
Abstract
A plasmid-less and marker-less strain with multi-copy integration of the arginase gene from Rummeliibacillus pycnus was constructed using Bacillus subtilis 168 as a host. A total of nine copies of the arg R.pyc cassettes, in which the R. pycnus arginase gene was fused with the strong promoter P43, were inserted into the recipient chromosome. These multiple insertions were completed via step-by-step integrations into designed (2 copies) and random (9 copies) sites, respectively. A strategy for random site integration was developed based on the construction of the arg R.pyc cassette sandwiched between “front” and “back” homologous arms which were randomly restricted from chromosomal DNA. An antibiotic resistance marker was applied in transformant selection and was eliminated via the Cre/lox system. Performance showed that the highest enzyme activity (14.5 U/mL) was obtained after culture in flasks, and this segregation stable strain could efficiently hydrolyze l-arginine with a 97.2% molar yield, showing potential application in the food industry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
l-Ornithine is a non-essential amino acid that plays important roles in metabolism. This amino acid is a drug component for mediating liver protection and treating liver disease (Jalan et al. 2007). l-Ornithine is an ideal nutritional supplement for athletes and bodybuilders, and this amino acid is effective for both relieving stress and improving sleep (Sugino et al. 2008). Therefore, great demand exists for l-ornithine in the food supplement and pharmaceutical industries. For these reasons, a simple method for l-ornithine production is needed.
Arginase (l-arginine amidohydrolase, EC 3.5.3.1) is a binuclear metalloenzyme that hydrolyzes l-arginine to l-ornithine and urea. Compared with other methods, the use of arginase hydrolysis has several advantages, including the independence on expensive equipment, specificity for substrate, and rapid production (Song et al. 2014). Currently, various arginases from different resources have been overexpressed in Escherichia coli (Song et al. 2014; Yu et al. 2013). Although these recombinant enzymes or cells showed l-ornithine biosynthesis abilities, the safety of engineered microorganisms and their products in the food industry presents growing concern by the public (He et al. 2016).
It is revealed that more than one-third of the industrial enzymes are produced by Bacillus strains (Meissner et al. 2015). Bacillus subtilis, regarded as the “Generally Recognized as Safe” (GRAS) organism for its lack of endotoxins and pathogenicity, has a long history of industrial use (Jeong et al. 1998; Sietske and Diderichsen 1991). Traditional methods of genetically engineering B. subtilis strains involve the introduction of a recombinant plasmid (Povelainen and Miasnikov 2007). However, this method has limitations, such as instability and usage of antibiotic resistance markers (Fleming and Patching 1994; Kastner et al. 2006). Chromosome integration may be an excellent way to overcome the disadvantages of the plasmids and remove the restrictions on the use of plasmid strains in industry. Usually, homologous recombination with the integration of target gene into desired chromosomal regions is applied in plasmid-less strain construction (Sauer et al. 2016). In addition, transposition and site-specific recombination are also utilized for the same integration strategy.
Homologous recombination can be performed through Campbell-type single-crossover recombination using circularized DNA plasmids with internal homologous fragments or through the use of double-crossover recombination with the target gene sandwiched by homologous flanking sequences (Yomantas et al. 2011). However, single-crossover recombination is unstable under non-selective conditions for the possible elimination of the inserted plasmid (Lee et al. 2016). Because of the low efficiency of single-crossover recombination, gene replacement by double-crossover has been designed for target gene integrations. Ectopic insertion with double-crossover leads to more stable recombinant strains.
With the increase of insertion DNA cassette copy number, high production of proteins could be obtained (Zhu et al. 2009). Several methods are available for multi-copy insertions of the target gene. One method uses PCR or isocaudamer enzymatic polymerization technology to construct tandem repeat sequences which are then integrated into target chromosome regions (He et al. 2016). The other one is using PCR or available plasmids to construct a set of DNA cassettes, which contain different homologous fragments and are then integrated into different chromosome regions with several steps (Yomantas et al. 2011). Amplification of the same gene in chromosomal DNA would certainly decrease the stability of the strains because of the potential of homologous chromosomal recombination. Directly repeated cassette insertion in one site would lead to internal chromosome deletions or loss by cyclization (Bron et al. 1991). In turn, integration in different sites may be more stable.
