
Abstract
Background
Plasmid-less, engineered Bacillus strains have several advantages over plasmid-carrier variants. Specifically, their stability and potential ecological safety make them of use in industrial applications. As a rule, however, it is necessary to incorporate many copies of a key gene into a chromosome to achieve strain performance that is comparable to that of cells carrying multiple copies of a recombinant plasmid.
Results
A plasmid-less B. subtilis JE852-based strain secreting glutamyl-specific protease (GSP-the protein product of the mpr gene from B. amyloliquefaciens) was constructed that exhibits decreased levels of other extracellular proteases. Ten copies of an mprB.amy cassette in which the GSP gene was placed between the promoter of the B. amyloliquefaciens rplU-rpmA genes and the Rho-independent transcription terminator were ectopically inserted into designated (3 copies) and random (7 copies) points in the recipient chromosome. The resulting strain produced approximately 0.5 g/L of secreted GSP after bacterial cultivation in flasks with starch-containing media, and its performance was comparable to an analogous strain in which the mprB.amy cassette was carried on a multi-copy plasmid.
Conclusion
A novel strategy for ectopically integrating a cassette into multiple random locations in the B. subtilis chromosome was developed. This new method is based on the construction of DNA fragments in which the desired gene, marked by antibiotic resistance, is sandwiched between "front" and "back" portions of random chromosomal DNA restriction fragments. These fragments were subsequently inserted into the targeted sites of the chromosome using double-cross recombination. The construction of a marker-free strain was achieved by gene conversion between the integrated marked gene and a marker-less variant carried by plasmid DNA, which was later removed from the cells.
Similar content being viewed by others

Background
Gram-positive bacteria are widely used for biotechnology applications, including vaccine delivery [1–3] and in situ production of anti-infective protectants [4] and microbicides [5]. These microorganisms serve as large-scale producers of nucleotides, vitamins, ribose, poly-γ-glutamic acids [6], absorbents [7], and insecticides [8]. Bacillus species are considered prospective cell-based factories for pharmaceutical proteins [9]. Currently, about 60% of commercially-available industrial enzymes are produced by selected and/or genetically-engineered Bacillus strains, most of which produce homologous proteins that are naturally secreted into the growth medium [6, 9–15].
Bacillus subtilis produces numerous extracellular proteolytic enzymes. The alkaline serine protease subtilisin and the neutral protease (gene products of aprE and nprE, respectively) often constitute more than 90% of the total extracellular protease activity [9, 16]. The contribution of glutamic acid-specific protease (GSP) does not normally exceed 2% [17]. B. subtilis GSP, encoded by the mpr gene, is synthesized as an inactive pre-pro-peptide. This precursor is subsequently processed by the Sip and Bpr proteases, and mature extracellular GSP have a length of 220 amino acids [17]. Though they were initially a subject of basic science investigation [18–20], some GSPs (from B. licheniformis in particular [21]) are now being utilized in commercial applications such as food production [22, 23].
A traditional approach to the genetic engineering of Bacillus strains involves the introduction of multi-copy-number recombinant plasmids [10]. However, the construction of plasmid-less strains has recently become more relevant and practical. The preference for plasmid-less Bacillus strains is due to the genetic instability of many recombinant plasmids [24, 25] and to official restrictions that concern the use of plasmid-carrying strains in large-scale industry in the First World [26]. Most often, the construction of plasmid-less Bacillus strains is performed by homologous recombination-mediated integration of the desired genes into the bacterial chromosome [10]. In some instances, specialized site-specific recombination [27] and transposition [28, 29] are used for the same integrative purposes.
Recombination-mediated DNA incorporation can be implemented through either Campbell-type single-crossover integration of plasmids based on specialized vectors carrying DNA sequences homologous to the Bacillus chromosome or through the use of ectopic insertion, i.e. double-cross recombination between the target in the chromosome and the homologous flanking sequences sandwiching the fragment of interest [10, 30, 31]. Both methods can be used for single-copy and multi-copy integrations [32–34]. Single-copy, plasmid-mediated integrants with inserted sequences bracketed by duplicated homologous regions are not stable under non-selective conditions due to the possible recombination-mediated elimination of the inserted plasmid [35]. Ectopic insertion(s) of a desired gene usually leads to significantly more stable recombinant strains. However, only a narrow set of well-characterized loci within the B. subtilis chromosome is normally used as targets for such insertions [10, 36, 37].
In this study, a recombinant, plasmid-less B. subtilis strain was developed that can efficiently produce and secrete GSP from B. amyloliquefaciens. Initially, three copies of the mpr gene were ectopically inserted into known B. subtilis genes encoding extracellular proteases. A novel, random integration methodology was then implemented to construct a stable strain with 10 mpr copies within the chromosome. Performance of the new strain was comparable to the strain carrying the mpr gene on a multi-copy plasmid, as exhibited by accumulation of the recombinant GSP in the media.
Results
Cloning and expression of B. amyloliquefaciens mpr on a B. subtilis plasmid
The nucleotide sequence of the mpr gene from B. amyloliquefaciens A-50 was not known. Primers for the amplification of mpr by PCR, mpr-F/R (the structures of the primers used in this study were presented in Additional file 1, Table S1), were therefore designed based on the available B. amyloliquefaciens ZB42 genome sequence (GenBank/EMBL accession number NC_009725) [38]. DNA amplicons of 972 base pairs (bp) in length were obtained and sequenced (GenBank accession number GU992366). The corresponding DNA sequence closely coincided with the mpr- containing sequence from B. amyloliquefaciens FZB42 (91% of identity) and covered the 909-bp open reading frame. An extended AG-rich block, including a B. subtilis Shine-Dalgarno sequence, AAGGAGG [39], was found upstream of the ATG codon of this ORF. The protein-coding ORF possessed 68% identity to well-characterized pre-pro-GSP from B. subtilis[17, 18, 20].
