
Abstract
The IPTG-inducible promoter family, Pgrac, allows high protein expression levels in an inducible manner. In this study, we constructed IPTG-inducible expression vectors containing strong Pgrac promoters that allow integration of the transgene at either the amyE or lacA locus or both loci in Bacillus subtilis. Our novel integrative expression vectors based on Pgrac promoters could control the repression of protein production in the absence and the induction in the presence of an inducer, IPTG. The β-galactosidase (BgaB) protein levels were 9.0%, 15% and 30% of the total cellular protein in the B. subtilis strains carrying single cassettes with the Pgrac01, Pgrac100 or Pgrac212 promoters, respectively. The maximal induction ratio of Pgrac01-bgaB was 35.5 while that of Pgrac100-bgaB was 7.5 and that of Pgrac212-bgaB was 9. The inducible expression of GFP and BgaB protein was stably maintained for 24 h, with the highest yield of GFP being 24% of cell total protein while the maximum amount of BgaB was found to be 38%. A dual integration of two copies of the gfp+ gene into the B. subtilis genome at the lacA and amyE loci resulted in a yield of about 40% of total cellular protein and a 1.74-fold increase in GFP compared with single-integrated strains containing the same Pgrac212 promoter. The capability of protein production from low to high levels of these inducible integrative systems is useful for fundamental and applied research in B. subtilis.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bacillus subtilis is a potential host strain for basic research and industrial applications (Ming-Ming et al. 2006; Stork et al. 2021; Toymentseva et al. 2012; Zhang et al. 2020; Zhou et al. 2019). This Gram-positive bacterium serves as a model system in fundamental research for studying metabolites, cell division, cell behavior, endospore and biofilm formation (Stork et al. 2021). B. subtilis has been considered a microbial factory for producing heterologous proteins, especially recombinant enzymes and biopharmaceuticals, because of its safety and physiological and biochemical characteristics (Guan et al. 2016). B. subtilis offers several advantages for large-scale fermentation, including a fast growth rate and high cell number, the capability for secretion of synthesized proteins into the culture medium (Guan et al. 2016; Phan et al. 2006; Su et al. 2020; Yang et al. 2020), as well as a relatively complete understanding of the molecular mechanisms of transcription, translation and protein processing (Cui et al. 2018). In biobased production, B. subtilis has also been used as an effective cellular chassis for building on. This bacterium can be transformed to be more suitable for industrial production using systems by metabolic engineering, which combines a thorough understanding of systematic biology, synthetic biology, and evolution-based engineering to construct optimal biosynthetic pathways in B. subtilis to produce bacterially synthesized compounds (Liao et al. 2021; Xiang et al. 2020).
The main challenge in using B. subtilis as a host organism for heterologous protein production is choosing an optimal expression system (Heravi et al. 2015). Some new strategies include optimizing promoters by enhancing their strength and modulating expression levels using constitutive or inducible promoters. The promoter is an essential element for the expression of a target gene, as it regulates the protein expression level by controlling the level of mRNA transcription (Cui et al. 2018). Different natural or artificial promoters have been tested and engineered, including inducible, constitutive, and auto-inducible promoters that can be used for efficient production of recombinant proteins in B. subtilis for many applications (Cui et al. 2018; Nguyen et al. 2007). Inducible promoters are commonly used to precisely express a target gene product under controlled conditions for a defined duration of fermentation. Expression systems with inducible promoters play significant roles in basic research to identify the function of essential genes in bacterial cells and also for the large-scale industrial production of recombinant proteins. These systems are especially useful for the synthesis of cytotoxic proteins, which are often desired for fine-tuning production (Kluge et al. 2018). These efficient, inducible expression vectors are essential elements for biotechnology applications and industrial manufacturing to convert low protein expression to high expression during fermentation (Castillo-Hair et al. 2019).
The main limitation of using plasmid-mediated expression systems in bacteria is their instability. This problem can be overcome by adding antibiotics to the fermentation medium, but the large-scale use of antibiotics in industrial production has been discouraged because of the risk of generating antibiotic-resistant bacteria with their adverse ecological and legal ramifications (Zhou et al. 2019). The integrative vectors allow the insertion of any gene into the B. subtilis genome by homologous recombination as an alternative way to avoid the disadvantage of host-independent plasmids (Härtl et al. 2001; Ming-Ming et al. 2006; Tran et al. 2020). The integrated gene can be stably maintained in the same copy number without using the selective pressure of antibiotics (Vázquez-Cruz et al. 1996). The integration site of the transgene may be constant in the bacterial chromosome while its physical location in the plasmid for maintenance may be varied for targeting different regions of the bacterial cell (Middleton and Hofmeister 2004). In addition, the presence of an integrated copy of the desired gene may be important for gene regulation (Middleton and Hofmeister 2004). Therefore, the creation of integrated strains has recently become more desirable and practical, especially for ensuring ecological safety in large-scale applications (Yomantas et al. 2011; Tran et al. 2020).
Over the past few decades, many integrative expression systems utilizing different promoters have been developed for the effective production of heterologous proteins in B. subtilis strains (Vavrová et al. 2010; Dong and Zhang 2014; Tran et al. 2020). Recently, some new, more efficient integrative systems have been reported, in which one copy or multiple copies of a target gene are inserted into the bacterial chromosome at different loci (Huang et al. 2017; Watzlawick and Altenbuchner 2019; Zhou et al. 2019). For example, the integrative plasmid-based NBP3510 promoter, a derivative of the stationary phase promoter Pylb (Yu et al. 2016), overexpressed recombinant proteins in B. subtilis from a single copy reporter cassette. When this promoter was used, the accumulation of foreign proteins was 43 percent for BgaB and 30 percent for sfGFP (Zhou et al. 2019). The lacI-T7 system (Castillo-Hair et al. 2019), an IPTG-inducible expression system derived from PT7lac, the repressor gene lacI, and T7 RNA polymerase, has superior performance in B. subtilis with a dynamic range of over 10,000, making it suitable for many applications in basic research and biotechnology. The limitation of incorporating a single copy of the target gene into the chromosome is the low product yield, and incorporation of gene copies at multiple loci is one of the best ways to achieve high yields and stable production (Watzlawick and Altenbuchner 2019). The CRISPR/Cas9 system was demonstrated to be an effective method for inserting five copies of the gan genes in the Pgut-ganA expression cassette into the B. subtilis chromosome to produce galactosidase in the presence of glucitol inducer, with protein activity equal to 10% of the cell's soluble proteins (Watzlawick and Altenbuchner 2019). The Pgrac01 promoter (formerly known as Pgrac) (Nguyen et al. 2007; Phan et al. 2006) and some inducer-free integrative Pgrac derivatives have shown high expression of recombinant proteins in B. subtilis. The integrative Pgrac212-bgaB construct in the absence of inducers produced 42% of total cell protein after 12 h of culture. The BgaB yield reached 53.4% with the dual integration of the Pgrac212-bgaB cassette at the amyE and lacA loci in the absence of an inducer (Tran et al. 2020). However, the strong inducible integrative expression systems for B. subtilis are unavailable to meet the need for studying the production of recombinant proteins and synthetic biology. In this research, we developed a diverse collection of potent IPTG-inducible integrative expression vectors based on Pgrac and its derivatives that control the protein production at different levels in B. subtilis.
