1.
The SUMOylation status of any given protein is a dynamic interplay between conjugation and deconjugation. In a pathway analogous to ubiquitination, activating E1 enzyme transfers SUMO onto E2 conjugating enzyme Ubc9, which catalyses SUMO conjugation to the substrate in conjunction with an E3 ligating enzyme. SUMO can be removed from substrates with the SENP family of SUMO-specific isopeptidases, six of which are known in mammals: SENP1-3 and SENP5-7 [16].
It is well known that ubiquitination, the covalent attachment of ubiquitin to a substrate, affects the location, function, and stability of modified proteins, thus playing a crucial role in nearly every biochemical pathway in eukaryotes [17]. Mono-ubiquitination has been shown to regulate receptor endocytosis and histone modification, while poly-ubiquitination plays diverse functions that are dependent on the type of ubiquitin chain linkages, including degradation of target proteins, DNA repair, and activation of signal transduction pathways [18]. In a similar but distinct way, SUMOylation alters the interactions of substrate proteins to change their localisation, stability, and/or activity. The functional consequences of SUMOylation vary greatly, depending on the SUMO target. SUMOylation is currently characterized best for a relatively small subset of nuclear proteins that regulate DNA replication and cell division. However, recent reports have unambiguously demonstrated that SUMOylation regulates hundreds of proteins, including those associated with the plasma membrane [19], many of which are present in neurons and are responsible for the regulation of synaptic transmission and stress response pathways.
Different studies have come to the conclusion that SUMO-1 participates predominantly in normal cell physiology and maintenance, whereas SUMO-2/3 are mostly involved in cell stress responses, which have been implicated in a wide range of clinically important neuropathologies including AD (for recent reviews, see [20–23]).
SUMOylation in AD
Post-translational modifications are important regulatory mechanisms for the structure and function of proteins. Several studies on AD have shown that various proteins are subjected to modifications such as phosphorylation, ubiquitination, and more recently, SUMOylation [24–28].
Altered levels of SUMOylation were confirmed in AD patients. Initially, SUMO-3 labeling was detected in postmortem brain sections from AD patients, especially in the hippocampus, which is the brain region responsible for learning and memory and the most affected in AD [29]. The protease SENP3, which participates both in the maturation of native SUMO and the de-SUMOylation process, was subsequently shown to be down-regulated in inferior parietal lobes of sporadic AD patients [30]. Furthermore, the SUMO conjugating enzyme Ubc9 has also been linked to AD. An analysis of genomic DNA from Korean patients with late-onset AD discovered variations of the Ubc9 gene (UBE2I) that might signify an increased risk of developing AD for the Korean population [31].
In animal models related to aging and AD, the changes in the expression of the main components of the SUMOylation pathway still remain unresolved. While decreased SUMO-1 and Ubc9 mRNA levels were observed in aged wild-type mice [32], unchanged Ubc9 and SENP1 protein levels were found in APP overexpressing mice [33]. Although global levels of SUMO-1 or SUMO-2/3 were not altered significantly in that model, several individual SUMO-2/3 bands decreased considerably. Ensuing studies showed increased SUMO-1 and SUMO-2/3 levels in APP overexpressing and aged mice [34, 35]. Adding to this controversy is the fact that decreased SUMO-2 levels were found in old APP mice (17 months), whereas increased SUMO-1, Ubc9, and SENP1 levels were observed in young APP mice (3 and 6 months) [36]. Consequently, further studies are required to comprehensively determine the in vivo changes in SUMOylation with aging and AD.
SUMOylation Targets in AD
Although the exact function of SUMOylation and the identity of disease-modified SUMO targets remain largely unknown, an increasing number of proteins associated with AD has been reported to be subjected to SUMOylation (Table 1). These proteins are intrinsically involved in AD pathophisiological mechanisms as well as the cellular pathways underlying the disease progression.
Tau Protein
AD induces high levels of tau protein expression, leading to increased formation of neurofibrillary tangles, whereby both 3R- and 4R-tau (functional points of interaction with microtubules) splice isoforms are observed [37]. Tau itself is a SUMO-1 target and the modified lysine has been identified as the K340 located within 4R-tau [25]. The interaction between tau and SUMO-1 was confirmed by an independent study, showing that the SUMO-1 immunoreactivity is co-localized with phosphorylated tau [38]. In addition, tau can also be ubiquitinated and degraded by the proteasome through both ubiquitin-dependent and ubiquitin-independent pathways [39, 40]. Recently, it has been reported that SUMOylation at lysine K340 stimulates tau phosphorylation and inhibits ubiquitination-mediated tau degradation, thus favoring its aggregation [41].
Amyloid Precursor Protein (APP)
The notion that the amyloidogenic pathway is the main contributor to AD has attracted increasing consent in recent years. Consequently, modulation of the cleavage of APP into Aβ could potentially reduce Aβ-induced toxicity and its associated cognitive deficits.