The mpr-carrier amplicon, flanked by artificial Bgl II sites (P1-bmp5 and P2-bmp2 were used as the primers), was cloned into the Bgl II site of the pHEA323 plasmid [40]. This placed it under the transcriptional control of the promoter (P rp ) of the rplU-rpmA genes from B. amyloliquefaciens A-50, which encode the L21 and L27 ribosomal proteins. In the resulting pHE52mpr recombinant plasmid, the cloned mpr gene became the central part of an artificial operon that was terminated by the Rho-independent transcription terminator (Ter) from the pheA gene of B. amyloliquefaciens A-50 (Figure 1). As was shown previously [40] and confirmed in the present study, the presence of Ter for the termination of efficient P rp -mediated transcription is conducive to the stable inheritance of pHEA323 and its derivatives (i.e., mprB.amy cassettes (P rp ➔mpr-Ter) in the pHE52mpr plasmid and/or integrated into the bacterial chromosome).

Structure of the pHE52 mpr plasmid carrying the mprB.amy cassette. P rp - promoter of the rplU-rpmA genes from B. amyloliquefaciens A-50; Ter - Rho-independent transcription terminator of the pheA gene. The Bgl II-site-ended (boldface) followed by 5'-portion of B. amyloliquefaciens A-50 mpr gene is shown. In this part, SD-sequence and ATG-codon are marked as boldface and capital letters, respectively. Two putative cre elements, which are homologous to the known [45–47] consensus sequence, are also indicated.
Mpr gene expression studies were performed with the B. subtilis strain JE852 serving as a recipient. This strain was a double mutant for genes encoding two major extracellular proteases (nprE512, aprE851), which simplified the assessment of recombinant GSP activity.
Initially, a plasmid-carrying, recombinant GSP-producing strain was constructed via the transformation of B. subtilis JE852 with pHE52mpr. The level of GSP accumulation was analyzed by the semi-quantitative skim milk method on media containing different carbon sources and by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of extracellular proteins. It was shown in these experiments that the expression of the mprB.amy cassette was under carbon catabolite control (CCC) in B. subtilis. Indeed, when glucose or maltose were added to the media, B. subtilis JE852/pHE52mpr grew well but did not form clear, hydrolytic zones around colonies on milk agar. On the other hand, during growth on medium containing soluble starch as the sole carbon source, abundant amounts of GSP accumulated and were easily distinguished from the other extracellular proteins by SDS-PAGE. The main mechanism of CCC in Bacillus has been well studied [41–44]. CCC is implemented through the binding of the CcpA-mediated regulatory protein complex to special DNA sites known as catabolite responsive elements (cre). This binding causes carbon catabolite repression (CCR) or activation (CCA), depending on the position of the cre. For example, when the regulatory complex binds to cre that is located downstream of the transcription initiation point, it evokes a transcription roadblock that leads to CCR of the corresponding genes [44]. Two putative catabolite responsive elements that were homologous to consensus cre sequences [44–46] were found in the N-terminal coding part of the B. amyloliquefaciens A-50 mpr gene by sequence analysis (Figure 1). It is possible that the CCR of mpr gene expression that we observed was caused by termination of transcription at these cre sites when they were bound to the CcpA-mediated regulatory complex. Moreover, the data suggest that a complicated regulatory network governs Bacillus extracellular proteolytic activity with CCR and that there may be changes in the control of enzyme biosynthesis, secretion, and/or maturation at different stages of bacterial growth [41, 42, 47].
Defining the mechanism of CCR modulation of GSP extracellular accumulation was outside the scope of the present paper. We showed that GSP production was significantly increased during fermentation of the B. subtilis JE852/pHE52mpr strain on TYS6C media, in which starch was the main carbon source. In this media, an enhanced biomass (growing up to an OD600 of around 40-50) and high level of extracellular GSP accumulation (up to approximately 0.5 g/L, as semi-quantitatively determined by SDS-PAGE, see Materials and methods) were detected. These results were obtained for the strain carrying multi-copy-number recombinant plasmids, suggesting that the integration of multiple copies of the mprB.amy -cassettes into the bacterial chromosome is indispensable for achieving comparably high GSP production levels in a plasmid-less Bacillus strain.
Ectopic insertion of mprB.amy cassettes into genes encoding known extracellular proteases
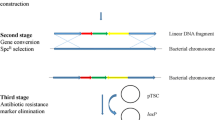
Ectopic insertion of several mprB.amy cassettes was performed to simultaneously inactivate known extracellular protease genes of B. subtilis: aprE, epr and nprB. The overall scheme of mprB.amy cassette insertion had three stages (see Figure 2 where the mprB.amy cassette insertion into the aprE851 allele of B. subtilis JE852 strain is shown as an example). First, a linear DNA fragment consisting of an antibiotic resistance (AntR) marker flanked with homologous arms was integrated into the corresponding chromosomal region via double-crossover recombination. Then, the AntR marker was exchanged for the mprB.amy cassette by gene conversion (for a review, see [48, 49]) between the chromosome and the autonomously replicating mprB.amy -carrying plasmid, and this was followed by plasmid curing and construction of the plasmid-less, targeted integrant.

Scheme of the targeted mprB.amy -cassette integration into the JE852 chromosome ( aprE851 is shown as an example of the target gene).
The aprE851 gene in B. subtilis JE852 was chosen as the first target gene for mprB.amy cassette insertion, primarily to prevent reversion of the mutant allele to the wild-type phenotype during the proposed long-term construction of a GSP-producing, plasmid-less strain. The CmR gene from pC194 [50] was used as the AntR marker for selective integration at the first stage. The linear fragment, used for aprE851 gene disruption, was constructed in vitro by overlapping PCR technique (see Materials and methods and Additional file 2, Figure S1 for details). The targeted integration of the CmR marker was followed by gene conversion using the EmR-marked recombinant plasmid pCBT(yhfO-mprB.amy -yhfN) and subsequent selection of the obtained EmR CmS clones, which were generated at a frequency of around 2%. Finally, the plasmid-less (EmS) variants were selected after bacterial cultivation in liquid erythromycin-free medium. All integration stages were assessed by PCR, and the chromosome structure of the B. subtilis JE852aprE851::mprB.amy strain was analyzed by PCR and using Southern hybridization.