Materials and methods
Strains, vectors and bacterial growth
The E. coli strain OmniMAX™ (Invitrogen) was used for all gene cloning and B. subtilis strain 1012 (Saito et al. 1979) was used for integration and protein expression. Cells of E. coli and B. subtilis were grown at 37 °C on Luria–Bertani (LB) agar plates or in LB broth with shaking at 200 rpm. The media were supplemented with appropriate antibiotics for selection as follows: ampicillin (Amp, 100 μg/mL), chloramphenicol (Cm, 10 μg/mL), neomycin (Neo, 10 μg/mL) and spectinomycin (Spc, 100 μg/mL). Plasmid pHT01 without target gene was used as a negative control. Inducible plasmids pHT01-bgaB, pHT10-gfp+ with reporter genes (bagB or gfp+) controlled by Pgrac01 were used as positive controls. The bacterial strains, plasmids, and oligonucleotides used in the present study are shown in Table 1. Bacterial growth was monitored by measuring OD600 using an S-20 spectrophotometer (Boeco, Germany).
Construction of IPTG-inducible integrative expression vectors
The first step in this procedure was the construction of the three basic inducible expression systems integrated at the amyE locus under control of the Pgrac01, Pgrac100, and Pgrac212 promoters, respectively named pHT1379, pHT1382 and pHT1380. To construct pHT1380, we used the integrative vector pHT1314 (from our lab’s collection) as a backbone. The neomycin resistance gene (neo) of pHT1314 was replaced by the spectinomycin-resistance gene (spc) by cutting pHT1314 with EcoRI and NotI and replacing the neo gene with the 993-bp spc sequence. The spc gene was amplified from pDG1728 (Guérout-Fleury et al. 1996) by PCR using the oligonucleotide pair ON979/ON980. The PCR product was isolated and incubated with Eco31I to optimize the T4 DNA ligase reaction and incorporate the spectinomycin-resistance gene into the pHT1380 vector. Next, the pHT1380 vector was used as the backbone to construct pHT1379 and pHT1382, in which the Pgrac212 promoter was replaced by Pgrac01, and Pgrac100, respectively. The inducible expression vectors, pHT1582, pHT1608 and pHT1619 were generated by introducing bgaB gene amplified by PCR using the oligonucleotide pair ON941/ON2134 and template pDH33-bgaB (Nguyen et al. 2007) into pHT1379, pHT1382 and pHT1380. Similarly, pHT2103, pHT2106 and pHT2107 were modified created by introducing gfp+ gene amplified by PCR using pHT10-gfp+ as template (Nguyen et al. 2007) with ON1277/ON1280 primers.
The inducible expression vector with the Pgrac212 promoter for integrating at lacA locus was constructed as follows. First, the Pgrac212 cassette amplified by PCR using ON873/ON872B and pHT264 was introduced into the backbone vector pHT1305 at BamHI and EcoRI resulting in the basic vector pHT1311. The gfp+ gene amplified from pHT10-gfp+ (Nguyen et al. 2007) with ON1277/ON1280 was inserted into the vector pHT1311 to make pHT2096. Vectors, pHT2096 and pHT2107 were used to generate the B. subtilis strain dually incorporated at both amyE and lacA loci. Figure 1 depicts the structural model for all inducible integrative vectors with Pgrac promoters used in this work.

Map of an integrative inducible expression vector and promoter sequences used in this study. a Map of vector pHT1619 carrying the Pgrac212 promoter between the 3’ and 5’ regions of the B. subtilis amyE gene. b Schematic representation of the cassettes that were inserted into the B. subtilis chromosome at amyE/lacA locus. c The promoter sequences of the Pgrac01, Pgrac100 and Pgrac212
Generation of B. subtilis recombinant strains
The transformation protocol to create the B. subtilis recombinant strains was described elsewhere (Phan et al. 2017). Competent B. subtilis 1012 cells in 10 ml of LS broth (Glucose 40%, L-Tryptophan 0.1%, casein 2%, yeast extract 10%, MgCl2 1 M, CaCl2 1 M, 10X S-Base + MgSO4) were shaken at 50 rpm for 2 h at 30 °C. Then, 100 µl of 0.1 M ethylene glycol tetraacetic acid (EGTA) was added to the flask, which was kept at room temperature (25 °C) for 10 min. Next, the prepared competent cells and recombinant plasmids were gently mixed in a 1.5-ml tube before shaking for 2 h at 200 rpm at 37 °C. Lastly, the cells were harvested by centrifugation at 8,000 rpm for 1 min, resuspended in 60 µl of the supernatant, and spread on LB agar plates containing suitable antibiotics. All plates were incubated overnight at 37 °C. The colonies of integrative B. subtilis strains were selected using PCR reactions with three specific primer pairs to confirm the integration (i) at 3'amyE or 3'lacA, (ii) at 5'amyE or 5'lacA, (iii) and the presence of target gene gfp+ or bgaB. PCR reactions with five pairs of primers (Fig. 2a) were also used to screen the B. subtilis strains that integrated two copies of the gfp+ gene into the genome at both the amyE and lacA loci.