Previously, a thermostable arginase from Rummeliibacillus pycnus SK 31.001 was found and its gene was sequenced (Huang et al. 2016). Characterization of the enzyme revealed that this enzyme could be a good candidate for l-ornithine biosynthesis. In this study, a recombinant plasmid-less and antibiotic resistance marker-less B. subtilis strain was constructed which can efficiently produce R. pycnus arginase. To improve the expression level of R. pycnus arginase, the arginase gene was fused with a P43 strong promoter to generate a P43-arg (arg R.pyc) expression cassette. Initially, the argI gene encoding B. subtilis arginase was knocked out to avoid the effects of this protein. Then, two copies of the expression cassettes were integrated into two designed regions in B. subtilis chromosome, respectively. Combined with the random integration method, a recombinant strain with nine copies of insertion cassettes was obtained. An antibiotic resistance marker was applied in transformant selection and was eliminated via the Cre/lox system. The performance of the new multi-copy strain showed that this marker-less and segregation stable recombinant stain could be a food-grade candidate for l-ornithine biosynthesis.
Materials and methods
Strains, plasmids, materials, and media
Strains and plasmids used in this study were listed in Table S1. All recombinant Bacillus strains were derived from B. subtilis 168.
R. pycnus was cultured in the medium which contained the following: 1% (w/v) glucose, 0.5% (w/v) peptone, 0.5% (w/v) yeast extract, 0.1% (w/v) K2HPO4, pH 6.5, 37 °C. E. coli and B. subtilis strains were grown at 37 °C in Luria Bertani (LB) medium (1% (w/v) NaCl, 1% Trypton, 0.5% (w/v) yeast extract, 1% (w/v) peptone) or LB with agar supplemented by antibiotics (spectinomycin (Spc, 100 mg/L), zeocin (Zeo 20 mg/L), or ampicillin (Amp, 100 mg/L)) when needed. Successful insertions of the arg R.pyc cassettes were confirmed by inoculation of recombinant cells on LB agar plates (containing 0.002% (w/v) phenol red, pH 6.0).
Seed cultures were prepared with inoculating colonies into 25-mL LB media in 250-mL flasks, and then 3% (v/v) seed cultures were inoculated into another 25-mL fresh LB medium for fermentation at 200-rpm rotary shaker at 37 °C.
Genetic engineering methods
Spizizen method was used for transformation of B. subtilis (Anagnostopoulos and Spizizen 1961).
Taq DNA polymerase, PrimeSTAR Max DNA polymerase, restriction endonuclease, SYBR Premix EX Taq™ II, and TA cloning vectors were purchased from TAKARA Co., Ltd. (Dalian, China). T4 DNA ligase, mini chromosome rapid isolation kit, DNA gel extraction kit, mini plasmid rapid isolation kit, oligonucleotide synthesis, and direct sequencing were accomplished by Sangon Biotech Co., Ltd. (Shanghai, China).
All the primers were listed in Table S2.
Enzyme activity assay
The arginase activity assay was determined using l-arginine as substrate. The reaction was performed in 50 mM NaOH-glycine buffer (pH 9.5) with 100 mM l-arginine and 1 mM Mn2+. The reaction was started by adding 200 μL fermentation broth to 1 mL reaction mixture at 80 °C. After incubation for 10 min, reaction was terminated with 200 μL 1.5 M trichloroacetic acid (TCA). Reaction without substrate added was taken as blank. Produced l-ornithine was measured by using HPLC (Zhang et al. 2013). One unit of arginase activity was defined as the amount of enzyme required to generate 1 μmol of l-ornithine per minute under the conditions above. Arginase activity was expressed as units per milliliter sample.
Construction of the BSKA-A2 (ΔamyE, ΔthrC::arg R.pyc) strain
To eliminate any effect of the arginase from B. subtilis 168, the arginase gene of B. subtilis 168 (argI) was knocked out. The ZeoR gene with two lox-sites (7Z6: lox72-zeo-lox66) and homologous arms were amplified from plasmid p7Z6 (primers—P1/P2). “Front” and “back” homologous arms (argIF, argIB) were amplified from the B. subtilis chromosome (primers—P3/P4, P5/P6). The linear DNA fragment, argIF-7Z6-argIB, was constructed with overlapping PCR and used for argI deletion via double-crossover recombination (shown in Fig. S1). Competent strains were selected on LB agar containing zeocin (100 mg/L) and then transformed with the plasmid pTSC. The zeo resistance gene could be deleted via the Cre/lox system (recombination between lox71 and lox66 by Cre recombinase) (Yan et al. 2008). For elimination of pTSC, transformants were patched on LB agar without antibiotic and incubated at 51 °C. The resulting strain colonies lost pTSC yielding the BSKA strain.