The same method, with modifications based on the nucleotide sequences of the target genes, was used for step-by-step ectopic insertion of the mprB.amy cassette into the epr and nprB genes, encoding two minor extracellular proteases of B. subtilis (see Materials and methods and Additional file 1, Table S1 for details). This process resulted in the desired B. subtilis strain, a JE852-based plasmid-less, marker-less strain, JE852(aprE851, epr, nprB)::mprB.amy , with three integrated mprB.amy cassettes.
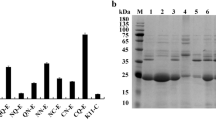
The dependence of GSP accumulation on the integrated cassette copy-number (N) was evaluated according to the semi-quantitative plate test based on casein hydrolysis (Figure 3) and using SDS-PAGE analysis of extracellular bacterial proteins (Figure 4). The results showed that GSP production was significantly lower than that of a recombinant strain that had multiple plasmid copies, even for the plasmid-less strain, which had three cassette insertions (N = 3). This finding suggested that the process of cassette amplification needed to be continued. However, simplifying the procedure to obtain many single-copy integrants and then combining the variants possessing segregation stability became an attractive option.

Cells plated on skim-milk test plates: The dependence of extracellular protease activity on the copy number of the integrated mprB.amy cassettes. 1.-clone of B. subtilis JE852; 2. and 3.-1 and 3 copies of mprB.amy-cassette integrated in the chromosome of JE852; 4.-clone of the plasmid-carrier strain B. subtilis JE852/pHE52mpr.

Southern hybridization of the chromosomal DNAs and SDS-PAGE of extracellular proteins secreted by the strains with different copy-number (N) of the integrated mprB.amy-cassettes. A – Hybridization with DNAs isolated from cells carrying (indicated as 1.; 2.; 3; etc. (N = 1: lane 1 (1.); N = 2: lane 2 (1.+2.); N = 4: lane 3 (1.+2.+3.+4.); and N = 7: lane 4 (1.+2.+3.+4.+5.+6.+7.) and lane 6 (1.+2.+3.+4.+5.+6.+8.). B SDS-PAGE of extracellular proteins secreted by the strains with: N = 0 (lane 2), N = 1 (lane 3), N = 2 (lane 4), N = 3 (lane 5), N = 9 (lane 8) and N = 10 (lane 9) mprB.amy cassettes in the chromosome or carrying the multi-copy-number recombinant plasmid pHE52mpr (lanes 6 and 10). Lanes 1 and 7-reference proteins with molecular mass given in kDa. The mature 220 amino acid B. amiloliquefaciens GSP has a molecular mass of about 23.7 kDa. The indicated extracellular GSP presented slightly lower electrophoretic mobility corresponding to 27-28 kDa; this was previously documented for the mature GSP from B. subtilis [17].
Integration of the mprB.amy cassette into random sites in the bacterial genome
A key aspect of the novel strategy presented here is the construction of DNA fragments in which the AntR-marked cassette (mprB.amy -AntR) is sandwiched between the "front" and "back" portions of randomly digested fragments of the recipient chromosome. The proposed scheme is presented in Figure 5. Initially, pHE52(mpr-CmR) was constructed (see Materials and methods). This plasmid carried the mprB.amy -CmR cassette that was bracketed by Pst I-sites and did not contain internal Bam HI-sites. The Pst I-generated mprB.amy -CmR cassette is marked as (a) in Figure 5. The Bam HI-generated DNA fragments of the B. subtilis JE852 chromosome ((b) fragments in Figure 5) were self-circularized by T4 ligase at a low DNA concentration and subsequently cleaved by Pst I. (b)-fragments in Figure 5 were a mixture of Pst I-site(s)-carrying (b1) and Pst I-site-free (b2) fragments. The (b1)-fragment with two internal Pst I-sites was shown in the Figure 5 for simplicity. The self-circularized (b2) fragments could not be linearized by Pst I and so would not be later linked with the (a)-fragment. In contrast, the self-circularized (b1)-fragments hydrolyzed by Pst I generated a mixture of Bam HI-site-carrier (c1) and Bam HI-site-free (c2) linear DNA fragments. The ligation of (c1)-fragments with (a)-fragment followed by Bam HI treatment caused formation of linear (Lin) fragments consisting of the cassette of interest sandwiched by "front" and "back" homologous arms. These (Lin)-fragments could participate in subsequent double-cross recombination-mediated integration into the bacterial chromosome. (c2)-fragments could be ligated with the (a)-fragment as well. These circular, recombinant DNAs, (Cir)-fragments, were resistant to Bam HI-mediated cleavage and could be integrated into the chromosome only via a single-cross Campbell-type recombination. It could be supposed that the number of (Cir)-mediated integrants would be more than (Lin)-mediated ectopic insertions [10]. At the same time, the (Cir)-mediated integrant with the cassette sandwiched between directly repeated (c2)-fragments (see Figure 5) could be rather unstable due to the possibility of recombination-dependent elimination of the cassette.

The general scheme of the mprB.amy -CmR-cassette integration into random points of the B. subtilis chromosome.
The success of the strategy led to the formation of about 250 CmR clones after transformation of the B. subtilis JE852 strain. These colonies were tested for their ability to grow on minimal media with glucose as the sole carbon source. Prototrophic CmR strains (190 colonies) were then tested for their segregation stability (see Materials and methods for details). Fifteen transformants that demonstrated 100% segregation stability after 60 generations were used in the experiments that followed. According to data from the literature [10], it could be supposed that the stable integrants were obtained due to the intrinsic ectopic insertions, whereas transformants that manifested decreased segregation stability were the result of Campbell-type integration.
According to experimental evaluation (including growth on skim milk plates and SDS-PAGE analysis of extracellular proteins), all 15 stable integrants produced and secreted GSP at slightly variable levels, and the levels corresponded to the presence of one mprB.amy cassette in the chromosome of B. subtilis JE852. Testing by Southern hybridization confirmed that these strains carried only one mprB.amy -CmR cassette integrated into different chromosomal loci (see Figure 4 where results for the corresponding marker-free mprB.amy -cassettes were presented). The strains from this set were designated, for example, B. subtilis JE852-(69xyz::mprB.amy -CmR). Here the number, 69 (84, 85, 114, etc. for the other strains), indicates the strain number in the laboratory collection, while the uniform three-letter appellation for all strains, xyz, indicates that the location of the cassette integration was not determined.