Representative PCR for the confirmation of the integrative B. subtilis strains. a Location of primers used for PCR to verify B. subtilis HT2107 colonies with double homologous recombination into amyE locus. b PCR products of template of B. subtilis HT2107 integrated at amyE locus run on 2% agarose gel: (1) products of PCR reaction with ON470/ON2010 (1558 bp), (2) products of PCR reaction with ON562/ON384 (751 bp), and (3) products of PCR reaction with ON979/ON469 (1827 bp). c PCR products of template of B. subtilis HT2107HT2096 integrated at both amyE and lacA loci on 2% agarose gel: (1) Products of PCR reaction with ON470/ON2010 (1558 bp), (2) products of PCR reaction with ON562/ON384 (751 bp), (3) products of PCR reaction with ON979/ON469 (1827 bp), (4) products of PCR reaction with ON1442/ON945 (747 bp), and (5) products of PCR reaction with ON946/ON1441 (713 bp). d Location of primers used for PCR to identify B. subtilis colonies with double homologous recombination into amyE and lacA loci
Determination of reporter protein expression
Single colonies of B. subtilis strains were inoculated into LB broth with suitable antibiotics (Table 1) and grown in a shaking incubator at 200 rpm at 37 °C to mid-log phase (OD600 of 0.8–1). Different concentrations of IPTG were added to each culture to induce the expression of the target gene. The optical density (OD) of each strain's culture varies with time and IPTG inducer concentration. Therefore, the amount of cells in each B. subtilis strain culture must be fixed in order to compare the levels of protein production induced by different IPTG concentrations. At the specified times (0 h—before induction and 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24 h—after induction), the OD600 of each culture was measured so that the volume of cells could be calculated using the rule of triangles to obtain OD600 = 1.2 (to measure protein activity) or OD600 = 2.4 (to conduct SDS-PAGE).
The GFP fluorescent measurements were prepared by adding 500 µl of PBS buffer (0.137 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, 0.0018 M KH2PO4; pH = 7.4) containing 200 µg/ml lysozyme to the samples and inoculated at 37 °C for 30 min. The mixtures were then centrifuged at 13,000 rpm for 5 min. The GFP fluorescent was measured by a microplate fluorometer (CLARIOstar Plus—BMG LabTech) at 470 ± 8 nm and 515 ± 8 nm (corresponding excitation and emission wavelength, respectively) after transferring 50 µl of each sample to a well of a NuncTM 384 plate. Finally, the GFP expression of each sample was calculated by dividing the relative fluorescent unit (RFU) by the sample's OD600 (dGFP/OD600) (Tran et al. 2017).
For measuring BgaB activity, samples were mixed with 480 µl lacZ buffer (0.06 M Na2HPO4, 0.04 M NaH2PO4, 0.01 M KCl, 1 mM MgSO4) containing 50 µg/ml lysozyme and centrifuged for 5 min at 13,000 rpm. Then, 10 µl of each sample was transferred to each well of a NuncTM 384 plate containing 40 µl of lacZ buffer. Each well was then added 12.5 µl of 1 mg/ml 4-Methylumbelliferyl -d- galactopyranoside (MUG) dissolved in dimethyl sulfoxide (DMSO). After 15 min of incubation at 55 °C, 15 µl of 1 M Na2CO3 ended the reaction. The fluorescent created by 4-methylumbelliferon, a product of MUG's hydrolized reaction, was then measured using a microplate fluorometer (CLARIOstar Plus—BMG LabTech). Consequently, the BgaB activity (MUG units) was determined as follows: (Vl/Vs) x F360/460/ (t x OD600). Vl: the sample lysis volume in lacZ buffer; Vs: the sample volume to the well plate; F360/460: the fluorescent signals determined by a microplate fluorometer; t: the inoculation time; OD600: the sample optical density (Tran et al. 2017).
For SDS-PAGE analysis, the B. subtilis strains were grown as described above and induced with different concentrations of IPTG. Different volumes of cell suspension equivalent to an OD600 of 2.4 (about 0.0853 ± 0.004 g cell biomass) were collected at 0 h (before induction) and at 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, and 24 h after induction. Cell pellets were lysed with 100 µl lysis buffer (25 mM SDS, 250 mM sucrose) and 2.5 µl lysozyme at 50 mg/mL. The mixtures were vortexed and then incubated for 5 min at 37 °C. Following that, 25 µl of 5X sample buffer was added. The samples were thoroughly mixed before being heated at 95 °C for 5 min and centrifuged at 13,000 rpm for 5 min. Aliquots of 8 µl of each sample were loaded to each well for SDS-PAGE (Phan et al. 2017). After electrophoresis, stain gels with a Coomassie staining solution and de-stain with a solution. Scan the gel using a conventional HP SCANJET G3110 scanner, and then calculate the percentage expression of each protein in each well of SDS-PAGE using the AlphaEaseFC 4.0 software.
Statistical methods
Each experiment was replicated three times under the same conditions. The protein activity units of each sample are shown as the mean ± standard deviation of the relative fluorescence units (RFU) for GFP and the methylumbelliferyl β-D- galactopyranoside (MUG) units for BgaB from three experiments. The relationships between β -galactosidase activity (y) and IPTG concentration (x) were fitted with third-degree polynomial equations using Excel.
Results
BgaB production under the control of Pgrac01, Pgrac100, and Pgrac212 promoters
The BgaB protein production levels in B. subtilis strains containing integrative constructs at the amyE locus were investigated after induction with IPTG. Samples of cell suspension were collected at the time of induction (0 h) and 4 h after induction. The B. subtilis strains carrying the Pgrac01—Pgrac100 and Pgrac212-bgaB constructs integrated in their amyE gene were named HT1582, HT1608, and HT1619, respectively. The B. subtilis strain carrying the replicative plasmid pHT01-bgaB under the control of the Pgrac01 promoter with bgaB as a reporter gene was used as the positive control, while pHT01 without the reporter gene was used as the negative control. The SDS-PAGE gel was scanned for the picture, and then the AlphaEaseFC 4.0 software was used to calculate the percentage of target proteins in total cellular proteins.
In comparison with the inducer-free integrative vector carrying the same cassette Pgrac01-bgab, the production of BgaB by HT1582 was approximately 82% of that of HT2170 (Fig. 3a). The BgaB expression level of HT1582 was about 66% of Bs/pHT01-bgaB in the presence of 1 mM IPTG for 4 h. The SDS-PAGE results revealed that Pgrac01 was the weakest promoter, with BgaB protein production accounting for only about 9% of total protein production (Fig. 3b). The BgaB yield accumulated to 15% in B. subtilis strain HT1608 (Pgrac100), and this increased to 30% in B. subtilis strain HT1619 (Pgrac212) after 4 h of induction with 1 mM IPTG. SDS-PAGE also confirmed the relatively low background expression of these integrative B. subtilis strains in non-induced cultures, where BgaB accumulation was undetectable in the HT1582 well and low BgaB levels were observed in the HT1608 and HT1619 wells (Fig. 3b).