The overexpression of SUMO-3 has been reported to reduce Aβ production via regulation of APP processing [29]. In this study, a partial resemblance between SUMOylation and ubiquitination was identified, whereby mono-SUMOylation and poly-SUMOylation exhibited different functional consequences. While an increased Aβ production was observed for mono-SUMOylation, poly-SUMOylation resulted in a decreased generation of this peptide.
Covalent modifications of APP K587 and K595 lysines by both SUMO-1 and SUMO-2 were reported to decrease Aβ aggregation levels in HELA cells overexpressing APP [42]. Even though the authors were unable to identify the precise mechanism behind this SUMO-mediated Aβ regulation, they tentatively assigned it to the unfavourable steric congestion between the protease and the APP, arising from the close spatial proximity of the SUMOylated lysines K587 and K595 to the β-secretase cleavage site, which are one and nine residues apart, respectively.
Further studies, investigating whether this SUMO-mediated APP regulation occurs in neurons under physiological and, more importantly, AD conditions, should reveal new information about APP SUMOylation and its potential medical applications.
β-Secretase (BACE1)
The beta-site amyloid precursor protein cleaving enzyme 1 (β-secretase or BACE1) mediates the initial and rate limiting step in the generation of Aβ. This enzyme is essential for the processing of APP and the formation of Aβ, which is evident from a lack of amyloid plaques in BACE1 knockout mice overexpressing human APP [43]. All three SUMO isoforms are capable to increase BACE1 accumulation and Aβ production [35]. Interaction between SUMO-1 and BACE1 was observed at a dileucine motif, which can regulate Aβ generation in an APP-independent manner, since a deletion of SUMO-1 did not change APP levels, but decreased both BACE1 and Aβ levels [35]. Subsequently, a positive feedback regulation was proposed for the production of Aβ in AD, whereas high concentrations of Aβ, or other parameters such as oxidative stress, increase SUMO-1 levels, which leads to an increased generation of Aβ via modulation of BACE1 levels.
The up-regulation of BACE by SUMO-3 overexpression has been previously demonstrated [20]. However, in this previous study the SUMO-3 modulation of not only BACE, but also APP and Aβ levels, did not require its conjugation to target proteins. The authors suggested that these increases may be due to altered protein turnover by the proteasome, changes in protein degradation pathways, or in protein trafficking.
Potential SUMO Targets in AD
In addition to SUMOylation of tau, APP, and BACE1, several other potential SUMO targets have been proposed, and their SUMOylation may play an important role in AD. The identities and functions of SUMO conjugates affected by high levels of Aβ or other AD-related perturbations in cell culture and/or transgenic mouse models still remain largely unknown. In the following sections, we discuss recent reports that are concerned with alternative SUMO substrates in the brain and the impact of their SUMOylation on AD.
Drp1 (Dynamin-Related Protein 1)
Drp1 is a GTPase enzyme that regulates mitochondrial dynamics and is involved in apoptosis via interactions with proteins from the Bcl-2 family, which mediate mitochondrial outer membrane permeabilization (MOMP). MOMP releases cytochrome c from mitochondria into the cytosol, where it activates caspases cascades and apoptosis [44]. Depending on the conditions, up- or down-regulation of Drp1 has been reported to be protective against apoptosis [45, 46].
SUMOylation plays a role in Drp1 regulation [47]. SUMOylation of Drp1 was initially reported to stabilise the protein, leading to an increase in the active pool of Drp1 present on the surface of mitochondria, thus increasing mitochondrial fragmentation [48]. A more recent study has confirmed Drp1 SUMOylation and identified the SUMOylation sites [49]. However, the findings that site-directed mutagenesis of the SUMOylatable lysines in Drp1 had no effect on protein stability, and that its recruitment to mitochondria was unaltered lead to the questioning of the functional roles of SUMOylated Drp1 [49]. These apparent differences remain to be fully resolved. Intriguingly, it has also been reported recently that Drp1 binds directly to both Aβ and phosphorylated tau, thus potentially leading to increased mitochondrial fragmentation [50].
Mitochondrial dysfunction is as a major factor in AD [51], and a significantly increased Drp1 GTPase activity in postmortem cortical tissue of AD patients has been reported [52]. In direct contrast, another study has reported significantly reduced Drp1 levels in the brains of patients suffering from AD [53]. Severely increased levels of cell stress induced by oxygen/glucose deprivation (OGD), an ischaemia model, cause SENP3 degradation and consequently increase the levels of protein SUMOylation, including the levels of Drp1 SUMOylation [54]. Even though these results suggest an involvement of SUMOylated Drp1 in AD, its precise role still remains to be defined.
Potassium Channels
Since AD is a disease that is primarily concerned with synaptic and cognitive dysfunction, proteins involved in synaptic plasticity and neuronal networking, such as ion channels regulating cellular excitability, are particularly interesting targets to investigate.