Step-by-step increase of the mprB.amy cassette chromosomal copy number
The set of strains with integrated mprB.amy -CmR cassettes was used to increase the occurrence of the mprB.amy gene in the genome of a strain that initially possessed three cassettes, B. subtilis JE852(aprE851, epr, nprB)::mprB.amy . For cassette amplification, extracted chromosomal DNA from one CmR strain was used for transformation of a marker-less strain carrying N copies of the mprB.amy -cassette (initially N = 3 in these experiments). Selection of CmR transformants led to the creation of a B. subtilis genome that contained N+1 copies of the gene encoding GSP. At the last step of this round of cassette amplification, the strain was rendered marker-less by gene conversion with pHE52mpr followed by plasmid curing. Then, the next CmR-marked cassette was inserted into the chromosome of the newly obtained strain, which carried N+1 cassettes.
The level of GSP secretion and the presence of all previously integrated cassettes in the bacterial genome were assessed at each stage of the cassette amplification process using Southern hybridization and SDS-PAGE analysis of extracellular proteins (Figure 4). As a rule, each subsequent generation displayed a slightly increased level of GSP accumulation in comparison to the previous generation, maintained the earlier integrated cassettes at their original positions in the bacterial genome and presented one novel hybridized DNA fragment that could be detected in the marker-less derivative of the corresponding donor strain.
Ultimately, a plasmid-less and marker-less strain carrying 10 copies of the mprB.amy cassette was obtained. This strain efficiently secreted GSP at the same level as the control, B. subtilis JE852/pHE52mpr.
Conclusion
Efficient production and secretion of B. amyloliquefaciens A-50 GSP by a recombinant plasmid-less B. subtilis strain was obtained. The mutant B. subtilis JE852 (nprE, aprE), which possessed significantly decreased levels of major extracellular proteases, was utilized as the initial recipient strain. The mprB.amy cassette, in which transcription of the mpr gene was controlled through a promoter that drives genes for ribosomal proteins in combination with a Rho-independent terminator, was expressed and stably maintained. Finally, the mprB.amy cassette was amplified by multiple ectopic insertions of the construct into the B. subtilis chromosome within known genes initially and then in random loci according to the methodology described above. The methods used for these insertions differed slightly but used the following general steps: (i) an AntR-marked linear DNA fragment was sandwiched between two arms that were homologous to a target in the bacterial genome, (ii) this fragment was incorporated using double homologous recombination, (iii) the marker was removed by gene conversion between the chromosome and an introduced plasmid and (iv) the incoming plasmid was eliminated from the cell.
It should be mentioned that the efficiency of iterative gene conversions using the same plasmid, pHE52mpr, decreased slightly with an increase in the copy number of the integrated cassettes. This efficiency was about (3 to 4)% when N = 3 or 4 but did not exceed 1% for strains with N = 9 or 10. Excisable markers that can be efficiently removed by different site-specific recombination events [27, 51] might be preferable for the amplification procedure.
The constructed plasmid-less strain, which has 10 chromosomal mprB.amy cassettes, displayed essentially the same GSP production level as the recombinant plasmid-carrying strain. According to data in the literature, there are likely 20-30 copies of the pSM19035 replicon-based plasmid in the recombinant plasmid-carrier strain [10, 52, 53]. This apparent incongruity has several explanations. First, the copy number of the recombinant mprB.amy -carrier plasmid could be lower than that of the vector, in particular, because of interference between plasmid replication and efficient intra-plasmid transcription. Second, expression levels of the same gene located in the chromosome vs. located on a plasmid could differ due to changes in the DNA curvature; dependence on restrained superhelical density is typical of protein-bound DNA molecules [54]. Third, P rp -mediated transcription of even ten copies of the mpr gene may be inherently efficient, such that the saturated translation/secretion machinery becomes the true bottleneck for extracellular GSP accumulation.
Segregation stability is a major factor that must be considered in the potential practical application of plasmid-less recombinant strains. As mentioned previously, only 10% of the clones that had a single-copy of the mprB.amy -carrying cassette integrated at random points within the bacterial chromosome possessed strong segregation stability. Amplification of the same cassettes in one genome could certainly decrease the strain's stability due to the potential for homologous intrachromosomal recombination. Recombination between directly repeated cassettes can lead to internal chromosomal deletions such that the strains, possessing essential genes in regions between the cassettes, have to be protected from these genomic rearrangements. In turn, recombination between inversely repeated cassettes leading to chromosomal inversions [55, 56] could be the basis of strain instability and, in particular, the decreased performance of the corresponding strain.
It seems useful to determine the integration points to finalize the construction of a set of stably-maintained single-copy cassette integrants. This determination could be performed using inverse PCR-based methods [57]. In this case, a task-oriented amplification of the cassettes could be performed to exclude the formation of inverted repeats and to localize essential genes between directly repeated cassettes.
It is possible that this strategy of ectopic multi-copy integration would be helpful for the construction of a broad range of plasmid-less, marker-less, recombinant Bacillus strains for microbial technology applications.
Materials and methods
Bacterial strains, plasmids, and culture conditions
Strains and plasmids used in the present study are shown in Additional file 3, Table S2. Cells of B. amyloliquefaciens and B. subtilis were grown at 37°C in liquid LB media or LB with agar [58] supplemented by antibiotics (chloramphenicol (Cm, 5-10 mg/L) or erythromycin (Em, 10 mg/L) when necessary.
Cells were plated on skim milk (20%) test plates for semi-quantitative detection of the total extracellular protease activity; activity was determined by the size of the clearance zone around each colony [17, 22].