BgaB expression by IPTG-inducible vectors with Pgrac promoter derivatives in B. subtilis 1012. a BgaB activities of surveyed B. subtilis trains; b SDS-PAGE analysis of the pellets of strains carrying vectors based on Pgrac01/Pgrac100/Pgrac212 integrated at amyE locus. Strains were cultured in triplicate and each experiment was repeated three times. Recombinant strains were cultured in LB broth with suitable antibiotics to mid-log phase, and cells were collected at 0 h (before induction) and 4 h (after induction). Bs/pHT01, B. subtilis strain containing pHT01 plasmid without bgab (negative control); Bs/pHT01-BgaB, B. subtilis strain containing pHT01-bgaB plasmid (positive control). HT2170, inducer-free integrative vector at amyE locus with Pgrac01 promoter and bgaB; HT1582, HT1608, HT1619, inducible integrative vector at amyE locus with Pgrac01, Pgrac100, Pgrac212 promoters, respectively. Error bars represent standard deviation. The relative levels of total intracellular protein were analyzed using Alpha Ease 4.0 software. The BgaB protein at about 79 kDa is indicated by the red dot
To determine the relative induction factor of Pgrac01, Pgrac100 and Pgrac212 promoters, we measured BgaB activities in the presence and absence of IPTG (Fig. 3a). Under induction with 1 mM IPTG, the highest activities of protein expression in the tested B. subtilis strains increased in the order of Pgrac01–Pgrac100–Pgrac212, in which the MUG units of Pgrac100 and Pgrac212 were 1.5 times and 1.8 times higher than those of Pgrac01, respectively. The best performance of HT1619 (Pgrac212) was 83,169 ± 3288 MUG units, while HT1582 (Pgrac01) had the lowest value with 50,342 ± 1690 MUG units (Fig. 3a). Despite the lowest protein expression level, the induction ratio obtained in the Pgrac01 integrative system was higher than in the other two systems. In the presence of 0.01 mM IPTG, the induction ratio of HT1582 (Pgrac01) was 7.6 and increased to 30 and 35.5 when the IPTG concentration was increased to 0.1 and 1.0 mM, respectively. Similarly, the variation of IPTG concentration in the cultures as 0.01, 0.1 and 1.0 mM, lead to the change of induction ratio of BgaB production for Pgrac100 and Pgrac212. For example, the induction ratio of the Pgrac100 integrative system was 5, 6, 7.5, while these ratios were 6, 8.5 and 9, respectively, for the Pgrac212 integrative system (Table 2). Thus, the integrative expression vectors could induce high expression levels of BgaB in B. subtilis. Next, to check the expression vectors with a different protein, we replaced the bgaB gene with the gfp+ gene under the control of Pgrac01, Pgrac100, Pgrac212 and measured GFP production in B. subtilis.
GFP expression levels under control of different Pgrac promoters
In comparing the inducible integrative vectors with the replicative plasmids, the GFP expression of the integrative strain, HT2103, was lower than that of the replicative strain, Bs/pHT10-gfp+, (positive control) with the same Pgrac01 promoter. After four hours of culture without inducer, the GFP production of HT2103 was about 52% RFU compared to Bs/pHT10-gfp + , and the value dropped to approximately 25% when the cultures were induced with 1 mM IPTG (Fig. 4b). The protein production of B. subtilis strains with integrative vectors was less than that of B. subtilis strains with replicative plasmids because of the limited number of copies in the former. However, the GFP production levels of integrative strains HT2106 and HT2107 carrying Pgrac100 and Pgrac212 promoters, respectively, increased significantly. The GFP production levels of HT2106 and HT2107 were 40% and 105%, respectively, of the replicative strain pHT10-gfp + carrying the Pgrac01 promoter (Fig. 4b). Similar to the BgaB production (Fig. 3b), the highest activities of GFP expression under induction with 1 mM IPTG increased in the order of Pgrac01—Pgrac100—Pgrac212, with the GFP accounting for 9%, 14%, and 22% of the total cellular protein in the integrative B. subtilis strains carrying the Pgrac01, Pgrac100, and Pgrac212 promoters, respectively (Fig. 4a).

GFP expression in B. subtilis 1012 constructs containing IPTG-inducible integrative vectors with Pgrac promoter derivatives. a SDS-PAGE showing GFP expression by constructs inserted at the amyE locus. B. subtilis strains with Pgrac01 (HT2103), Pgrac100 (HT2106), or Pgrac212 (HT2107). b GFP activities of all strains with vectors integrated at amyE locus. All strains were cultured in triplicate and each experiment was repeated three times. The recombinant strains were cultured in LB broth with suitable antibiotics to mid-log phase. Cells were collected at 0 h (before induction) and 4 h (after induction). Bs/pHT01, B. subtilis strain containing independent plasmid pHT01 without reporter protein (negative control); Bs/pHT10-gfp+, B. subtilis strain containing pHT01-gfp+ (positive control). Error bars represent standard deviation. The amounts of total intracellular protein were assessed using Alpha Ease 4.0 software. The GFP protein at about 27 kDa is indicated by the red dot
After four hours of non-induction without IPTG, B. subtilis strain HT2103 had a background expression level of 3,700 ± 216 RFU; B. subtilis strain HT2106 had a level of 17,980 ± 451 RFU, and B. subtilis strain HT2107 had a level of 19,720 ± 1,206 RFU. The RFUs of these strains increased to 53,030 ± 2,495; 91,560 ± 966 and 232,510 ± 4,997, respectively, after 4 h of induction with 1 mM IPTG (Fig. 4b). The value of the induction factor increased as IPTG concentration increased; the induction factor of HT2103 (Pgrac01-gfp+) was 1.45 in the culture with 0.01 mM IPTG, 8.01 in the culture with 0.1 mM IPTG, and 14.3 in the culture with 1.0 mM IPTG. Similarly, in the presence of 0.01, 0.1, and 1.0 mM IPTG, the induction rates of HT2106 (Pgrac100-gfp+) were 1.9, 4.1, and 5.1, respectively, while the ratios for HT2107 (Pgrac212-gfp+) were 1.9, 10.0, and 11.8, respectively (Table 2). These results confirmed that our novel integrative systems were able to control multiple levels of protein production in B. subtilis. These new inducible integrated systems created a range of expression vectors from low to high for use in various applications in industry as well as research.
