
Abstract
Two coordination polymers, namely [Cu(L)2]·(NO3)2 (1) and [Co(L)(chdc)]·2H2O (2) [(chdc = 1,4-cyclohexanedicarboxylate, L = 1,3-bis-(5,6-dimethylbenzimidazole-1-yl-methylene)-benzene], based on a flexible 5,6-dimethylbenzimidazole ligand have been synthesized and characterized by physico-chemical and spectroscopic methods and single-crystal diffraction. Complex 1 shows a 2D (4,4) network structure linked by L ligands while complex 2 displays a 1D ladder-like chain bridged by L and chdc ligands. The catalytic activities of both complexes for the degradation of methyl orange have been investigated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Current interest in the crystal engineering of metal–organic coordination polymers not only results from their diverse applications in porous materials, optical devices, magnetism, microelectronics, and heterogeneous catalysis, but also from their intriguing variety of molecular architectures and topologies [1–6]. In the rational design and synthesis of metal–organic coordination compounds, several factors need to be taken into consideration, such as the preferred coordination geometry of the metals, the functionality, flexibility and symmetry of the organic ligands and the template effects of structure-directing agents [7, 8]. Among various organic ligands, the flexible bis(benzimidazole) derivatives, which can satisfy the coordination needs of the metal centers and consequently generate more robust and intricate networks, have attracted much attention and have been widely used as a classical N-based ligands. We and other groups have devoted much effort to investigate the coordination modes of these ligands, the geometric conformation of the metal centers and the different properties of the resulting coordination compounds [9–15]. In a series of benzimidazole derivatives, the most prominent compound is 5,6-dimethylbenzimidazole, which also serves as an axial ligand for cobalt in vitamin B12 [16]. Bis(5,6-dimethylbenzimidazole)alkanes act as flexible bridging ligands, frequently giving coordination polymers that can provide more information on the influence of the benzimidazoles on the structures and properties of the resulting complexes. However, to the best of our knowledge, such coordination polymers based on the flexible bis(5,6-dimethylbenzimidazole) ligands with various metal salts have only been scarcely studied [17].
The development of metal–organic frameworks (MOFs) into heterogeneous catalysts could yield several significant advantages, such as enhanced catalyst stability due to the spatial separation of individual catalytic sites within the framework [18]. MOFs can possess high porosities in the absence of any inaccessible bulk volume (dead volume), and most evidently their pore size(s), and thus their substrate shape and size selectivity can be systematically tailored by employing different organic linkers [19].
Our particular interest is in the catalytic purification of contaminated water. Much attention in this area has been focused on the advanced oxidation processess (AOPs) [20, 21], which uses Fenton-like chemistry to oxidize contaminants of concern and is largely dependent on metal-based catalysts [22–24]. This technique has the potential to completely mineralize organic compounds. As a part of our ongoing studies on metal coordination polymers of flexible bis(benzimidazole) ligands, herein we report the synthesis and characterization of Cu(II) and Co(II) coordination polymers and their catalytic activity for the degradation of methyl orange as a model organic contaminant.
Experimental
All commercially available chemicals were of reagent grade and used as received without further purification. The ligand L (see Scheme 1) was synthesized according to the literature method [25]. Elemental analyses were obtained on a Perkin-Elmer automatic analyzer. IR spectra were recorded on a Nicolet FTIR Avatar 360 spectrophotometer in the 4,000–400 cm−1 region using KBr pellets. Powder X-ray diffraction measurements were executed on a D/MAX 2500PC X-ray diffractometer using Cu-Kα radiation (λ = 0.1542 nm) in the 2θ range of 5°–50° with a step size of 0.02° and a scanning rate of 10° min−1. The absorption of methyl orange solution was measured with a Shanghai Jingke 722 N spectrophotometer at the maximum wavelength of 506 nm.