The linear DNA fragment, arg R.pyc -7S6 (7S6: lox72-spc-lox66), used for integration into the B. subtilis chromosome was constructed with overlapping PCR (shown in Fig. S1) and then sandwiched by “front” and “back” homologous arms. P43, arginase gene, and SpcR gene with two lox-sites were amplified from B. subtilis chromosome, R. pycnus chromosome, and plasmid p7S6 (primers—P7/P8, P9/P10, P11/P12), respectively. The BSKA-A2 (ΔamyE, ΔthrC::arg R.pyc) strain was completed via step-by-step insertion of two arg R.pyc cassettes into the amyE and thrC genes of the BSKA strain. The overall scheme for the arg R.pyc cassette integration is shown in Fig. 1 (where the insertion of the arg R.pyc cassette into B. subtilis amyE was taken as an example). First, a linear DNA fragment, the arg R.pyc cassette ligated with a resistance selection marker, was flanked with homologous arms. Second, linear DNA fragments were integrated into the corresponding chromosomal region via double-crossover recombination. Finally, the resistance marker cassette was eliminated by the Cre/lox system leaving a 34-bp lox72 site in the chromosome. This step was followed by plasmid pTSC curing and led to construction of a marker-less and plasmid-less R. pycnus arginase-producing strain. The constructed strain was used as the recipient for the next integration using the same method. Step-by-step ectopic insertion resulted in the desired BSKA-A2 (ΔamyE, ΔthrC::arg R.pyc) strain with 2 integrated arg R.pyc cassettes.

Scheme for the arg R.pyc cassette integration into designed chromosomal region (insertion at amyE site was taken as an example)
Random integration of the arg R.pyc cassette
The random integration was based on the competent strain of BSKA. A total of 10 μg BSKA chromosomal DNA were digested by BamHI followed by self-circularization with T4 ligase. The reaction mixture was then digested by EcoRI, and the products were purified by agarose extraction. Purified DNA products (5 μg) were then ligated with the DNA fragments of EcoRI-generated arg R.pyc -7S6 (5 μg), which was amplified from the linear DNA fragment arg R.pyc -7S6 constructed above (primers—P21/P22). The ligation mixture was digested by BamHI, and the digested products were used for transformation of BSKA. Finally, N copies of the arg R.pyc cassettes were integrated into the chromosomal regions.
Segregation stability test
SpcR recombinant strains
More than 100 individual colonies of recombinant strains were tested for SpcR. Cells were plated on an LB agar plate containing 100 mg/L of spectinomycin and cultured for 12 h. Generated SpcR colonies were inoculated on another plate. Each round of inoculation counted for one generation. Strains which generated 100 SpcR colonies among the 100, tested after 20 generations, were considered to be stable. The stable strains were then inoculated into 25-mL LB medium and cultivated at 37 °C. The strains with the highest enzyme activity were selected for the next experiments.
Marker-less recombinant strains
The stability assessment was carried out by transferring single colonies of the marker-less recombinant strains into 25-mL LB medium. After culturing for 24 h at 37 °C, 3% (v/v) cultures were inoculated into another 25-mL fresh LB medium and the enzyme activities were determined. Each round of inoculation counted for one generation and the determinations were performed for a few days.
Further increase of the arg R.pyc cassette copy number
The strain that initially possessed 2 arg R.pyc cassettes, BSKA-A2 (ΔamyE, ΔthrC::arg R.pyc), was used as the first recipient. Chromosomal DNA of a stable SpcR strain was extracted and used for transformation of BSKA-A2. B. subtilis genome with increased copy number was obtained via selection of SpcR transformants. Recombinant strain with SpcR deletion was used as the next recipient, and another stable SpcR strain was considered to be a new donor of chromosomal DNA for copy number increase. In the last round of the arg R.pyc cassette amplification, a marker-less strain with multi-copy ectopic insertions of the target gene was obtained.
The level of arginase expression and the presence of integrated cassettes in the genome were detected by using real-time quantitative PCR (RT-qPCR) and SDS-PAGE analysis.
RT-qPCR analysis of B. subtilis integrated cassettes
PCR was prepared based on Bust n’ Grab method reported previously (Harju et al. 2004). Genomic DNA was used as the template for the copy numbers of arg R.pyc detection with SYBR Green I dye. Primers (PR1/PR2) were designed from the sequence of arg R.pyc and utilized to quantify the arg R.pyc copy number. Reaction conditions were established as recommended by SYBR Premix EX Taq™ II manual. Each reaction mixture contained 5 μL of template DNA (1 ng/μL). Concentrations of genomic DNA were measured by a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). Serial 10-fold dilutions of BSKA-A1 genomic DNA were used to establish the standard curve. To ensure the accuracy, PCR product amplified from BSKA-A1 was sequenced. BSKA was taken as the negative control. In addition, determination of genomic DNA concentration was necessary, as the aim was to obtain a different copy concentration at the same genomic DNA level. All RT-qPCR reactions were performed in triplicate on CFX96 real-time PCR detection system (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Protein analysis
The cells harboring R. pycnus arginase were harvested from the culture medium (1 mL) by centrifugation. Harvested cells were resuspended with 200 μL Tris-HCl buffer (50 mM, pH 7.5). Then, 20 μL lysozyme (1 mg/mL) was added into the cell suspensions and incubated at 37 °C for 1 h and cell debris was removed by centrifugation. The mixture containing 80 μL crude enzyme and 20 μL SDS-PAGE loading buffer (5×) was incubated in boiling water for 10 min. Total 5 μL prepared protein sample was loaded onto each lane of the SDS-PAGE. Purified recombinant R. pycnus arginase overexpressed in E. coli BL21 (DE3) was taken as a control.