Influence of induction time on the production of BgaB and GFP
The experiments above demonstrated the expression capability of inducible vectors based on Pgrac01 and Pgrac derivatives inserted into the B. subtilis genome. Next, in order to provide the induction time needed for overproduction of a recombinant protein in B. subtilis, we followed the production of BgaB and GFP for 24 h. Two B. subtilis strains HT2107 and HT1619 carrying the Pgrac212 promoters were grown in LB broth to mid-log phase, which corresponds to an OD600 of 0.8–1.0. Cells were harvested at different times: 0 h, before induction, and at 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22 and 24 h after induction. The Pgrac212 promoter was chosen for this experiment because of its enhanced expression of the target protein.
The results verified the capacity of the inducible integrative vectors constructed in this study for consistently high protein production (Fig. 5a, b). The expression of GFP and BgaB protein was maintained after 24 h of induction with 1.0 mM IPTG, in which the highest yield of GFP obtained at 8 h was 24% of cell total protein while the maximum amount of BgaB was found to be 38% at 24 h (data after subtracting the background at 0 h). The BgaB expression level steadily rose with increasing incubation time. The BgaB yield and its activity obtained at 24 h were 1.42 times and 1.73 times higher than that obtained at 4 h, respectively (Fig. 5b, c). BgaB production by the B. subtilis strain containing the chromosomally integrated Pylb-bgaB cassette was maintained during 18 h of culture (Zhou et al. 2019). Similarly, the production of BgaB by the inducer-free integrative vector with Pgrac212 promoter was also found to be maintained over a 12 h culture period (Tran et al. 2020).

Effect of time and IPTG concentration on the ability to produce recombinant protein of B. subtilis 1012 strains with Pgrac212. a The SDS-PAGE of pellets of strains HT1619 at different times of induction; b The SDS-PAGE of pellets of strains HT2107 at different times of induction; c Protein activity of HT1619 and HT2107 at different times of induction; d GFP activity of HT2107 (RFU) and β-galactosidase activity (MUG) of HT1619 are shown as a function of IPTG concentration. All strains were cultured in triplicate and each experiment was repeated three times under the same conditions. The recombinant strains were cultured in LB broth without antibiotics to mid-log phase. Cells were collected at 0 h before induction and at 2, 4, 6, 8, 10, 12, 14, 16, 18, 20 and 24 h after induction with 1.0 mM IPTG. Bs/pHT01, B. subtilis strain containing independent plasmid pHT01 without reporter protein (negative control). Error bars represent standard deviation. The amounts of total intracellular protein were assessed with Alpha Ease 4.0 software. The BgaB and GFP proteins at 79 kDa and 27 kDa, respectively, are indicated by red dots. The polynomial lines represent the correlation function
There was a significant decrease in both GFP yield and GFP activity of the HT2107 strain when the culture time was continued longer than ten hours (Fig. 5a, c). The GFP activity showed the highest value of 252,510 ± 12,853 RFU at four hours even though the GFP yield was only 70% of that at six hours and eight hours. The reduction of GFP levels when the cells enter the middle/late stationary phase may be related to the degradation of GFP in the cytoplasm of B. subtilis. The differences could be explained by the nature of the target protein on the reporter stability. The variations in BgaB and GFP production were also observed when evaluating protein expression in various integrative B. subtilis strains with the Pgrac01 and Pgrac100 promoters, in which BgaB expression is more stable than GFP expression (data not shown).
We demonstrated that the stability of expression controlled by these systems in B. subtilis strains could be manipulated using various types of recombinant protein. For example, BgaB expression was consistent for 24 h after induction, whereas GFP production peaked at 4 h. Therefore, four-hour cultures of B. subtilis strain 1012 were used to determine the influence of IPTG concentration on the expression level of recombinant protein in subsequent experiments.
Influence of inducer IPTG concentration on protein production levels
In order to fine-tune the correlation of IPTG concentration with BgaB and GFP protein production levels, we cultured B. subtilis strains in LB broth without antibiotics at 37 °C with or without a range of IPTG concentrations for induction. IPTG concentrations ranging from 0 to 500 μM were used in this study. After four hours of induction, we measured the OD600 of each cell suspension. The cell volume, defined as 1.2 × OD600, was assumed to correspond to protein activity.
The newly designed vectors with Pgrac212 promoter effectively controlled the rate of protein expression in B. subtilis (Fig. 5d). In the absence of IPTG, the LacI inhibitor protein acts as a repressor of transcription from the Pgrac promoter. When IPTG is added to the culture, it binds to LacI and allows transcription to occur, which results in the production of the target genes, gfp+ and bgaB. In this experiment, the relationship between β-galactosidase activity and IPTG concentration was fitted with a third-degree polynomial equation, in which the β-galactosidase activity (MUG units) changed as a function of y and the independent variable x were related as:
Similarly, the GFP activity and IPTG concentrations were fitted to the equation:
In general, protein expression was sensitive to changes in IPTG concentration, the protein production level rising along with the increase in inducer concentration. These results suggested that the rate of gene transcription, and hence the amount of protein synthesized could be regulated by using the appropriate IPTG concentration. However, a decline in cell viability was observed for B. subtilis cultures grown under highly induced conditions: > 1000 μM IPTG (data not shown). It was demonstrated that the adjustment of IPTG concentration may influence the final performance and growth of E.coli strains (Dvorak et al. 2015), and the same may be true for B. subtilis strains. The potential toxicity of IPTG may affect the survival of bacterial cells (Dvorak et al. 2015).
Increasing the recombinant protein production with multiple integration cassettes
Our systems deliver critical elements for basic research, biotechnology, and industrial applications. However, the ectopic integrative vectors suffer from the disadvantage of low product yield because the gene is present as only a single copy. This limitation can be overcome by the integration of many target genes into the B. subtilis genome at various sites (Watzlawick and Altenbuchner 2019). The integrative strains with multiple loci are expected to show stable, high production of recombinant proteins (Watzlawick and Altenbuchner 2019; Tran et al. 2020). In this study, we created B. subtilis 1012 strains with the gfp+ gene under the control of the Pgrac212 promoter integrated at both the lacA and amyE loci to overexpress the GFP protein. First, recombinant B. subtilis cells harboring vector pHT2107 incorporated at the amyE locus were generated by double homologous recombination in the presence of spectinomycin. Next, competent cells of HT2107 were used to integrate the second vector, pHT2096, by homologous recombination at the lacA locus in the presence of neomycin. As a result of this construction, these B. subtilis strains have two copies of the gfp+ gene in the genome–one at the amyE and one at the lacA site–which stably produces GFP protein at high levels. The recombinant B. subtilis strains, HT2107HT2096 with two copies of gfp+ gene by dual incorporation, were verified by PCR (Fig. 2c, d). The GFP activity level of HT2107HT2096 was measured as dimensionless relative fluorescence units (RFUs) and the expression as GFP protein was evaluated by SDS-PAGE.