The fermentation media TYS6C that was used for GSP production was composed of the following: 2% tryptone, 3% yeast extract, 6% soluble starch, 2% corn steep liquor (CSL), 0.1% CaCl2 (added after autoclaving), and 1% CaCO3 (added after sterilization) at pH 7.0. A final concentration of 10 mg/L Em was added to the media for cultivation of the plasmid-carrying strain. B. subtilis strains were cultured for 48 hours on a rotary shaker (at 220 rpm) at 37°C in 750-mL flasks containing 30 mL of media. Seed cultures were standardized by the preparation of freezer stock (-70°C) cultures in 20% glycerol. Then, 0.15 mL of the seed culture from the glycerol stock was used to inoculate 30 mL of TYS6C media in a single 750-mL flask. Samples for SDS-PAGE were taken after 48 hours of bacterial cultivation.
TYS6 media was the same as TYSC media, but without the CSL component. TYS6 media with 2%-4% glucose or maltose was used as the test media for generating CCR conditions.
Standard genetic engineering methods
Transformation of B. subtilis was performed using the method described by Spizizen [59].
Treatment of recombinant DNA and Southern hybridization were carried out in accordance with conventional protocols [60]. Chromosomal DNA of B. subtilis strains was hydrolyzed by Eco RI overnight, separated by electrophoresis in agarose and hybridized with biotinilated, mpr-containing PCR fragments that were amplified with mprF/mprR primers using pHE52mpr as a template. The Biotin DecaLabel™ Kit and Biotin Chromogenic Detection Kits (Fermentas, Lithuania) were used to label and detect DNA.
Preparations of restriction enzymes, T4 DNA ligase and DNA polymerase I Klenow fragments from Fermentas were used. Taq DNA polymerase (Fermentas) or AccuTaqLA DNA polymerase (Sigma, USA) were used for PCR in accordance with the manufacturers' instructions. The structures of all primers used in the present study are listed in Additional file 1, Table S1.
Construction of the pHE52mpr and pHE52(mpr-CmR) plasmids
The pairs of primers mprF/mprR and P1-bmp5/P2-bmp2 were used for PCR-mediated amplification and then for cloning of the mpr gene from the chromosomal DNA of B. amyloliquefaciens A-50. The amplicons, generated in PCR with P1-bmp5/P2-bmp2 as the primers, were treated with Bgl II and inserted into the Bgl II site of the pHEA323 plasmid [40] to form the pHE52mpr plasmid. The CmR gene from the pC194 plasmid [50] was cloned into a Bgl II-site of the pHE52mpr plasmid located just downstream of the mpr gene (with coordinate (2,174) in Figure 1). As a result, the pHE52(mpr-CmR) plasmid carrying the mprB.amy -CmR cassette was obtained. The mprB.amy and mprB.amy -CmR cassettes had the mutual DNA fragments not only in proximal part, but in distal part, as well. The later included B. amyloliquefaciens DNA fragment of the pHE52mpr plasmid (about 1,800 bp in length) consisted of pheA gene and Ter. So, pHE52mpr plasmid could be efficiently used for gene conversion resulting in substitution of mprB.amy -CmR cassette integrated in the chromosome by the marker-less mprB.amy -cassette from the plasmid (see below).
Construction of the JE852(aprE851, epr, nprB)::mprB.amy strain
The strain JE852(aprE851, epr, nprB)::mprB.amy was constructed via step-by-step ectopic integration of three copies of the mprB.amy cassette into the aprE851, epr and nprB genes of the JE852 strain. For each integration, two target-specific DNA molecules were constructed: (i) linear CmR-carrier DNA fragments for the target gene inactivation and (ii) mprB.amy -carrier plasmids for gene conversion.
As for integration into the aprE851 gene, the linear DNA fragment, yhfO'-CmR-'yhfN, was constructed in vitro by overlapping PCR, as shown in Additional file 2, Figure S1. The final DNA amplicon was treated with Eco RI and cloned into a pCB20-based [52] plasmid for the construction of pCBT(yhfO-CmR-yhfN). The later recombinant plasmid was used as a vector for the in vitro substitution of the CmR-marker by the Pst I-generated mprB.amy -cassette from pHE52mpr (Figure 1). The obtained pCBT(yhfO-mprB.amy -yhfN) plasmid was used for in vivo gene conversion, which resulted in construction of the JE852aprE851::mprB.amy strain (Figure 2).
A linear DNA fragment for integration into the epr gene was designed using Pr7/Pr8 as the primers for PCR-mediated amplification of the B. subtilis 168 chromosome. Insertion of the Pst I-generated amplicon with the CmR gene from pC194 (the primers-Pr9/Pr10) was between two Pst I-sites in the epr gene. Two auxiliary plasmids, pCBT-epr and pCBT(epr::CmR), were obtained for construction of this linear fragment. The latter plasmid served as a vector for the cloning of the mprB.amy cassette from pHE52mpr, resulting in pCBT(epr-mpr 52). The linear epr::CmR DNA fragment and pCBT(epr-mpr 52) were used for integration of the second copy of the mprB.amy cassette and construction of the JE852(aprE851, epr)::mprB.amy strain.
The third integration was based on the linear DNA fragment, nprB::CmR, carrying the nprB gene (the primers-Pr11/Pr12) disrupted by a Hind III-generated CmR-carrier amplicon from pC194 (primers-Pr9/Pr10) that was inserted into the unique Hind III site in the structural part of nprB. Construction of this fragment was provided through formation of the auxiliary plasmid pCBT(nprB::CmR). This plasmid was used later as a vector for cloning of the Pst I-generated mprB.amy cassette instead of CmR disrupted of nprB and construction of pCBT(nprB-mpr 52). It was possible so long as the Pr9/Pr10 were designed for bracketing the CmR-marker by (Hind III-PstI)/(Pst I-Hind III) sites. The linear DNA fragment, nprB::CmR, and the pCBT(nprB-mpr 52) plasmid were used for construction of the JE852(aprE851, epr, nprB)::mprB.amy strain that possessed three copies of the mprB.amy -cassette in the targeted loci of the bacterial chromosome.
Construction of DNA fragments for random integration of the mprB.amy -cassette
A total of 5 μg of chromosomal DNA from B. subtilis JE852 was exhaustively hydrolyzed by Bam HI, followed by self-circularization of the linear DNA fragments by treatment with T4 ligase in 1 mL of reaction mixture. This DNA was then digested by Pst I and ligated with 5 μg of Pst I-generated mprB.amy -CmR-cassette from pHE52(mpr-CmR) that had been purified from low melting agarose. The ligation mixture was digested by Bam HI, and about 1 μg of the total DNA was used for the transformation of B. subtilis JE852.