Biosynthesis of l-ornithine with the constructed B. subtilis
Biosynthesis of l-ornithine was determined under optimal conditions. A total volume of 100 mL of the conversion mixture consisted of 100 g/L l-arginine, 2 mM Mn2+, and 0.3 g harvested cells. The reaction was performed in a shaking water bath at pH 9.0 and 40 °C for 12 h. The concentrations of the product and substrate were analyzed by HPLC at the given time intervals.
Results
Ectopic insertion of arg R.pyc cassettes into designed sites
Preciously, a thermostable arginase from R. pycnus SK 31.001 has been characterized. The nucleotide sequence of the arginase gene was amplified from the R. pycnus chromosome, and primers were designed based on the available gene sequence (GenBank accession number KP702726). DNA amplicons covering a 906-bp open reading frame were sequenced, and the result coincided with the arginase gene. To improve the production level of the R. pycnus arginase, a strong promoter P43 from B. subtilis was fused with the arginase gene to generate an arg R.pyc expression cassette, which was taken as a monomeric fragment. Furthermore, to eliminate the effect of the B. subtilis arginase, the gene encoding arginase (argI) was knocked out.
Several genes have been reported for target gene integration (Härtl et al. 2001), such as amyE, thrC, lacA, nprB, and rib. In this study, amyE and thrC were chosen as the target integration sites. Ectopic insertions of arg R.pyc were performed to simultaneously inactivate the target integration site genes. The rapid confirmation of a recombinant B. subtilis was made by inoculation of cells on LB agar plates containing 0.002% phenol red (shown in Fig. 2). Ammonia, produced by the arginase and the urease reactions from l-arginine, mediated the pH increase. The alkaline drift in pH produced a red coloration around colonies exposed to phenol red. Furthermore, a higher expression level of arg R.pyc would lead to a redder and larger coloration region.

Inoculation of recombinant strains on LBAP agar: the dependence of arginase production. 1 BSKA, 2 B. subtilis 168, 3 BSKA-A1, 4 BSKA-A2
Random integration into the bacterial genome
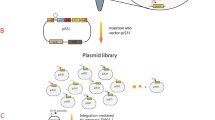
The key step of the random integration strategy was the construction of arg R.pyc -7S6 sandwiched by “front” and “back” homologous arms which were digested from the recipient chromosome. The strategy of random integration was shown in Fig. 3. During the random integration, linear BamHI-generated fragments consisting of “front” and “back” homologous arms and an arg R.pyc cassette were used for double-crossover recombination into the chromosome. These circular fragments which were resistant to BamHI cleavage were integrated into the chromosome via single-crossover recombination.

Scheme for the arg R.pyc cassette integration into random chromosomal region. Random integration includes three stages and there would be two kinds of recombination during the gene conversion
To select stable recombinant strains, a segregation stability experiment was carried out. Successful random integrations led to form more than 100 SpcR clones; however, only 20 transformants performed 100% segregation stability were used for further experiments.
Further increase of the arg R.pyc cassette copy number
The three strains with the highest enzyme activity from the random integrations were selected from the 20 transformants showing 100% segregation stability. These selected strains were designated with labels, such as BSKA-B1 (1 × 1 , 1 × 2 … 1x N :: arg R.pyc -7S6). The number, B1 (B2, B3 for the other strains), indicated the strain number, N and x indicated the copy numbers and unsure loci in the BSKA chromosome. For the arg R.pyc cassette amplification, extracted chromosomal DNA from one SpcR strain was used for transformation of a marker-less strain (Parsons et al. 2014; Würtele et al. 2003). BSKA-A2 (ΔamyE, ΔthrC::arg R.pyc) was designated as the first recipient, which was initially integrated with two copies of the arg R.pyc cassettes. Selection of a SpcR transformant led to a strain with increased copy number. The resistance gene was then eliminated via the Cre/lox system. The newly constructed marker-less strain was used as the next recipient, and the chromosomal DNA from another SpcR strain was applied in the next transformation. With the step-by-step increase of the integration of the arg R.pyc cassette, marker-less strains (BSKA-C1, BSKA-C2, and BSKA-C3) with multi-copy insertions of the arg R.pyc cassettes were obtained.