Potassium (K+) channel abnormalities have been reported in both neural and peripheral tissue of AD patients; in particular, K+ channel expression was found to be reduced in postmortem brains [55]. Recent studies have suggested a relationship between Aβ toxicity and the modulation of K+ channels in both microglial and neuronal cells. For example, the chronic activation of large-conductance calcium-activated K+ channels has proven effective in the cognition recovery of triple transgenic animals for AD (3xTg), as evaluated by the object recognition test [56]. The implication of voltage-dependent K+ channels (Kv) has also been studied in models of AD, where the increased expression of Kv1.4, Kv2.1, and Kv4.2 subunits in Aβ-treated rats could be partially responsible for the memory impairment detected in these animals [57].
SUMOylation has been reported to play an important role in controlling membrane potential by modulating a wide range of ion channels, including K+ channels [19]. Recent studies have implicated the modulation of Kv channels by SUMOylation as a potential therapeutic pathway. Qi and colleagues (2014) have reported that SENP2 (deSUMOylation enzyme) deficiency, results in hyper-SUMOylation of Kv7 channels, causing the development of spontaneous seizures, cardiac abnormalities, and sudden unexpected death by epilepsy in mice [58]. Another study of particular importance showed that SUMOylation can modulate the voltage-dependent activation of Kv2.1 channels in the membrane of cultured rat hippocampal neurons, where SUMO-1 modification of Kv2.1 channels suppresses neuronal excitability as a result of a shift in the V1/2 for the activation to more depolarized potentials [59]. This study could be a link between the SUMOylation of Kv2.1 and others subunits and Aβ toxicity, as Aβ-treated rats show enhanced expression of these channels.
Glutamate Receptors
The kainate receptor subunit GluK2 was also identified as a SUMO substrate in cultured neurons [60]. SUMOylation of GluK2 at K886 lysine is required for agonist-induced endocytosis of GluK2-containing kainate receptors. SUMOylation of GluK2 is enhanced by agonist-induced PKC phosphorylation of GluK2, and this phospo-SUMOylation switch is a crucial determinant of kainate receptor-induced LTD at mossy fiber synapses [61–63]. The expression and localization of kainate receptors in AD has not yet been studied in detail, but significantly increased levels of kainate receptor binding have been observed in the frontal cortex of AD patients, and a positive correlation between kainate binding sites and the senile plaque number in deep cortical layers has been established. In addition, immunohistochemical analyses have identified a decrease of GluK5/6/7 subunits in vulnerable regions such as the CA1 hippocampal area [64].
SUMO modification is moreover essential for α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) trafficking [65]. In neuronal cultures, SUMOylation is required for the insertion of the GluA1 AMPAR subunit during glycine-induced increase in AMPAR surface expression (chemical LTP), and even though the specific SUMO substrate proteins that regulate these pathways have not yet been identified, a mediating role has been assigned to SUMOylation of AMPA receptor insertion during LTP [66]. Since AD is characterized by decreased AMPAR activation and endocytosis [67], SUMOylation-induced up-regulation of AMPAR trafficking may potentially be able to counterbalance synaptic dysfunction and loss.
Glutamate Transporters
AD strongly affects synaptic transmission, which depends under physiological conditions on the uptake of glutamate, especially in glial cells, via a process mediated by the high affinity excitatory aminoacid transporters (EAAT) [68–70].
During the course of AD, the glial glutamate transporters are reduced substantially [71–73]. Down-regulation of the glial glutamate transporters EAAT1/GLAST and EAAT2/GLT-1 has been observed in the hippocampus of a 3xTg mouse model of AD [74] and also following a single intracerebroventricular injection of Aβ1–40 [75]. Nevertheless, the expression of EAAT2/GLT-1 in astrocytes in the medial prefrontal cortex remained unaltered during the progression of AD in the same transgenic model [76].
The loss of these transporters accelerates neurodegeneration via glutamate-dependent excitotoxicity [77], and the internalisation and degradation of glutamate transporters is likely to be a consequence of the post-translational modification of these transporters on the cell surface by processes including SUMOylation.
Evidence for nuclear localization of a SUMOylated EAAT2/GLT-1 fragment in vivo, leading to neuronal toxicity, has been acquired from an animal model of amyotrophic lateral sclerosis (ALS). Other studies from the same group have shown that SUMO-1 modification of EAAT2 in cultured astrocytes can prevent the transporter to reach the membrane and subsequently exert its function while inhibiting SUMOylation induced EAAT2 translocation from intracellular compartments to the plasma membrane, resulting in an increased glutamate uptake [78, 79].
Given the neuroprotective potential of inhibiting glutamate transporter internalisation and increasing its fuction at the plasma membrane, it is tempting to speculate that the SUMO-mediated modifications in glutamate transporters observed in ALS might be extended to other neurodegenerative diseases, such as AD.
NOS2 (iNOS)
Nitric oxide (NO) promotes a variety of physiological processes and is generated by nitric oxide synthases (NOS), a family of enzymes present in most cells. Three distinct NOS isoforms, differing with respect to localization, regulation, catalytic properties, and inhibitor sensitivity have been identified: neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3).