The ligand L
Synthesis of the coordination polymers
Coordination polymer 1, [Cu(L)2]·(NO3)2: A mixture of Cu(NO3)2·2.5H2O (0.233 g, 1.0 mmol), L (0.39 g, 1.0 mmol), H2O (10 mL) and ethanol (2 mL) was placed in a Teflon-lined stainless steel vessel, heated to 140 °C for 72 h under autogenous pressure and then cooled to room temperature at a rate of 5°/h. Blue crystals of 1 suitable for X-ray diffraction were obtained in 46 % yield based on Cu(NO3)2·2.5H2O. Calcd. for C52H52O6N10Cu (Fw = 976.58): C 64.0, H 5.4, N 14.3 %; found C 63.7, H 5.6, N 14.6 %. FTIR (KBr pellet, cm–1): 3,442(m), 3,126(m), 2,905(m), 1,629(m), 1,515(m), 1,380(s), 1,320(s), 1,300(w), 1,206(m), 1,024(m), 857(m), 709(m).
Coordination polymer 2, [Co(L)(chdc)]·2H2O: A mixture of Co(CH3COO)2·4H2O (0.249 g, 1.0 mmol), L (0.39 g, 1.0 mmol), 1,4-cyclohexanedicarboxylic acid (0.172 g, 1.0 mmol), H2O (8 mL) and DMF (2 mL) was placed in a Teflon-lined stainless steel vessel, heated to 140 °C for 72 h under autogenous pressure and then cooled to room temperature at a rate of 5°/h. Purple crystals of 2 suitable for X-ray diffraction were obtained in 37.5 % yield based on Co(OAc)2·4H2O. Calcd. for C34H40N4O6Co (Fw = 659.65): C 61.9, H 6.1, N 8.5 %; found C 61.7, H 6.3, N 8.7 %. FTIR (KBr pellet, cm–1): 3,419(s), 2,930(m), 1,569(m), 1,498(s), 1,444(s), 1,365(m), 1,276(m), 1,213(m), 1,022(w), 842(m), 711(w).
Catalytic experiments
The catalytic activities of 1 and 2 were tested using 200 mL aqueous methyl orange solution (10 mg/L) containing 2 mL H2O2 (30 %) and 60 mg of 1 or 2 as catalyst [26]. The experiments were performed at 45 °C, and the pH was adjusted to 3 by dropwise addition of 0.1 mol/dm3 H2SO4 or NaOH as required. Samples of the mixture were withdrawn and analyzed periodically using a Shanghai Jingke 722 N UV–vis spectrophotometer at 506 nm. This procedure was also done in the absence of catalyst as a control experiment under the same conditions.
X-ray crystallography
The selected crystal of 1 or 2 was mounted on the tip of a glass rod at room temperature. The crystallographic data collections were carried out on a Bruker Smart 1000 CCD diffractometer with graphite-monochromated Mo–Kα radiation (λ = 0.71073 Å) and ω − 2θ scan mode at 293 K. All absorption corrections were applied using the SADABS program [27]. The structures were solved by direct methods and refined anisotropically by the full-matrix least-squares technique using the Bruker SHELXTL program package [28]. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms of water were located on a difference Fourier map, while other hydrogen atoms were included in calculated positions and refined with isotropic thermal parameters riding on the corresponding parent atoms. Crystallographic data and experimental details for structural analysis of 1 and 2 are summarized in Table 1. However, the crystal size of complex 2 was small and the crystal was also not of good quality, which might lead to decay during data collection; hence, the wR 2 parameter of 2 is high. In the crystal structure of 2, one lattice water molecule is normal and the other solvent water molecule with partial occupation sites has been isotropically treated; final difference Fourier maps were flat, if excluding a residual electron density peak with a height of ca. 2 e Å−3, which is located toward the carboxylate C27 atom (2.09 Å), but this spurious peak could not be modeled as a disordered position. This may be caused by residual absorption artefacts.