Presence of the integrated cassettes in the genome and the level of arginase expression at each step were determined by RT-qPCR and SDS-PAGE. As shown in Fig. 4a and Fig. 4b, the copy numbers of insertion cassettes in each strain were determined, and BSKA-A1 strain was used as a standard. Three strains with the highest enzyme activity containing two, two, and three copies of the arg R.pyc cassettes were constructed via random integration. As mentioned previously, these three strains were segregation stable and could be used as the chromosomal DNA donors for transformation of a marker-less strain. Ultimately, with further increase of copy numbers, three plasmid-less and marker-less strains carrying four, six, and nine copies of the arg R.pyc cassettes were obtained. To estimate the level of arginase expression, SDS-PAGE analysis was carried out. As shown in Fig. 4c, the molecular weight of the subunit of R. pycnus arginase was determined to be 33 kDa (lane 3). It was reported that the molecular mass of B. subtilis arginase was 32 kDa (Yu et al. 2013). Though the argI gene encoding B. subtilis arginase was knocked out, there was still a light band around 33 kDa (lane 1). There was no arginase activity assayed from BSKA (see below) which revealed that the background band around 33 kDa was not caused by arginase. Compared with this light band, the bands in lanes 4 to 11 were much more obvious and indicated that the arginase expression level increased with the increase of copy number. The strain with nine copy insertions of the arg R.pyc cassettes showed the highest arginase expression level.

RT-qPCR analysis of the transformant chromosome and SDS-PAGE of recombinant strains with different arg R.pyc copy numbers. a Standard curve for arg R.pyc DNA concentrations. The standard curve was determined with 10-fold serial dilutions of BSKA-A1 genomic DNA (RT-qPCR results were shown in the small figure). b Copy number determination for each strains. BSKA was taken as a negative control. c SDS-PAGE analysis of recombinant strains. Lane M marker, 1 BSKA, 2 B. subtilis 168, 3 purified arginase, 4 BSKA-A1, 5 BSKA-A2, 6 BSKA-B1, 7 BSKA-B2, 8 BSKA-B3, 9 BSKA-C1, 10 BSKA-C2, 11 BSKA-C3
Performance of the multi-copy strains
All multi-copy strains were constructed as described above. A set of strains, including BSKA (0 copy), BSKA-A1 (1 copy), BSKA-A2 (2 copies), BSKA-B3 (3 copies), BSKA-C1 (4 copies), BSKA-C2 (6 copies), and BSKA-C3 (9 copies), were used to evaluate the effect of the arg R.pyc cassettes on arginase expression in shake flask culture. As shown in Fig. 5a, the growth curves of B. subtilis 168 and BSKA-C3 were assayed. Both B. subtilis 168 and BSKA-C3 reached the highest OD600 value around 14 h. Then, the growth of recombinant strains was measured by determining OD600 values after culture for 14 h at 37 °C. The strains with 1–3 copies showed the same growth trend and slightly lower than the wild-type strain (B. subtilis 168). However, with copies higher than three, there was a gradual decrease in growth with the increase of arg R.pyc cassettes. The total enzyme activity reached a peak value of 14.5 U/mL with strain BSKA-C3, which harbored nine copies of the arg R.pyc cassettes (shown in Fig. 5b). Strain BSKA-C3 showed approximately 3-fold and 18-fold higher activity than the single copy strain BSKA-A1 and B. subtilis 168, respectively. Segregation stability of the marker-less strains was determined. As shown in Fig. 5c, the enzyme stability of the recombinant strains was tested for ten generations and the results suggested that stable multi-copy strains were obtained with the insertion of the arg R.pyc cassettes into designed and random regions in B. subtilis chromosome. Whole-cell conversion was performed under optimal conditions with the nine copy recombinant strain BSKA-C3. As shown in Fig. 5d, the highest conversion rate of l-arginine was reached at 8 h. At this point, 73.8 g/L l-ornithine was obtained with a molar yield of 97.2%. However, there was a slight decrease when reaction time was more than 8 h. This decrease might be a result of consumption of l-ornithine by the cells. Within 8 h, most of the substrate was hydrolyzed with only 2.6 g/L residue.