In the brain, NOS2 expression has been well characterized in astrocytes, microglia, and—to a lesser extent—in endothelial cells. Higher concentrations of NOS2-generated NO can interact with numerous substances, participating in the pathology of inflammatory diseases [80]. Tissue and neuronal analyses of AD patients revealed that Aβ is able to promote NOS2 expression and NO production in microglia cells and reactive astrocytes [81, 82]. Aβ also stimulates the release of pro-inflammatory cytokines such as interleucine 1β (IL-1β) and the tumoral necrosis factor (TNF-α), which induce NO and peroxynitrite synthesis, causing protein and lipid modifications, mitochondrial damage, apoptosis, and increased formation of Aβ [83, 84].
Pro-inflammatory conditions induced by treatment of cultured astrocytes with lipopolysaccharide resulted in decreased SUMO-1, Ubc9, and SENP1 mRNA levels [32]. In the same study, decreased NOS2 promoter activity was observed for an overexpression of SUMO-1, Ubc9, and SENP1. This regulatory effect was assigned to modifications of the transcription factor C/EBPβ, a protein involved in the transcription of NOS2. Since NOS2 contributes to the disease progression in a variety of neurological diseases including AD, an anti-inflammatory role was accordingly suggested for SUMO-1 in the brain. Although an increase in SENP1 expression might be expected to promote inflammation (by reducing global SUMOylation levels), this enzyme is also involved in SUMO-1 maturation [85].
A recent study from our group demonstrated that Aβ1–42 exposure, concomitant with increasing reactive astrogliosis, decreases Ubc9 protein levels and SUMO-1 conjugates in cultured astrocytes [86]. Overexpression of constitutively active SUMO-1, but not a conjugation-deficient SUMO-1, was found to prevent the up-regulation and morphological reactivity of glial fibrillary acidic protein (GFAP). These results suggest that astrocytes require SUMO-1 conjugation to remain non-reactive under Aβ-induced astrogliosis.
Signaling Pathways
Several other proteins, known to be involved in signaling pathways crucial to the pathophysiology of AD, have been identified as SUMO conjugation targets. However, previous studies have used heterologous cells, and the question of whether SUMOylation of these proteins also occurs in brain cells remains to be addressed.
The cAMP-responsive element binding protein (CREB) is a transcription factor that plays a key role in the induction of synaptic plasticity and memory. CREB is SUMOylated by SUMO-1 at lysines K285 and K304, resulting in a stabilization and nuclear translocation of the transcription factor [87]. Changes in CREB SUMOylation may accordingly be involved in AD as a contributing factor to synaptic loss and cognitive impairment.
Glycogen synthase kinase 3β (GSK-3β) is a serine/threonine protein kinase involved in several physiological processes, e.g. glycogen metabolism and gene transcription. GSK-3β also plays a pivotal role in the pathogenesis of both sporadic and familial forms of AD, and it is one of the main kinases associated to the hyperphosphorylation of tau and plaque-associated microglial-mediated inflammatory responses [88]. Recently, GSK-3β has also been identified as a SUMO target [89]. SUMO-1 conjugation at K292 lysine promotes GSK-3β nuclear localization and protein stability, resulting in the stimulation of cell apoptosis. In human APP transfected cells, GSK-3β/NF-κB signaling pathways regulate BACE1 transcription, which could lead to an increased generation of Aβ [90]. Therefore, SUMOylation of GSK-3β may represent another promising target for AD therapy.
The c-Jun N-terminal kinase (JNK) signaling pathway is strongly associated with a variety of different stress stimuli and cell death. In recent years, it has been associated with the regulation of APP cleavage and tau hyperphosphorylation in AD [91, 92]. SUMO-1 and Ubc9 overexpression promote increased H2O2-induced JNK activation, whereas SENP1 overexpression leads to a significant dose-dependent prevention of JNK activation [93]. An interaction between SUMO-1 and the phosphorylated (active) form of JNK, which leads to cell death pathways, was also established. Although further studies are required, these results suggest that SUMOylation of JNK may play an important role in the pathophysiology of AD.
Conclusions and Future Perspectives
Pharmacological interventions that modulate protein SUMOylation could represent potential therapeutic approaches for treating neurodegenerative diseases. Considering that the involvement of SUMOylation in AD is a relatively new topic, several unanswered questions need to be addressed before SUMOylation can be established as a therapeutic target for AD. For instance, the significance of the SUMO-modification of proteins for the fate of neurons and glial cells in AD patients remains to be determined. Due to the low endogenous abundance of SUMO conjugates, most SUMOylation studies have relied on cell culture experiments, overexpressing SUMO signalling components and/or target proteins. Thus, a significant part of the current knowledge on the SUMO conjugation pathway originates from overexpression systems, which suffer from some inherent limitations. For several SUMO targets that have been identified using these systems, the SUMO effects on endogenous proteins have yet to be determined. Before any conclusions can be drawn and translated into clinical approaches, results from these studies need to be verified under physiological/pathophysiological conditions in intact organs.