The GFP activities of B. subtilis strains containing two copies of the gfp+ gene inserted into the genome at the lacA and amyE loci were almost twice as high as in strains containing only a single integrated gene at lacA or amyE with the same promoter. The new dual-integration system of HT2107HT2096 could strictly control the expression of the target protein, GFP, through addition of IPTG at a specific concentration. The ratio of the GFP activity of HT2107HT2096 after induction with 1 mM IPTG to the activity without inducer was 32.2-fold after 2 h of induction, and 6.7-fold after 4 h (Fig. 6b). The background expression level of the dual-integrated B. subtilis strain with two copies of gfp+ gene was not significantly different from that of the single-integrated strains with one copy of the gfp+ gene at 0 h in the absence of IPTG (Fig. 6b). The multiple integrative strain harboring the inducible Pgrac212 promoter accumulated GFP at a level of about 40% of total protein. The protein bands on SDS-PAGE (Fig. 6a) showed that the GFP level in the double-integration strains was increased 1.74-fold compared to single-integration strains containing the same promoter. Similarly, BgaB expression in the inducer-free integrative B. subtilis strains with Pgrac212 cassettes incorporated at both lacA and amyE loci, showed similar results with a yield of BgaB of up to 53.4% of total protein. The dual-expressing construct was 1.3-fold higher than the single-integrative strains with the same Pgrac212 auto-inducible promoter (Tran et al. 2020).

GFP expression levels in B. subtilis strains with Pgrac212 promoters. HT2107 and HT2096 contained one copy of gfp+ gene at amyE locus or lacA locus, respectively. HT2107HT2096 contained two copies of gfp+ gene at both amyE and lacA loci. a SDS-PAGE separations; b GFP activities. Cells were cultured in LB broth with inducer IPTG (0.01; 0.1; 1 mM) or without inducer IPTG (0 mM). Cells were collected after four hours of induction and GFP activity and protein yield were measured. Data are means of three independent experiments. The level of total intracellular protein was determined with Alpha Ease 4.0 software. GFP protein at about 27 kDa is indicated by the red dot
In conclusion, the transformation procedure to create the recombinant B. subtilis 1012 strains for the overproduction of the heterologous protein by dual gene integration is feasible and effective. The multiple integrations increase the target gene copy number, thus increasing expression levels. These multiple-integrating vectors can overcome the disadvantage of low expression levels of single-copy transformants.
Discussion
Previous research showed that bacterial strains containing IPTG-inducible plasmids with the Pgrac100 promoter could produce BgaB protein levels up to 30% of total cellular protein (Phan et al. 2012). The protein expression levels were 4.5-fold lower than those of IPTG inducer-free expression plasmids generated by deleting the lacI gene (Tran et al. 2017). The inducer-free integrative Pgrac212-bgaB construct has been reported to sustain a production level of 42% of total cell protein after 12 h of culture. The BgaB yield reached 53.4% with the dual-integration Pgrac212-bgaB cassette at both the amyE and lacA loci without inducer (Tran et al. 2020). Thus, the Pgrac promoter and its derivatives could be effectively used for the expression of recombinant proteins in B. subtilis, either as a replicative plasmid or an inducer-free integrative vector. However, whether these promoters can control the expression of protein in an inducible integrative manner has not been investigated.
In this paper, we developed a collection of inducible integrative expression vectors based on the Pgrac01 promoter and its derivatives, including Pgrac100 and Pgrac212, and investigated protein expression levels to determine the functionality of the different constructs for different purposes. The BgaB protein yields were 9%, 15% and 30%, while the GFP protein levels were 9.0%, 14% and 22% of the total intracellular protein in the inducible integrative B. subtilis strains carrying Pgrac01, Pgrac100 and Pgrac212 promoters, respectively. Developing integrative vectors based on strong promoters is an effective method for increasing the target protein's expression level. Pgrac100 was derived from Pgrac01 due to the variations of -35, -15, and the UP element. These modifications improve the efficiency of transcriptional machinery initiation by increasing the affinity between RNA polymerase and the promoter region. Consequently, the cell produces more mRNA from the target gene, enhancing its ability to produce the target protein (Phan et al. 2015). The promoter Pgrac212 is optimized from Pgrac01 with a 13-nucleotide distance between lacO and the ribosome binding site (RBS). This allows the ribosome to bind to the mRNA without the stem-loop structure at the 5'phosphate terminus interfering. Thus, mRNA is protected from RNase attack, extending its half-life to greater than 60 min (Phan et al. 2012). All of these modifications have been shown to increase the expression level of the target protein relative to Pgrac01.
In addition, these expression systems could strictly control protein expression in B. subtilis. The low leakiness is essential for excitable systems where the production of target protein must be controlled under different cellular programs, especially for rapid initial cell growth with toxic proteins (Castillo-Hair et al. 2019). After four hours of induction with 1 mM IPTG, the MUG units of the B. subtilis strain containing the Pgrac212-bgaB cassette were about 9.0 times higher than in the absence of IPTG. The amount of protein produced by the B. subtilis strain containing the Pgrac212-gfp+ cassette was 11.8-fold higher. After induction with 1 mM IPTG, the target protein's expression was sustained for 24 h.
The protein expression levels were sensitive to changes in IPTG concentration, and protein activity steadily increased along with the increase in inducer concentration. Our new inducible integrative systems were shown to be as efficient as some recently published systems. For example, the Pylb promoter could efficiently control the expression of β-galactosidase (BgaB) and super-folded green fluorescent protein (sfGFP), and the resulting levels of these two proteins were 43% and 30% of total cellular protein, respectively (Zhou et al. 2019). We concluded that our integrative inducible vectors containing Pgrac family promoters could be used as an effective means for recombinant protein production for various purposes.
Using an inducible integrative vector has proven to be a workable solution for controlling the level and stability of recombinant protein production in B. subtilis. However, the protein expression levels of integrative B. subtilis strains were usually lower than those of plasmid strains with the same promoter because of the limited number of gene copies. In this study, the fluorescence of cells containing the integrative vector HT2103 was about 52% of that from the independent plasmid pHT10-gfp+ after 4 h of culture without inducer and these values dropped to approximately 25% when the cultures were induced with 1 mM IPTG (Fig. 4b). Because the pMTLBs72 plasmid has been shown to be stably inherited as 6 units per chromosome (Titok et al. 2003), it was used as a template to create pHT plasmids. As a result, when both strains contain pHT systems with the same promoter, a single copy of a gene in an integrative strain has a higher capacity for expression than a plasmid strain.