Performance of the constructed multi-copy integration strains. a Growth curve of BS (B. subtilis 168) and BSKA-C3. b Total enzyme activity (black bar) and growth determination (red bar) of each recombinant strain. Each strain was estimated after cultured at 37 °C for 14 h. c Segregation stability of the constructed marker-less strains (9 copies (▲), 6 copies (●), 4 copies (■)). The stability of each strain was estimated by enzyme activity of every generation. d Bioconversion for l-ornithine production (l-arginine (●), l-ornithine (■)). The conversion was performed in a shaking water bath at pH 9.5 and 40 °C for 12 h
Discussion
A number of biotransformation process running on a commercial scale revealed that biocatalysis had been a standard technology in the food and chemicals industry. One hundred and thirty-four industrial biotransformation list by Straathof et al. (2002) showed that more than 50 industrial biotransformations were used for whole-cell catalysis. B. subtilis is an attractive host for the production and secretion of heterologous proteins. We had tried to utilize different Sec-type and Tat-type signal peptides to lead the secretion of R. pycnus arginase; however, it has no success yet. It is not clear why the cytoplasmic protein could not be transported with different signal peptides. It was hypothesized that the N terminal regions of this cytoplasmic protein containing order-promoting amino acids might easily form the structure which hindered its secretion (Wang et al. 2013). Therefore, in the present studies, intracellular expression of arginase was chosen.
Arginase from R. pycnus SK 31.001 has been characterized and showed that this thermostable enzyme could be applied in l-ornithine biosynthesis (Huang et al. 2016). Introduction of a recombinant plasmid might be a traditional method for expression of arginase. However, structural and segregational instability of plasmids in B. subtilis was well-documented (Shoham and Demain 1991; Fleming and Patching 1994). Compared with the expression with recombinant plasmid, chromosome integration might be a good choice to overcome these disadvantages. To ensure that expression level of the R. pycnus arginase, a strong promoter P43 from B. subtilis was fused with the arginase gene to generate an arg R.pyc expression cassette. A strategy of multi-copy insertions was used to integrate the arg R.pyc cassettes into different designed and random sites in the host chromosome. Southern hybridization might be the most widely used method for copy number determination. However, this method was time-consuming, semi-quantitative, and laborious (Pushnova et al. 2000). Therefore, an RT-qPCR method was established, and the genomic DNA (Zhu et al. 2009) was used directly as the template.
Multi-copy insertions were separated into three parts including designed site insertion, random insertion, and further increase of the argR.pyc cassette copy number. There were two kinds of homologous recombination during the random insertions. It was supposed that the number of circular-mediated integrations might be more than linear fragment insertions (Schumann 2007), while double-crossover recombination (linear insertions) would be rather stable than single-crossover recombination (circular-mediated integrations) for the recombination-dependent elimination of the cassette in the B. subtilis expression system (Schumann 2007). These might explain why only 20 transformants performed 100% segregation stability among the generated more than 200 clones. Random integration of target gene could be easily obtained; however, the copy numbers would be unexpectedly high (most of the integrations were approximately 1–2 copies) (Yomantas et al. 2011). The highest three copies of the arg R.pyc cassettes were constructed via random integration. Combination with the designed site insertion and the random insertion led to a further increase of the argR.pyc cassette copy number. According to the reported studies, there were likely 50–100 copies in a plasmid carrier strain (Wu and Wong 1999). Moreover, 52 copies could be also obtained with repeated cassette insertions into the target chromosome region (Zhu et al. 2009). However, plasmid and repeated cassette insertion might be unstable in B. subtilis host (Fleming and Patching 1994). Recombination between repeated cassettes would lead to internal chromosomal deletions through cyclization (He et al. 2016). In some cases, instability might reduce the profitability of fermentations or even make the strain non-viable. Segregation stability was a major factor for the application of recombinant strains that must be taken into consideration (Yomantas et al. 2011). BSKA-C3 with nine copy insertions of arg R.pyc cassettes was constructed. Compared with these reported strains, the copy number of chromosomal integration strain in this study was low. However, segregation stability determination of the marker-less strain showed that BSKA-C3 was relatively stable. Moreover, this constructed plasmid-less strain displayed essentially the same arginase activity (14.5 U/mL) level as the recombinant B. subtilis strain carrying expression plasmid with inserted Bacillus amyloliquefaciens argI gene (which showed nearly 26-fold higher enzyme activity than the wild-type B. subtilis 168 strain) (Wang et al. 2015). The fermentation of recombinant strain was based on the LB medium; then, higher activity could be obtained with the optimization of medium composition. High concentration of residual substrate might make product separation difficult. Whole-cell conversion performed with BSKA-C3 led to a molar yield of 97.2%, which was higher than the reported yield (Song et al. 2014; Wang et al. 2015). In addition, it seemed useful to estimate the random integration sites for the construction of stable recombinant strains. This determination could be carried out by inverse PCR with a further study (Tchetina and Newman 1995).
l-Ornithine could be used as a food additive, so food safety must be taken into consideration by food industry. Previous studies were focused on the bioconversion of l-ornithine production using whole-cell recombinant E. coli or the purified arginase (Song et al. 2014; Yu et al. 2013). In contrast to the E. coli expression system, B. subtilis is a GRAS organism that has been used in the food industry for a long time. In this study, a multi-copy, non-antibiotic resistant B. subtilis strain was constructed. This segregation stable recombinant strain showed excellent catalytic activity and would be a good candidate for l-ornithine production in the food industry.