Some of the studies reviewed here, focusing on SUMOylation in AD models and potential SUMO targets in AD, suggest that SUMO conjugation could be a toxic stress response to affected cells, whereas others suggest that it could be part of an protective endogenous response. This is also the case for other neurodegenerative diseases, e.g. brain ischaemia and Parkinson’s disease (for reviews, see [19, 94]). In this regard, it is still not entirely clear whether the inhibition or the activation of SUMOylation could be a therapeutic target for AD. Despite previous reports on the activation and inhibition of SUMOylation pathways [95, 96], studies showing these effects in the context of neurodegenerative diseases still remain elusive. Nevertheless, in the event that this physiological pathway is confirmed to hold neuroprotective potential, several obstacles will have to be circumvented prior to the establishment of a therapeutic approach that could have significant clinical implications.
SUMO modification has been reported for several substrates, the majority of which is located within the nucleus, where SUMOylation plays a key role e.g. in DNA repair and transcription. In addition, increasing numbers of SUMO substrates are being identified in compartments outside the nucleus, where SUMOylation can regulate e.g. synaptic plasticity. Both nuclear and extranuclear SUMO targets have been implicated in a number of physiological and pathological processes. Due to the vast amount and the diversity of substrate proteins, a therapy based on SUMOylation inhibition or activation would have very little specificity, making it difficult to predict the overall effects of global SUMOylation. Such therapies most likely exhibit numerous potential problems and side effects, which could be overcome by the development of substrate- and target-specific SUMOylation inhibitors or activators.
Ubiquitination and phosphorylation, which are well known to be affected in AD, have been the main focus of several studies and various drug discovery efforts. Following these studies, SUMOylation is now emerging as a central post-translational protein modification, the targeting of which could become a therapeutic strategy for AD. Whereas the ubiquitination system comprises several enzymes that may serve as potential drug targets, only few components comprise the SUMOylation pathway. Since all SUMOylation events are mediated by only one E1 and one E2 enzyme, the diversity of E3 s that form the SUMOylation complex may provide a mechanism for subtle, substrate-specific regulation of SUMOylation. The E3 s responsible for regulating the SUMOylation efficiency of disease-related proteins could be potentially desirable targets of such interventions, because they should offer greater selectivity than globally altering SUMOylation through targeting the SUMO E2 enzyme (Ubc9). A growing body of evidence suggests a molecular cross-talk between SUMOylation and ubiquitination/phosphorylation. Ultimately, a deeper understanding of this cross-talk should enable the design of effective strategies for AD and other neurodegenerative diseases.
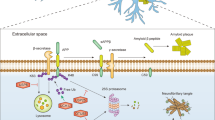
Before SUMOylation can be consolidated as a novel and effective therapeutic target for AD, SUMOylated proteins need to be confirmed in neurons and further characterized in terms of SUMOylation sites and molecular consequences. Although the pathology in AD and other neurodegenerative diseases is rather complex, the use of simple models and the development of SUMO-defective mutants might be helpful in dissecting molecular mechanisms. Apart from APP and tau, the two major AD proteins, it has been proposed that SUMOylation dysfunction of multiple substrates could be involved in disease progression (Fig. 2). Since synaptic dysfunction and the resulting cognitive loss is the primary cause of dementia, it is feasible to expect that synaptic SUMO targets, such as kainate and AMPA receptors, as well as EAAT2-type glutamate transporters, are likely to be the focus of future investigation aiming at the development of AD drugs. The comprehension of functional consequences of SUMOylation on the proteins reviewed here and on other potential targets, will provide mechanistic insights into the orchestration of SUMOylation in the context of controlling neuronal function and survival in AD.