Results and discussion
Synthesis and spectroscopic characterization
The coordination polymer 1 was obtained by hydrothermally reacting Cu(NO3)2·2.5H2O with ligand L in a mole ratio of 1:1 in water/ethanol (5:1); complex 2 was prepared by hydrothermal reaction of Co(OAc)2·4H2O, ligand L and 1,4-cyclohexanedicarboxylic acid at a molar ratio of 1:1:1 in water/DMF (4:1). Both of the complexes are stable in air and insoluble in common solvents such as water, methanol and ethanol. The elemental analyses for 1 and 2 were in good agreement with the theoretical requirements of their compositions (X-ray analysis results). In the FTIR spectra of 1 and 2, the bands at 1,515, 1,206 and 1,024 cm−1 for 1 and 1,498, 1,213, 1,022 cm−1 for 2 could be assigned to the vibrations of the benzimidazolyl rings in the ligand L. In 1, the C=N stretching vibration of the benzimidazolyl groups (1,515 cm−1) is blue-shifted by 19 cm−1 compared with that (1,496 cm−1) in the free ligand, consistent with coordination of L to Cu(II). For complex 2, a strong band at 3,419 cm−1 may be assigned to the stretching vibrations (υOH) of lattice water molecules, and the broad shape of this band suggests the existence of H-bonds. The asymmetric and symmetric stretching vibrations of the carboxylate groups were observed at 1,720 and 1,410 cm−1, respectively, for free 1,4-cyclohexanecarboxylic acid, while for complex 2 these bands were observed at 1,569 and 1,365 cm−1, respectively, with a separation Δυ[υas(COO) − υs(COO)] of 204 cm−1, indicating monodentate coordination of the carboxylate to the metal center [29].
Crystal structure of complex 1
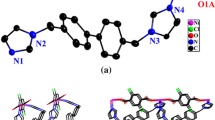
Crystallographic analysis indicates that 1 is a 2D coordination polymer. The molecule has twofold symmetry with a center of inversion located at the Cu(II) atom. The asymmetric unit consists of half of a Cu(II) atom, one L ligand and one nitrate anion. Selected bond distances and angles for 1 are listed in Table 2. As shown in Fig. 1, each Cu(II) center is coordinated by four N atoms from four different L ligands, giving square planar geometry. The Cu–N bond lengths vary from 1.990(2) to 2.002(3) Å, and the bond angles are from 89.46(10)° to 90.54(10)°, all in the normal range for analogous CuN4 complexes [30].

Coordination environment of the Cu(II) atom in 1. Hydrogen atoms were omitted for clarity, and symmetry transformations were used to generate equivalent atoms: A: 1 − x, −y, 1 − z; B: x, 0.5 − y, 0.5 + z; C: 1 − x, 0.5 + y, 1.5 − z
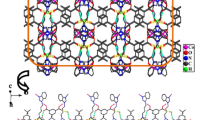
The Cu(II) atoms in 1 are bridged by L in bis-monodentate mode to form a 1D infinite chain, and these chains are further linked by other L in same coordination mode to form a 2D (4,4) network, which contains a 48-member rhombic grid with Cu(II) atoms at each corner and L ligands at each edge; the length of the edges is defined by a Cu···Cu distance of 11.886(9) Å. The ligands L are all neutral and adopt anti conformation, which have dihedral angles between the mean planes of the two benzimidazole rings of L of 57.392(71)°. The NO3 − anions lie in each rhombic grid to balance the charge and stabilize the crystal structure, (Fig. 2), giving an overall [Cu(L)2](NO3)2 stoichiometry.

2D (4,4) net connected by L ligands in 1
Crystal structure of complex 2
Single-crystal X-ray diffraction reveals that complex 2 crystallizes in monoclinic space group P-1 and displays a 1D ladder-like chain structure. Selected bond distances and angles for 2 are listed in Table 3. The asymmetric unit consists of one Co(II) atom, one L ligand, one 1,4-chdc dianion and two lattice water molecules, giving the formula [Co(L)(chdc)]·2H2O. The Co(II) center is coordinated by two nitrogen atoms from two different L ligands (Co1–N1 = 2.029(4), Co1–N3 = 2.028(4) Å), plus two carboxylate oxygen atoms from two distinct chdc ligands, giving a distorted tetrahedral CoO2N2 coordination geometry (Fig. 3). The Co–N bond lengths vary from 2.028(4) to 2.029 (4) Å, the Co–O bond lengths lie in the range of 1.935(5)–1.938(5) Å and the coordination bond angles range from 103.16(17)° to 121.83(18)°, all in the normal ranges for related CuN2O2 complexes [31].