Similarly, although both the integrative vector, HT1582, and the replicative plasmid, pHT01-bgaB, (positive control) carried the Pgrac01 promoter, the MUG units of HT1582 were 11.5% of Bs/pHT01-bgaB in culture without IPTG and reached 66% with induction (Fig. 3a). To overcome this disadvantage, we introduced multiple copies into the genome at the amyE and lacA loci to increase expression levels. Employing the CRISPR/Cas9 method, a B. subtilis transformant with five copies of chromosomally integrated ganA was constructed that produced β-galactosidase efficiently during forty generations (Watzlawick and Altenbuchner 2019). The effort to develop a dual-incorporated system of the bgaB gene in the B. subtilis genome was justified by the results with the inducer-free promoter, Pgrac212, which produced BgaB up to 53.4% of total cellular proteins (Tran et al. 2020). Our IPTG-inducible system employing the Pgrac212 promoter could strictly control the expression of two copies of the gfp+ gene inserted into the B. subtilis genome at both lacA and amyE loci, resulting in a GFP accumulation of about 40% of intracellular protein.
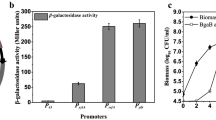
Under some experimental conditions, different levels of protein expression are required. To meet this need, the range of protein expression levels attainable has been estimated by determining the best promoter for each demand. Figure 7 shows the spectrum of protein production capacity based on protein activity. It can be seen that the protein expression controlled by our system steadily increases from low to high levels. Therefore, these constructs are expected to offer an assortment of suitable options for expressing target proteins for different needs. The inducible Pgrac promoter and its derivatives could be used as efficient elements in various expression systems, including replicative plasmids and integrative vectors. A series of our integrative vectors with a wide range of protein expression levels, besides inducer-free expression vectors, could be useful for various biotechnology research applications and industrial production.

Range of protein expression levels in B. subtilis 1012 containing inducible integrated vectors controlled by the Pgrac promoter and its derivatives. Data were collected from 4-h cultures without inducer (0 mM IPTG) or with inducer (1 mM IPTG)
Data availability
All data generated or analyzed during this study are included in this published article.
References
Castillo-Hair SM, Fujita M, Igoshin OA, Tabor JJ (2019) An engineered B. subtilis inducible promoter system with over 10 000-fold dynamic range. ACS Synth Biol 8:1673–1678. https://doi.org/10.1021/acssynbio.8b00469
Cui W, Han L, Suo F, Liu Z, Zhou L, Zhou Z (2018) Exploitation of Bacillus subtilis as a robust workhorse for production of heterologous proteins and beyond. World J Microbiol Biotechnol 34:145. https://doi.org/10.1007/s11274-018-2531-7
Dong H, Zhang D (2014) Current development in genetic engineering strategies of Bacillus species. Microb Cell Factor 13:63. https://doi.org/10.1186/1475-2859-13-63
Dvorak P, Chrast L, Nikel PI, Fedr R, Soucek K, Sedlackova M, Chaloupkova R, de Lorenzo V, Prokop Z, Damborsky J (2015) Exacerbation of substrate toxicity by IPTG in Escherichia coli BL21(DE3) carrying a synthetic metabolic pathway. Microb Cell Factor 14:201. https://doi.org/10.1186/s12934-015-0393-3
Guan C, Cui W, Cheng J, Liu R, Liu Z, Zhou L, Zhou Z (2016) Construction of a highly active secretory expression system via an engineered dual promoter and a highly efficient signal peptide in Bacillus subtilis. New Biotechnol 33:372–379. https://doi.org/10.1016/j.nbt.2016.01.005
Guérout-Fleury A-M, Frandsen N, Stragier P (1996) Plasmids for ectopic integration in Bacillus subtilis. Gene 180:57–61. https://doi.org/10.1016/S0378-1119(96)00404-0
Härtl B, Wehrl W, Wiegert T, Homuth G, Schumann W (2001) Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol 183:2696–2699. https://doi.org/10.1128/JB.183.8.2696-2699.2001
Heravi KM, Watzlawick H, Altenbuchner J (2015) Development of an anhydrotetracycline-inducible expression system for expression of a neopullulanase in B. subtilis. Plasmid 82:35–42. https://doi.org/10.1016/j.plasmid.2015.10.002
Huang K, Zhang T, Jiang B, Yan X, Mu W, Miao M (2017) Overproduction of Rummeliibacillus pycnus arginase with multi-copy insertion of the arg R.pyc cassette into the Bacillus subtilis chromosome. Appl Microbiol Biotechnol 101:6039–6048. https://doi.org/10.1007/s00253-017-8355-9
Kluge J, Terfehr D, Kück U (2018) Inducible promoters and functional genomic approaches for the genetic engineering of filamentous fungi. Appl Microbiol Biotechnol 102:6357–6372. https://doi.org/10.1007/s00253-018-9115-1
Liao C, Ayansola H, Ma Y, Ito K, Guo Y, Zhang B (2021) Advances in enhanced menaquinone-7 production from Bacillus subtilis. Front Bioeng Biotechnol. https://doi.org/10.3389/fbioe.2021.695526
Middleton R, Hofmeister A (2004) New shuttle vectors for ectopic insertion of genes into Bacillus subtilis. Plasmid 51:238–245. https://doi.org/10.1016/j.plasmid.2004.01.006
Ming-Ming Y, Wei-Wei Z, Xi-Feng Z, Pei-Lin C (2006) Construction and characterization of a novel maltose inducible expression vector in Bacillus subtilis. Biotechnol Lett 28:1713–1718. https://doi.org/10.1007/s10529-006-9146-z
Nguyen HD, Schumann W (2006) Establishment of an experimental system allowing immobilization of proteins on the surface of Bacillus subtilis cells. J Biotechnol 122:473–482. https://doi.org/10.1016/j.jbiotec.2005.09.012
Nguyen HD, Phan TTP, Schumann W (2007) Expression vectors for the rapid purification of recombinant proteins in Bacillus subtilis. Curr Microbiol 55:89–93. https://doi.org/10.1007/s00284-006-0419-5