Putative SUMO targets in AD. SUMO stimulates tau phosphorylation at the Lys340 site in HEK293 cells (1). In a transgenic mice model of ALS, increased levels of SUMO lead to glutamate transporter EAAT2 internalization (2). In a LPS-induced inflammatory model, SUMO can decrease NOS2 expression through modulation of its promoter C/EBPβ (3). SUMOylation can modulate K+ channel activity (4) and membrane expression of glutamate receptors (5). Both increased and decreased SUMO levels may reduce Aβ aggregation, which depends on the SUMOylated target protein (6). Increased SUMO levels lead to BACE1 accumulation and augment Aβ production in APP/PS1 double transgenic mice (7). SUMO can target lysines 587 and 595 in APP, where the proximity to the site of β-secretase cleavage could block this protease action, thus inhibiting the production of Aβ in HEK293 and HeLa cells (8)
References
Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimer’s Dement 9:63–75
Van Dam D, De Deyn PP (2006) Drug discovery in dementia: the role of rodent models. Nat Rev Drug Discov 5:956–970
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185
Herrup K (2015) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18:794–799
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362:329–344
Spires-Jones TL, Hyman BT (2014) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82:756–771
Grundke-Iqbal I (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8:101–112
West S, Bhugra P (2015) Emerging drug targets for Aβ and tau in Alzheimer’s disease: a systematic review. Br J Clin Pharmacol 80:221–234
Thornton E, Vink R, Blumbergs PC, Heuvel CVD (2006) Soluble amyloid precursor protein α reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res 1094:38–46
Harris ME, Wang Y, Pedigo NW Jr, Hensley K, Butterfield DA, Carney JM (1996) Amyloid β peptide (25–35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem 67:277–286
Butterfield DA, Lauderback CM (2002) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid-β-peptide associated free radical oxidative stress. Free Radic Biol Med 32:1050–1060
Demuro A, Parker I, Stutzmann GE (2010) Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 285:12463–12468
Masters CL, Selkoe DJ (2012) Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer’s disease. Cold Spring Harb Perspect Med 2:a006262
Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44:181–193
Yeh ET (2009) SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem 284:8223–8227
Grabbe C, Husnjak K, Dikic I (2011) The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol 12:295–307
Park CW, Ryu KY (2014) Cellular ubiquitin pool dynamics and homeostasis. BMB Rep 47:475–482
Silveirinha V, Stephens GJ, Cimarosti H (2013) Molecular targets underlying SUMO-mediated neuroprotection in brain ischemia. J Neurochem 127:580–591
Dorval V, Fraser PE (2007) SUMO on the road to neurodegeneration. Biochem Biophys Acta 1773:694–706
Anderson DB, Wilkinson KA, Henley JM (2009) Protein SUMOylation in neuropathological conditions. Drug News Perspect 22:255–265
Wilkinson KA, Nakamura Y, Henley JM (2010) Targets and consequences of protein SUMOylation in neurons. Brain Res Rev 64:195–212
Sarge KD, Park-Sarge OK (2011) SUMO and its role in human diseases. Int Rev Cell Mol Biol 288:167–183
Atkin G, Paulson H (2014) Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci 7:63
Dorval V, Fraser PE (2006) Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and α-synuclein. J Biol Chem 281:9919–9924
Martin L, Latypova X, Terro F (2011) Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem Int 58:458–471
Riederer BM, Leuba G, Vernay A, Riederer IM (2011) The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp Biol Med 236:268–276
Tenreiro S, Eckermann K, Outeiro TF (2014) Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci 7:42
Li Y, Wang H, Wang S, Quon D, Liu YW, Cordell B (2003) Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc Natl Acad Sci USA 100:259–264
Weeraratna AT, Kalehua A, Deleon I, Bertak D, Maher G, Wade MS et al (2007) Alterations in immunological and neurological gene expression patterns in Alzheimer’s disease tissues. Exp Cell Res 313:450–461
Ahn K, Song JH, Kim DK, Park MH, Jo SA, Koh YH (2009) Ubc9 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population: a genetic association study. Neurosci Lett 465:272–275
Akar CA, Feinstein DL (2009) Modulation of inducible nitric oxide synthase expression by sumoylation. J Neuroinflamm 6:12
McMillan LE, Brown JT, Henley JM, Cimarosti H (2011) Profiles of SUMO and ubiquitin conjugation in an Alzheimer’s disease model. Neurosci Lett 502:201–208
Yang QG, Wang F, Zhang Q, Xu WR, Chen YP, Chen GH (2012) Correlation of increased hippocampal Sumo3 with spatial learning ability in old C57BL/6 mice. Neurosci Lett 518:75–79
Yun SM, Cho SJ, Song JC, Song SY, Jo SA, Jo C, Yoon K, Tanzi RE, Choi EJ, Koh YH (2013) SUMO1 modulates Aβ generation via BACE1 accumulation. Neurobiol Aging 34:650–662
Nisticò R, Ferraina C, Marconi V, Blandini F, Negri L, Egebjerg J, Feligioni M (2014) Age-related changes of protein SUMOylation balance in the AβPP Tg2576 mouse model of Alzheimer’s disease. Front Pharmacol 5:1–9