Segregation stability test
About 102 cells from overnight cultures of the B. subtilis JE852-(Nxyz::mprB.amy -CmR) strains were inoculated into 10 mL of fresh LB medium, cultivated for 20 generations and cloned. One hundred individual colonies were tested for Cm resistance. Strains that generated 100% CmR clones after 20 generations were tested for stability after 40 generations and then again after 60 generations. Finally, JE852-(Nxyz::mprB.amy -CmR) strains, which generated 100 CmR colonies among the 100 that were tested after 60 generations, were considered to be stable and were used as donors of chromosomal DNA for increasing the mprB.amy -cassette copy-number.
Protein analysis
SDS-PAGE was conducted using Laemmli's method [61] for the evaluation of GSP accumulation in the culture supernatants of B. subtilis strains. Gels were stained with Coomassie R-250 and scanned to estimate the protein content with the TotalLab v. 2.01 computer software for determine the portion of GSP among the secreted proteins. Total extracellular protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad, USA) in accordance with the manufacturer's instructions. In addition, the known concentrations of the commercially available carbonic anhydrase from bovine erythrocytes (Sigma) with Mw 29 kDa were used for SDS-PAGE followed by staining and scanning the gel for comparative evaluation of GSP production. Both independent methods gave, practically, coincident results.
Abbreviations
- AntR:
-
antibiotic resistance marker
- AprE:
-
alkaline serine protease subtilisin
- bp:
-
base pair(s)
- CCA:
-
carbon catabolite activation
- CCC:
-
carbon catabolite control
- CCR:
-
carbon catabolite repression
- Cm:
-
chloramphenicol
- CmR:
-
Cm resistance
- cre :
-
catabolite responsive element
- CSL:
-
corn steep liquor
- Em:
-
erythromycin
- EmR:
-
Em resistance
- GSP:
-
glutamyl-specific protease, the mpr gene protein product
- marker-less strain:
-
a bacterial strain that does not carry AntR in its genome
- NprE:
-
neutral protease
- PCR:
-
polymerase chain reaction
- P rp :
-
promoter of the B. amyloliquefaciens A-50 rplU-rpmA genes
- Ter:
-
transcription terminator of the B. amyloliquefaciens pheA gene
- mprB.amy cassette:
-
expression cassette where the structural portion of the B. amyloliquefaciens A-50 mpr gene is sandwiched between P rp and Ter
- SDS-PAGE:
-
sodium dodecyl sulphate polyacrilamide gel electrophoresis
- /:
-
denotes a plasmid-carrying strain.
References
Oggioni MR, Ciabattini A, Cuppone AM, Pozzi G: Bacillus spores for vaccine delivery. Vaccine. 2003, 21 (Suppl 2): S96-101.
Samuelson P, Gunneriusson E, Nygren PA, Stahl S: Display of proteins on bacteria. J Biotechnol. 2002, 96 (2): 129-154. 10.1016/S0168-1656(02)00043-3.
Seegers JF: Lactobacilli as live vaccine delivery vectors: progress and prospects. Trends Biotechnol. 2002, 20 (12): 508-515. 10.1016/S0167-7799(02)02075-9.
Magliani W, Conti S, Frazzi R, Pozzi G, Oggioni M, Polonelli L: Engineered commensal bacteria as delivery systems of anti-infective mucosal protectants. Biotechnol Genet Eng Rev. 2002, 19: 139-156.
Giomarelli B, Provvedi R, Meacci F, Maggi T, Medaglini D, Pozzi G, Mori T, McMahon JB, Gardella R, Boyd MR: The microbicide cyanovirin-N expressed on the surface of commensal bacterium Streptococcus gordonii captures HIV-I. Aids. 2002, 16 (10): 1351-1356. 10.1097/00002030-200207050-00006.
Schallmey M, Singh A, Ward OP: Developments in the use of Bacillus species for industrial production. Can J Microbiol. 2004, 50 (1): 1-17. 10.1139/w03-076.
Abdel-Mawgoud AM, Aboulwafa MM, Hassouna NA: Characterization of surfactin produced by Bacillus subtilis isolate BS5. Appl Biochem Biotechnol. 2008, 150 (3): 289-303. 10.1007/s12010-008-8153-z.
Yue C, Sun M, Yu Z: Improved production of insecticidal proteins in Bacillus thuringiensis strains carrying an additional cry1C gene in its chromosome. Biotechnol Bioeng. 2005, 92 (1): 1-7. 10.1002/bit.20396.
Westers L, Westers H, Quax WJ: Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta. 2004, 1694 (1-3): 299-310. 10.1016/j.bbamcr.2004.02.011.
Schumann W: Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol. 2007, 62: 137-189.
Vary PS, Biedendieck R, Fuerch T, Meinhardt F, Rohde M, Deckwer WD, Jahn D: Bacillus megaterium - from simple soil bacterium to industrial protein production host. Appl Microbiol Biotechnol. 2007, 76 (5): 957-967. 10.1007/s00253-007-1089-3.
Jones A, Lamsa M, Frandsen TP, Spendler T, Harris P, Sloma A, Xu F, Nielsen JB, Cherry JR: Directed evolution of a maltogenic α-amylase from Bacillus sp. TS-25. J Biotechnol. 2008, 134 (3-4): 325-333. 10.1016/j.jbiotec.2008.01.016.
Khemakhem B, Ali MB, Aghajari N, Juy M, Haser R, Bejar S: Engineering of the α-amylase from Geobacillus stearothermophilus US100 for detergent incorporation. Biotechnol Bioeng. 2009, 102 (2): 380-389. 10.1002/bit.22083.
Ahlawat S, Mandhan RP, Dhiman SS, Kumar R, Sharma J: Potential application of alkaline pectinase from Bacillus subtilis SS in pulp and paper industry. Appl Biochem Biotechnol. 2008, 149 (3): 287-293. 10.1007/s12010-007-8096-9.