Phan TTP, Nguyen HD, Schumann W (2006) Novel plasmid-based expression vectors for intra- and extracellular production of recombinant proteins in Bacillus subtilis. Protein Expr Purif 46:189–195. https://doi.org/10.1016/j.pep.2005.07.005
Phan TTP, Nguyen HD, Schumann W (2012) Development of a strong intracellular expression system for Bacillus subtilis by optimizing promoter elements. J Biotechnol 157:167–172. https://doi.org/10.1016/j.jbiotec.2011.10.006
Phan TTP, Tran LT, Schumann W, Nguyen HD (2015) Development of Pgrac100-based expression vectors allowing high protein production levels in Bacillus subtilis and relatively low basal expression in Escherichia coli. Microb Cell Factories 14:72. https://doi.org/10.1186/s12934-015-0255-z
Phan T, Huynh P, Truong T, Nguyen H (2017) A generic protocol for intracellular expression of recombinant proteins in Bacillus subtilis. In: Burgess-Brown NA (ed) Heterologous gene expression in Ecoli. Springer, New York, pp 325–334
Saito H, Shibata T, Ando T (1979) Mapping of genes determining nonpermissiveness and host-specific restriction to bacteriophages in Bacillus subtilis Marburg. Mol Gen Genet MGG 170:117–122. https://doi.org/10.1007/BF00337785
Stork DA, Squyres GR, Kuru E, Gromek KA, Rittichier J, Jog A, Burton BM, Church GM, Garner EC, Kunjapur AM (2021) Designing efficient genetic code expansion in Bacillus subtilis to gain biological insights. Nat Commun 12:5429. https://doi.org/10.1038/s41467-021-25691-4
Su Y, Liu C, Fang H, Zhang D (2020) Bacillus subtilis: a universal cell factory for industry, agriculture, biomaterials and medicine. Microb Cell Factories 19:173. https://doi.org/10.1186/s12934-020-01436-8
Titok MA, Chapuis J, Selezneva YV, Lagodich AV, Prokulevich VA, Ehrlich SD, Jannière L (2003) Bacillus subtilis soil isolates: plasmid replicon analysis and construction of a new theta-replicating vector. Plasmid 49:53–62. https://doi.org/10.1016/S0147-619X(02)00109-9
Toymentseva AA, Schrecke K, Sharipova MR, Mascher T (2012) The LIKE system, a novel protein expression toolbox for Bacillus subtilis based on the liaI promoter. Microb Cell Factories 11:143. https://doi.org/10.1186/1475-2859-11-143
Tran DTM, Phan TTP, Huynh TK, Dang NTK, Huynh PTK, Nguyen TM, Truong TTT, Tran TL, Schumann W, Nguyen HD (2017) Development of inducer-free expression plasmids based on IPTG-inducible promoters for Bacillus subtilis. Microb Cell Factories 16:130. https://doi.org/10.1186/s12934-017-0747-0
Tran DTM, Phan TTP, Doan TTN, Tran TL, Schumann W, Nguyen HD (2020) Integrative expression vectors with Pgrac promoters for inducer-free overproduction of recombinant proteins in Bacillus subtilis. Biotechnol Rep 28:e00540. https://doi.org/10.1016/j.btre.2020.e00540
Vavrová Ľ, Muchová K, Barák I (2010) Comparison of different Bacillus subtilis expression systems. Res Microbiol 161:791–797. https://doi.org/10.1016/j.resmic.2010.09.004
Vázquez-Cruz C, Ochoa-Sánchez JC, Olmedo-Alvarez G (1996) Pulse-Field gel-electrophoretic analysis of the amplification and copy-number stability of an integrational plasmid in Bacillus subtilis. Appl Microbiol Biotechnol 46:55–60. https://doi.org/10.1007/s002530050782
Watzlawick H, Altenbuchner J (2019) Multiple integration of the gene ganA into the Bacillus subtilis chromosome for enhanced β-galactosidase production using the CRISPR/Cas9 system. AMB Express. https://doi.org/10.1186/s13568-019-0884-4
Xiang M, Kang Q, Zhang D (2020) Advances on systems metabolic engineering of Bacillus subtilis as a chassis cell. Synth Syst Biotechnol 5:245–251. https://doi.org/10.1016/j.synbio.2020.07.005
Yang H, Ma Y, Zhao Y, Shen W, Chen X (2020) Systematic engineering of transport and transcription to boost alkaline α-amylase production in Bacillus subtilis. Appl Microbiol Biotechnol 104:2973–2985. https://doi.org/10.1007/s00253-020-10435-z
Yomantas YA, Abalakina EG, Golubeva LI, Gorbacheva LY, Mashko SV (2011) Overproduction of Bacillus amyloliquefaciens extracellular glutamyl-endopeptidase as a result of ectopic multi-copy insertion of an efficiently-expressed mpr gene into the Bacillus subtilis chromosome. Microb Cell Factories 10:64. https://doi.org/10.1186/1475-2859-10-64
Yu X, Xu J, Liu X, Chu X, Wang P, Tian J, Wu N, Fan Y (2016) Identification of a highly efficient stationary phase promoter in Bacillus subtilis. Sci Rep 5:18405. https://doi.org/10.1038/srep18405
Zhang L, Wei D, Zhan N, Sun T, Shan B, Shan A (2020) Heterologous expression of the novel α-helical hybrid peptide PR-FO in Bacillus subtilis. Bioprocess Biosyst Eng 43:1619–1627. https://doi.org/10.1007/s00449-020-02353-1
Zhou C, Ye B, Cheng S, Zhao L, Liu Y, Jiang J, Yan X (2019) Promoter engineering enables overproduction of foreign proteins from a single copy expression cassette in Bacillus subtilis. Microb Cell Factories 18:111. https://doi.org/10.1186/s12934-019-1159-0
Funding
This work was partially funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under Grant Number 106-NN.02–2015.24. The funding agencies had no role in the design, execution, or interpretation of this work. Phuong Thi Bich Chu was funded by Vingroup Joint Stock Company and supported by the Domestic PhD Scholarship Programme of Vingroup Innovation Foundation (VINIF), Vingroup Big Data Institute (VINBIGDATA), code VINIF.2020.TS.29.
Author information
Authors and Affiliations
Contributions
HDN, TTPP and WS conceived the research and designed experiments. PTBC, TTTN and TTTT conducted experiments. PTBC, HDN analyzed data. PTBC, HDN and WS wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chu, P.T.B., Phan, T.T.P., Nguyen, T.T.T. et al. Potent IPTG-inducible integrative expression vectors for production of recombinant proteins in Bacillus subtilis. World J Microbiol Biotechnol 39, 143 (2023). https://doi.org/10.1007/s11274-023-03566-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-023-03566-8