The coordination environment around Co(II) center in 2. Hydrogen atoms and lattice water molecules were omitted for clarity, and symmetry transformations were used to generate equivalent atoms: D = −x, −y + 1, −z. E = 1 + x, y, z.)
Each L ligand adopts anti conformation to cross-link Co2(chdc)2 units in which the chdc dianionic ligands show bis-monodentate coordination mode, giving a 1D ladder-like chain (Fig. 4). The adjacent non-bonding Co···Co distances are 8.213(4) Å (Co1···Co1D) and 10.487(5) Å (Co1D···Co1E) (symmetry codes: D = −x, −y + 1, −z; E = 1 + x, y, z), respectively. Different from 1, the dihedral angle between the mean planes of the two benzimidazole rings of L is 61.89(11)°. In addition, O–H···O hydrogen bonding interactions between the lattice water and uncoordinated carboxylate O atoms of chdc further stabilize the ladder-like structure. The distances of O1w–H1wA···O4D (symmetry code: D = −x, 1 − y, −z) and O1w–H1wB···O2 are 2.796(9) and 2.775(10) Å, respectively.

1D ladder-like chain connected by L and 1,4-chdc ligand in 2
X-ray powder diffraction (XRPD)
Complexes 1 and 2 were also characterized by X-ray powder diffraction (XRPD) at room temperature (Figs. 5, 6). It is clear that the peak positions in the experimental patterns are well matched to the corresponding simulated patterns generated by the Mercury Program [32] using the single-crystal X-ray diffraction data, confirming the phase purities of 1 and 2. The differences in peak intensifies for the simulations may be due to the preferred orientation of the powder samples.

XRPD pattern of 1

XRPD pattern of 2
Catalytic degradation of methyl orange
Recently, a new advanced oxidation technology based on Fenton-like reactions has been developed to degrade azo dyes such as methyl orange [33]. However, the uncatalyzed reaction efficiency is extremely low. Fenton-like reaction consists of producing ·OH radicals from H2O2 in presence of the metal coordination polymers 1 or 2 as catalyst. It is possible to represent the MO degradation by a simplified schema [23].
The methyl orange degradation experiments were carried out using UV–vis spectrophotometer, and the results are depicted in Fig. 7 (The vertical axis shows the remaining percentage of methyl orange at time t.). As shown, within the first 10 min the control experiment proceeded rapidly and then nearly ceased when the remaining percentage of methyl orange was about 81 %. For the equivalent reaction with complex 1 as catalyst, the concentration of methyl orange decreased steadily within 60 min, and finally the remaining percentage was 62 %. In contrast, using complex 2 as catalyst, the concentration of methyl orange continued to decrease until 120 min, and the final remaining percentage reduced to 13 %. Compared with the control experiment, both complexes have a positive impact on degradation of methyl orange, while their different catalytic activities may be due to their distinct metal centers or molecular structures [17].

The experiments on the degradation of methyl orange [C 0 (mg/L) is the initial concentration of methyl orange and C (mg/L) is the concentration of methyl orange at reaction time t]
Conclusion
In summary, two coordination polymers of a benzimidazole ligand, having different structural types, have been synthesized hydrothermally. Complex 1 shows a 2D network structure connected by the benzimidazole ligands, with nitrate located in the grid center to balance the charge, while complex 2 shows a 1D ladder-like chain connected by benzimidazole ligands and chdc dianions. In both complexes, the benzimidazole ligands L all adopt anti conformation but with different dihedral angles between the two benzimidazole rings, revealing the flexibility of this ligand. Both complexes, and especially 2, show higher catalytic activity compared with those of complexes reported in the literature for the degradation of methyl orange [23].