Inouye K, Kusano M, Hashida Y, Minoda M, Yasukawa K: Engineering, expression, purification, and production of recombinant thermolysin. Biotechnol Annu Rev. 2007, 13: 43-64.
Ferrari E, Jarnagin AS, Schmidt BF: Commercial production of extracellular enzymes. Bacillus subtilis and other Gram-positive bacteria. Edited by: Sonenshein AL, Hoch JA, Losick R. 1993, American Society for Microbiology, Washington DC, 917-937.
Park CH, Lee SJ, Lee SG, Lee WS, Byun SM: Hetero- and autoprocessing of the extracellular metalloprotease (Mpr) in Bacillus subtilis. J Bacteriol. 2004, 186 (19): 6457-6464. 10.1128/JB.186.19.6457-6464.2004.
Sloma A, Rudolph CF, Rufo GA, Sullivan BJ, Theriault KA, Ally D, Pero J: Gene encoding a novel extracellular metalloprotease in Bacillus subtilis. J Bacteriol. 1990, 172 (2): 1024-1029.
Barbosa JA, Saldanha JW, Garratt RC: Novel features of serine protease active sites and specificity pockets: sequence analysis and modelling studies of glutamate-specific endopeptidases and epidermolytic toxins. Protein Eng. 1996, 9 (7): 591-601. 10.1093/protein/9.7.591.
Okamoto H, Fujiwara T, Nakamura E, Katoh T, Iwamoto H, Tsuzuki H: Purification and characterization of a glutamic-acid-specific endopeptidase from Bacillus subtilis ATCC 6051; application to the recovery of bioactive peptides from fusion proteins by sequence-specific digestion. Appl Microbiol Biotechnol. 1997, 48 (1): 27-33. 10.1007/s002530051010.
Svendsen I, Breddam K: Isolation and amino acid sequence of a glutamic acid-specific endopeptidase from Bacillus licheniformis. Eur J Biochem. 1992, 204 (1): 165-171. 10.1111/j.1432-1033.1992.tb16619.x.
Madsen JS, Qvist KB: Hydrolysis of milk protein by a Bacillus licheniformis protease specific for acidic amino acid residues. J Food Science. 1997, 62 (3): 579-582. 10.1111/j.1365-2621.1997.tb04435.x.
Fox PF, Grufferty MB: Exogenous enzymes in dairy technology. Food Enzymology. Edited by: Pox PF. 1991, London: Elsevier Applied Science, 1: 219-269.
Gruss A, Ehrlich SD: The family of highly interrelated single-stranded deoxyribonucleic acid plasmids. Microbiol Rev. 1989, 53 (2): 231-241.
Jannière L, Bruand C, Ehrlich SD: Structurally stable Bacillus subtilis cloning vectors. Gene. 1990, 87 (1): 53-61. 10.1016/0378-1119(90)90495-D.
European Council Directives. (90/220/EEC of 23 April 1990 on the deliberate release into the environment of genetically modified organisms; 98/81/EC of 26 October 1998 amending Directive 90/219/EEC on the contained use of genetically modified microorganisms).
Lee CA, Auchtung JM, Monson RE, Grossman AD: Identification and characterization of int (integrase), xis (excisionase) and chromosomal attachement sites of the integrative and conjugative element ICEBs1 of Bacillus subtilis. Mol Microbiol. 2007, 66 (6): 1356-1369.
Petit M-A, Bruand C, Jannière L, Ehrlich SD: Tn10-derived transposons active in Bacillus subtilis. J Bacteriol. 1990, 172 (12): 6736-6740.
Provvedi R, Maggi T, Oggioni MR, Manganelli R, Pozzi G: Selection and characterization of a promoter for expression of single-copy recombinant genes in Gram-positive bacteria. BMC Biotechnology. 2005, 5: 3-10.1186/1472-6750-5-3.
Vazquez-Cruz C, Ochoa-Sanchez JC, Olmedo-Alvarez G: Pulse-field gel-electrophoretic analysis of the amplification and copy-number stability of an integrational plasmid in Bacillus subtilis. Appl Microbiol Biotechnol. 1996, 46 (1): 55-60. 10.1007/s002530050782.
Middleton R, Hofmeister A: New shuttle vectors for ectopic insertion of genes into Bacillus subtilis. Plasmid. 2004, 51 (3): 238-245. 10.1016/j.plasmid.2004.01.006.
Leenhouts KJ, Kok J, Venema G: Campbell-like integration of heterologous plasmid DNA into the chromosome of Lactococcus lactis subsp. lactis. Appl Environ Microbiol. 1989, 55 (2): 394-400.
Leenhouts KJ, Bolhuis A, Venema G, Kok J: Construction of a food-grade multiple-copy integration system for Lactococcus lactis. Appl Microbiol Biotechnol. 1998, 49 (4): 417-423. 10.1007/s002530051192.
Petit M-A, Mesas JM, Noirot P, Morel-Deville F, Ehrlich SD: Induction of DNA amplification in the Bacillus subtilis chromosome. EMBO J. 1992, 11 (4): 1317-1326.
Leenhouts KJ, Kok J, Venema G: Stability of Integrated Plasmids in the Chromosome of Lactococcus lactis. Appl Environ Microbiol. 1990, 56 (9): 2726-2735.
Shimotsu H, Henner DJ: Construction of a single-copy integration vector and its use in analysis of regulation of the trp operon of Bacillus subtilis. Gene. 1986, 43 (1-2): 85-94. 10.1016/0378-1119(86)90011-9.
Härtl B, Wehrl W, Wiegert T, Homuth G, Schumann W: Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol. 2001, 183 (8): 2696-2699. 10.1128/JB.183.8.2696-2699.2001.
Chen XH, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, Morgenstern B, Voss B, Hess WR, Reva O, Junge H, Voigt B, Jungblut PR, Vater J, Süssmuth R, Liesegang H, Strittmatter A, Gottschalk G, Borriss R: Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol. 2007, 25 (9): 1007-1014. 10.1038/nbt1325.
Band L, Henner DJ: Bacillus subtilis requires a "stringent" Shine-Dalgarno region for gene expression. DNA. 1984, 3 (1): 17-21. 10.1089/dna.1.1984.3.17.