References
Anagnostopoulos C, Spizizen J (1961) Requirements for transformation in Bacillus subtilis. J Bacteriol 81(5):741–746
Bron S, Holsappel S, Venema G, Peeters BPH (1991) Plasmid deletion formation between short direct repeats in Bacillus subtilis is stimulated by single-stranded rolling-circle replication intermediates. Mol gen Genet 226(1–2):88–96. doi:10.1007/BF00273591
Fleming GT, Patching DJW (1994) Plasmid instability in an industrial strain of Bacillus subtilis grown in chemostat culture. J Ind Microbiol 13(2):106–111. doi:10.1007/BF01584107
Harju S, Fedosyuk H, Peterson KR (2004) Rapid isolation of yeast genomic DNA: bust n’ grab. BMC Biotechnol 4(1):1–6. doi:10.1186/1472-6750-4-8
Härtl B, Wehrl W, Wiegert T, Homuth G, Schumann W (2001) Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol 183(8):2696–2699. doi:10.1128/JB.183.8.2696-2699.2001
He W, Mu W, Jiang B, Yan X, Zhang T (2016) Food-grade expression of d-Psicose 3-epimerase with tandem repeat genes in Bacillus subtilis. J Agr Food Chem 64(28):5701–5707. doi:10.1021/acs.jafc.6b02209
Huang K, Zhang T, Jiang B, Mu W, Miao M (2016) Characterization of a thermostable arginase from Rummeliibacillus pycnus SK31.001. J Mol Catal B-Enz. doi:10.1016/j.molcatb.2016.11.020
Jalan R, Wright G, Davies NA, Hodges SJ (2007) L-Ornithine phenylacetate (OP): a novel treatment for hyperammonemia and hepatic encephalopathy. Med Hypotheses 69(5):1064–1069. doi:10.1016/j.mehy.2006.12.061
Jeong KJ, Park IY, Kim MS, Kim SC (1998) High-level expression of an endoxylanase gene from Bacillus sp. in Bacillus subtilis DB104 for the production of xylobiose from xylan. Appl Microbiol Biotechnol 50(1):113. doi:10.1007/s002530051264
Kastner S, Perreten V, Bleuler H, Hugenschmidt G, Lacroix C, Meile L (2006) Antibiotic susceptibility patterns and resistance genes of starter cultures and probiotic bacteria used in food. Syst Appl Microbiol 29(2):145–155. doi:10.1016/j.syapm.2005.07.009
Lee SH, Kim HJ, Shin YA, Kim KH, Lee SJ (2016) Single crossover-mediated markerless genome engineering in Clostridium acetobutylicum. J Microbiol Biotechnol 26(4):725–729. doi:10.4014/jmb.1512.12012
Meissner L, Kauffmann K, Wengeler T, Mitsunaga H, Fukusaki E, Büchs J (2015) Influence of nitrogen source and pH value on undesired poly(γ-glutamic acid) formation of a protease producing Bacillus licheniformis strain. J Ind Microbiol Biot 42(9):1203–1215. doi:10.1007/s10295-015-1640-7
Parsons JB, Frank MW, Eleveld MJ, Schalkwijk J, Broussard TC, Jonge MID, Rock CO (2014) A thioesterase bypasses the requirement for exogenous fatty acids in the plsX deletion of Streptococcus pneumoniae. Mol Microbiol 96(1):28–41. doi:10.1111/mmi.12916
Povelainen M, Miasnikov AN (2007) Production of xylitol by metabolically engineered strains of Bacillus subtilis. J Bacteriol 128(1):24–31. doi:10.1016/j.jbiotec.2006.09.008
Pushnova EA, Geier M, Yu SZ (2000) An easy and accurate agarose gel assay for quantitation of bacterial plasmid copy numbers. Anal Biochem 284(1):70–76. doi:10.1006/abio.2000.4668
Sauer C, Syvertsson S, Bohorquez LC, Cruz R, Harwood CR, van Rij T, Hamoen LW (2016) Effect of genome position on heterologous gene expression in Bacillus subtilis: an unbiased analysis. ACS Synth Biol 5(9):942–947. doi:10.1021/acssynbio.6b00065
Schumann W (2007) Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol 62(62):137–189. doi:10.1016/S0065-2164(07)62006-1
Shoham Y, Demain AL (1991) Kinetics of loss of a recombinant plasmid in Bacillus subtilis. Biotechnol Bioeng 37(10):927–935. doi:10.1002/bit.260371006