Goedert M, Spillantini MG, Cairns NJ, Crowther RA (1992) Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8:159–168
Takahashi KM, Ishida M, Komano H, Takahashi H (2008) SUMO-1 immunoreactivity co-localizes with phospho-Tau in APP transgenic mice but not in mutant Tau transgenic mice. Neurosci Lett 441:90–93
David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG (2002) Proteasomal degradation of tau protein. J Neurochem 83:176–185
Shimura H, Schwartz D, Gygi SP, Kosik KS (2004) CHIPHsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 279:4869–4876
Luo HB, Xia YY, Shu XJ, Liu ZC, Feng Y, Liu XH et al (2014) SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci USA 111:16586–16591
Zhang YQ, Sarge KD (2008) Sumoylation of amyloid precursor protein negatively regulates Aβ aggregate levels. Biochem Biophys Res Commun 374:673–678
Luo Y, Bolon B, Damore MA, Fitzpatrick D, Liu H, Zhang J, Yan Q, Vassar R, Citron M (2003) BACE1 (β-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol Dis 14:81–88
Jourdain A, Martinou JC (2009) Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol 41:1884–1889
Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15:5001–5011
Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ et al (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 16:59–68
Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U et al (2011) Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev 67:103–118
Harder Z, Zunino R, McBride H (2004) Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14:340–345
Figueroa-Romero C, Iñiguez-Lluhí JA, Stadler J, Chang CR, Arnoult D, Keller PJ, Hong Y, Blackstone C, Feldman EL (2009) SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J 23:3917–3927
Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20:2495–2509
Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9:505–518
Manczak M, Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21:2538–2547
Wang X, Su B, Lee HG, Li X, Perry G et al (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103
Guo C, Hildick KL, Luo J, Dearden L, Wilkinson KA, Henley JM (2013) SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J 32:1514–1528
Alkon DL (1999) Ionic conductance determinants of synaptic memory nets and their implications for Alzheimer’s disease. J Neurosci Res 58:24–32
Wang L, Kang H, Li Y, Shui Y, Yamamoto R, Sugai T, Kato N (2015) Cognitive recovery by chronic activation of the large-conductance calcium-activated potassium channel in a mouse model of Alzheimer’s disease. Neuropharmacol 92:8–15
Campolongo P, Ratano P, Ciotti MT, Florenzano F, Nori SL, Marolda R, Palmery M, Rinaldi AM, Zona C, Possenti R, Calissano P, Severini C (2013) Systemic administration of substance P recovers beta amyloid-induced cognitive deficits in rat: involvement of Kv potassium channels. PLoS One 8:e78036
Qi Y, Wang J, Bomben VC, Li DP, Chen SR, Sun H, Xi Y, Reed JG, Cheng J, Pan HL, Noebels JL, Yeh ET (2014) Hyper-SUMOylation of the Kv7 potassium channel diminishes the M-current leading to seizures and sudden death. Neuron 83:1159–1171
Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SA (2011) SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J Gen Physiol 137:441–454
Martin S, Nishimune A, Mellor JR, Henley JM (2007) SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature 447:321–325
Konopacki FA, Jaafari N, Rocca DL, Wilkinson KA, Chamberlain S et al (2011) Agonist-induced PKC phosphorylation regulates GluK2 SUMOylation and kainate receptor endocytosis. Proc Natl Acad Sci USA 108:19772–19777
Chamberlain SE, Gonzalez-Gonzalez IM, Wilkinson KA, Konopacki FA, Kantamneni S et al (2012) SUMOylation and phosphorylation of GluK2 regulate kainate receptor trafficking and synaptic plasticity. Nat Neurosci 15:845–852
Wilkinson KA, Konopacki F, Henley JM (2012) Modification and movement: phosphorylation and SUMOylation regulate endocytosis of GluK2-containing kainate receptors. Commun Integr Biol. 5:223–226
Lee HG, Zhu X, Ghanbari HA, Ogawa O, Raina AK, O’Neill MJ, Perry G, Smith MA (2002) Differential regulation of glutamate receptors in Alzheimer’s disease. Neurosignals 11:282–292
Craig TJ, Jaafari N, Petrovic MM, Rubin PP, Mellor JR et al (2012) Homeostatic synaptic scaling is regulated by protein SUMOylation. J Biol Chem 287:22781–22788
Jaafari N, Konopacki FA, Owen TF, Kantamneni S, Rubin P, Craig TJ, Wilkinson KA, Henley JM (2013) SUMOylation is required for glycine-induced increases in AMPA receptor surface expression (ChemLTP) in hippocampal neurons. PLoS One 8:e52345
Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R (2006) AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 52:831–843
Struzynska L, Chalimoniuk M, Sulkowski G (2005) Changes in expression of neuronal and glial glutamate transporters in lead-exposed adult rat brain. Neurochem Int 47:326–333
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Bonde C, Sarup A, Schousboe A, Gegelashvili G, Zimmer J, Noraberg J (2003) Neurotoxic and Neuroprotective effects of the glutamate transporter inhibitor DL-threo-beta-benzyloxyaspartate (DL-TBOA) during physiological and ischemia-like conditions. Neurochem Int 43:371–380
Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB (2010) M.R.C. Cognitive Function Ageing Neuropathology Study Group, astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 31:578–590
Tian G, Kong Q, Lai L, Ray-Chaudhury A, Lin CL (2010) Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer’s disease. J Neurochem 113:978–989
Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR (2011) Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging 32:553.e1–553.e11
Cassano T, Serviddio G, Gaetani S, Romano A, Dipasquale P, Cianci S, Bellanti F, Laconca L, Romano AD, Padalino I, Laferla FM, Nicoletti F, Cuomo V, Vendemiale G (2011) Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol Aging. 33:1121.e1–1121.e12
Piermartiri TCB, Figueiredo CP, Rial D, Duarte FS, Bezerra SC, Mancini G, De Bem AF, Prediger RDS, Tasca CI (2010) Atorvastatin prevents hippocampal cell death, neuroinflammation and oxidative stress following amyloid-β1–40 administration in mice: evidence for dissociation between cognitive deficits and neuronal damage. Exp Neurol 226:274–284
Kulijewicz-Nawrot M, Syková E, Chvátal A, Verkhratsky A, Rodríguez JJ (2013) Astrocytes and glutamate homoeostasis in Alzheimer’s disease: a decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neurol 5:273–282
Beart PM, O’Shea RD (2007) Transporters for l-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150:5–17
Foran E, Bogush A, Goffredo M, Roncaglia P, Gustincich S, Pasinelli P, Trotti D (2011) Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia 59:1719–1731
Foran E, Rosenblum L, Bogush A, Pasinelli P, Trotti D (2014) Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia 62:1241–1253
Alderton WK, Cooper CE, Knowles RG (2001) Nitric oxide synthases: structure, function and inhibition. Biochem J 357:593–615
Goodwin JL, Kehrli ME Jr, Uemura E (1997) Integrin Mac-1 and β-amyloid in microglial release of nitric oxide. Brain Res 768:279–286
Akama KT, Albanese C, Pestell RG, Van Eldik LJ (1998) Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFkβ-dependent mechanism. Proc Natl Acad Sci USA 95:5795–5800
Rossi F, Bianchini E (1996) Synergistic induction of nitric oxide by β-amyloid and cytokines in astrocytes. Biochem Biophys Res Commun 225:474–478
Combs CK, Karlo JC, Kao SC, Landreth GE (2001) β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 21:1179–1188
Melchior F, Schergaut M, Pichler A (2003) SUMO: ligases, isopeptidases and nuclear pores. Trends Biochem Sci 28:612–618
Hoppe JB, Rattray M, Tu H, Salbego CG, Cimarosti H (2013) SUMO-1 conjugation blocks β-amyloid induced astrocyte reactivity. Neurosci Lett 546:51–56
Comerford KM, Leonard MO, Karhausen J, Carey R, Colgan SP, Taylor CT (2003) Small ubiquitin-related modifier-1 modification mediates resolution of CREB-dependent responses to hypoxia. Proc Natl Acad Sci USA 100:986–991
Hooper C, Killick R, Lovestone S (2008) The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 104:1433–1439
Eun Jeoung L, Sung Hee H, Jaesun C, Sung Hwa S, Kwang Hum Y, Min Kyoung K et al (2008) Regulation of glycogen synthase kinase 3β functions by modification of the small ubiquitin-like modifier. Open Biochem J 2:67–76
Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W (2013) Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 123:224–235
Colombo A, Bastone A, Ploia C, Sclip A, Salmona M, Forloni G, Borsello T (2009) JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol Dis 33:518–525
Ploia C, Antoniou X, Sclip A, Grande V, Cardinetti D, Colombo A, Canu N, Benussi L, Ghidoni R, Forloni G, Borsello T (2011) JNK plays a key role in tau hyperphosphorylation in Alzheimer’s disease models. J Alzheimer’s Dis 26:315–329
Feligioni M, Brambilla E, Camassa A, Sclip A, Arnaboldi A, Morelli F, Antoniou X, Borsello T (2011) Crosstalk between JNK and SUMO signaling pathways: deSUMOylation is protective against H2O2-induced cell injury. PLoS One 6:e28185
Eckermann K (2013) SUMO and Parkinson’s disease. Neuromol Med 15:737–759
Boggio R, Colombo R, Hay RT, Draetta GF, Chiocca S (2004) A mechanism for inhibiting the SUMO pathway. Mol Cell 16:549–561
Madu IG, Namanja AT, Su Y, Wong S, Li YJ, Chen Y (2013) Identification and characterization of a new chemotype of noncovalent SENP inhibitors. ACS Chem Biol 8:1435–1441
Acknowledgments
The authors would like to thank Dr. Ulrich F. J. Mayer at Mayer Scientific Editing (www.mayerscientificediting.com) for his assistance during the preparation of this manuscript. The authors would like to apologize to all researchers whose work could not be included in this article due to imposed space limitations. This study was supported financially by a grant from the CAEN-ISN, as well as an IBRO Return Home and a Newton Advanced Fellowship (H.C.). W.C.M. would like to thank CAPES for a postgraduate studentship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Additional information
Special Issue: In honor of Dr. Philip Beart.
Rights and permissions
About this article

Cite this article
Martins, W.C., Tasca, C.I. & Cimarosti, H. Battling Alzheimer’s Disease: Targeting SUMOylation-Mediated Pathways. Neurochem Res 41, 568–578 (2016). https://doi.org/10.1007/s11064-015-1681-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1681-3