Iomantas YAV, Abalakina EG, Yampolskaya TA, Bachina TA, Polanuer BM, Kozlov YI: Method for producing shikimik acid. US Patent. 2002, No. 6,436,664 B1
Görke B, Stülke J: Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol. 2008, 6 (8): 613-624. 10.1038/nrmicro1932.
Lorka GL, Chung YJ, Barabote RD, Weyler W, Schilling CH, Saier MH: Catabolite repression and activation in Bacillus subtilis: dependency on CcpA, HPr, and HprK. J Bacteriol. 2005, 187 (22): 7826-7839. 10.1128/JB.187.22.7826-7839.2005.
Singh KD, Schmalisch MH, Stülke J, Görke B: Carbon catabolite repression in Bacillus subtilis: quantitative analysis of repression exerted by different carbon sources. J Bacteriol. 2008, 190 (21): 7275-7284. 10.1128/JB.00848-08.
Fujita Y: Carbon catabolite control of metabolic network in Bacillus subtilis. Biosci Biotechnol Biochem. 2009, 73 (2): 245-259. 10.1271/bbb.80479.
Miwa Y, Nakata A, Ogiwara A, Yamamoto M, Fujita Y: Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res. 2000, 28 (5): 1206-1210. 10.1093/nar/28.5.1206.
Miwa Y, Fujita Y: Involvement of two distinct catabolite-responsive elements in catabolite repression of Bacillus subtilis myo-inositol (iol) operon. J Bacteriol. 2001, 183 (20): 5877-5884. 10.1128/JB.183.20.5877-5884.2001.
Sharipova M, Balaban N, Kayumov A, Kirillova Y, Mardanova A, Gabdrakhmanova L, Leshchinskaya I, Rudenskaya G, Akimkina T, Safina D, Demidyuk I, Kostrov S: The expression of the serine proteinase gene of Bacillus intermedius in Bacillus subtilis. Microbiol Res. 2008, 163 (1): 39-50. 10.1016/j.micres.2006.03.003.
Vary P: Development of genetic engineering in Bacillus megaterium. Biotechnology. 1992, 22: 251-310.
Steinmetz M, Richter R: Easy cloning of mini-Tn10 insertions from the Bacillus subtilis chromosome. J Bacteriol. 1994, 176 (6): 1761-1763.
Horinouchi S, Weisblum B: Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J Bacteriol. 1982, 150 (2): 815-825.
Pomerantsev AP, Sitaraman R, Galloway CR, Kivovich V, Leppla SH: Genome engineering in Bacillus anthracis using Cre recombinase. Infect Immun. 2006, 74 (1): 682-693. 10.1128/IAI.74.1.682-693.2006.
Ceglowski P, Boitsov A, Karamyan N, Chai S, Alonso JC: Characterization of the effectors required for stable inheritance of Streptococcus pyogenes pSM19035-derived plasmids in Bacillus subtilis. Mol Gen Genet. 1993, 241 (5-6): 579-585. 10.1007/BF00279900.
Avakov AS, Bolotin AP, Kolibaba LG, Sorokin AV, Shemiakina TM, Paberit M, Raîk Kh, Aaviksaar A: Cloning and expression in Bacillus subtilis of the gene for neutral protease of Bacillus brevis. Mol Biol (Mosk). 1990, 24 (4): 1001-1009.
Dillon SC, Dorman CJ: Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat Rev Microbiol. 2010, 8 (3): 185-195. 10.1038/nrmicro2261.
Schofield MA, Agbunag R, Miller JH: DNA inversions between short inverted repeats in Escherichia coli. Genetics. 1992, 132 (2): 295-302.
Toda T, Tanaka T, Itaya M: A method to invert DNA segments of the Bacillus subtilis 168 genome by recombination between two homologous sequences. Biosci Biotechnol Biochem. 1996, 60 (5): 773-778. 10.1271/bbb.60.773.
Zimenkov DV, Skorokhodova AYu, Katashkina JI, Minaeva NI, Savrasova EA, Biryukova IV, Doroshenko VG, Akhverdyan VZ, Mashko SV: E. coli chromosome regions that are more preferable for gene insertion, when the phage Mu-driven system is used for integration. Biotechnology in Russia. 2004, 6: 1-22.
Miller JH: Experiments in Molecular Genetics. 1972, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press
Anagnostopoulos C, Spizizen J: Requirement for transformation in Bacillus subtilis. J Bacteriol. 1961, 81 (5): 741-746.
Sambrook J, Russel DW: Molecular cloning: Laboratory manual. 2001, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 3
Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970, 227 (5259): 680-685. 10.1038/227680a0.
Acknowledgements
The authors would like to acknowledge Prof. Y.I. Kozlov, who tragically passed away in 2007. He had considerable experience in the field of genetically engineered microorganisms, contributed significantly to the initiation of the present study and gave us abundant, as well as helpful, scientific advice. In addition, we are grateful to Dr. I.L. Tokmakova for useful discussion of the manuscript and the creative drawing of the most complicated part of the presented Figures.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YAVY designed the methods and performed the multi-copy number integrations at random sites of the B. subtilis chromosome. EAG designed and constructed the recombinant DNA used in this study and drafted the manuscript. LIG tested the level of extracellular GSP accumulation by protein electrophoresis and edited the manuscript. LYG performed the Southern hybridization experiments. SVM coordinated the work and amended the manuscript. All authors read and approved the final version of the manuscript.
Electronic supplementary material
12934_2011_578_MOESM2_ESM.PDF
Additional file 2: Figure S1. Construction of the linear DNA fragment used for the JE852aprE::mprB.amystrain construction. (PDF 479 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yomantas, Y.A., Abalakina, E.G., Golubeva, L.I. et al. Overproduction of Bacillus amyloliquefaciens extracellular glutamyl-endopeptidase as a result of ectopic multi-copy insertion of an efficiently-expressed mpr gene into the Bacillus subtilis chromosome. Microb Cell Fact 10, 64 (2011). https://doi.org/10.1186/1475-2859-10-64
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-10-64