Sietske ADB, Diderichsen B (1991) On the safety of Bacillus subtilis and B. amyloliquefaciens: a review. Appl Microbiol Biotechnol 36(1):1–4. doi:10.1007/BF00164689
Song W, Niu P, Chen X, Liu L (2014) Enzymatic production of L-ornithine from L-arginine with recombinant thermophilic arginase. J Mol Catal B-Enz 110:1–7. doi:10.1016/j.molcatb.2014.09.005
Straathof AJ, Panke S, Schmid A (2002) The production of fine chemicals by biotransformations. Curr Opin Biotech 13(6):548–556. doi:10.1016/S0958-1669(02)00360-9
Sugino T, Shirai T, Kajimoto Y, Kajimoto O (2008) L-Ornithine supplementation attenuates physical fatigue in healthy volunteers by modulating lipid and amino acid metabolism. Nutr res 28(11):738–743. doi:10.1016/j.nutres.2008.08.008
Tchetina E, Newman EB (1995) Identification of Lrp-regulated genes by inverse PCR and sequencing: regulation of two mal operons of Escherichia coli by leucine-responsive regulatory protein. J Bacteriol 177(10):2679–2683
Wang G, Chen H, Zhang H, Song Y, Chen W (2013) The secretion of an intrinsically disordered protein with different secretion signals in Bacillus subtilis. Curr Microbiol 66(6):566–572. doi:10.1007/s00284-013-0315-8
Wang M, Xu M, Rao Z, Yang T, Zhang X (2015) Construction of a highly efficient Bacillus subtilis 168 whole-cell biocatalyst and its application in the production of L-ornithine. J Ind Microbiol Biotechnol 42(11):1427–1437. doi:10.1007/s10295-015-1672-z
Wu SC, Wong SL (1999) Development of improved pUB110-based vectors for expression and secretion studies in Bacillus subtilis. J Biotechnol 72(1–2):185–195. doi:10.1016/S0168-1656(99)00101-7
Würtele H, Little KCE, Chartrand P (2003) Illegitimate DNA integration in mammalian cells. Gene Ther 10(21):1791–1799. doi:10.1038/sj.gt.3302074
Yan X, Yu HJ, Hong Q, Li SP (2008) Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl Environ Microb 74(17):5556–5562. doi:10.1128/AEM.01156-08
Yomantas YA, Abalakina EG, Golubeva LI, Gorbacheva LY, Mashko SV (2011) Overproduction of Bacillus amyloliquefaciens extracellular glutamyl-endopeptidase as a result of ectopic multi-copy insertion of an efficiently-expressed mpr gene into the Bacillus subtilis chromosome. Microb Cell Factories 10(1):1–10. doi:10.1186/1475-2859-10-64
Yu JJ, Park KB, Kim SG, Oh SH (2013) Expression, purification, and biochemical properties of arginase from Bacillus subtilis 168. J Microbiol 51(2):222–228. doi:10.1007/s12275-013-2669-9
Zhang T, Guo Y, Zhang H, Mu W, Miao M, Jiang B (2013) Arginase from Bacillus thuringiensis SK 20.001: purification, characteristics, and implications for L-ornithine biosynthesis. Process Biochem 48(4):663–668. doi:10.1016/j.procbio.2013.02.023
Zhu T, Guo M, Tang Z, Zhang M, Zhuang Y, Chu J, Zhang S (2009) Efficient generation of multi-copy strains for optimizing secretory expression of porcine insulin precursor in yeast Pichia pastoris. J Appl Microbiol 107(3):954–963. doi:10.1111/j.1365-2672.2009.04279.x
Acknowledgments
This work was financially supported by the 863 Project of China (No. 2013AA102102) and the Fundamental Research Funds for the Central Universities (SKLF-ZZA-201509).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 541 kb).
Rights and permissions
About this article

Cite this article
Huang, K., Zhang, T., Jiang, B. et al. Overproduction of Rummeliibacillus pycnus arginase with multi-copy insertion of the arg R.pyc cassette into the Bacillus subtilis chromosome. Appl Microbiol Biotechnol 101, 6039–6048 (2017). https://doi.org/10.1007/s00253-017-8355-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8355-9