Supplementary materials
CCDC 877560 and 877561 contain the supplementary crystallographic data for complexes 1 and 2, respectively. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Paz FAA, Klinowski J, Vilela SMF, Tomé JPC, Cavaleiro JAS, Rocha J (2012) Chem Soc Rev 41:1088–1110
Ma JF, Li SL, Lan YQ, Ma JC, Su ZM (2010) Cryst Growth Des 10:1161–1170
Hu TL, Li JR, Liu CS, Shi XS, Zhou JN, Bu XH, Ribas J (2006) Inorg Chem 45:162–173
Lee J, Farha OK, Roberts J, Scheidt KA, Nguyen ST, Hupp JT (2009) Chem Soc Rev 38:1450–1459
Wang H, Wang YY, Yang GP, Wang CJ, Wen GL, Shi QZ, Batten SR (2008) CrystEngComm 10:1583–1594
O’Keeffe M, Yaghi OM (2012) Chem Rev 112:675–702
Li CP, Du M (2011) Chem Commun 47:5958–5972
Stock N, Biswas S (2012) Chem Rev 112:933–969
Contreras R, Flores-Parra A, Mijangos E, Téllez F, López-Sandoval H, Barba-Behrens N (2009) Coord Chem Rev 253:1979–1999
Wang XL, Yang S, Liu GC, Hou LL, Lin HY, Tian AX (2011) Transition Met Chem 36:891–896
Jing W, Ren Z, Dai M, Chen Y, Lang JP (2011) CrystEngComm 13:5111–5118
Wang XL, Zhang JX, Hou LL, Zhang JW, Liu GC, Lin HY (2011) J Chem Crystallogr 41:1579–1585
Jiang H, Liu YY, Ma JF, Zhang WL, Yang J (2008) Polyhedron 27:2595–2602
Hartog R, Harvey MR, Hummel JJA, van der Pol ST, Mutikainen I, van Albada GA, Bouwman E (2011) Inorg Chim Acta 376:664–670
Cai YP, Su CY, Zhang HX, Zhou ZY, Zhu LX, Chan ASC, Liu HQ, Kang BS (2002) Z Anorg Allg Chem 628:2321–2328
Marwaha SS, Sethi RP, Kennedy JF (1983) Enzyme Microb Technol 5:361–482
Zhang SY, Zhang ZJ, Shi W, Zhao B, Cheng P (2010) Inorg Chim Acta 363:3784–3789
Corma A, García H, Llabrés i Xamena FX (2010) Chem Rev 110:4606–4655
Li JR, Sculley J, Zhou HC (2012) Chem Rev 112(2):869–932
Ahmad M, Teel AL, Watts RJ (2010) J Contam Hydrol 115:34–45
Ahmed B, Limem E, Abdel-Wahab A, Nasr B (2011) Ind Eng Chem Res 50:6673–6680
Liu TF, Cui GH, Jiao CH, Li CS, Deng XC (2011) Chin J Inorg Chem 27:1417–1422
Jiao CH, He CH, Geng JC, Cui GH (2012) Transition Met Chem 37:17–23
Cui GH, He CH, Jiao CH, Geng JC CrystEngComm. doi:10.1039/c2ce25264c
Aakeröy CB, Desper J, Elisabeth E, Helfrich BA, Levin B, Urbina JF (2005) Z Kristallogr 220:325–332
Devi LG, Kumar SG, Reddy KM, Munikrishnappa C (2009) J Hazard Mater 164:459–467
Sheldrick GM (1997) SADABS, program for empirical absorption correction of area detector data. University of Gottingen, Germany
Sheldrick GM (1997) SHELXS 97, program for crystal structure solution. University of Göttingen, Germany
Deacon GB, Phillips RJ (1980) Coord Chem Rev 33:227–250
van Albada GA, Smeets WJJ, Veldman N, Spek AL, Reedijk J (1999) Inorg Chim Acta 290:105–112
Crane JD, Sinn E, Tann B (1999) Polyhedron 18:1527–1532
Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J (2006) J Appl Crystallogr 39:453–457
Ocampo AM (2009) Persulfate activation by organic compounds, Thesis for the Doctorate of Washington State University
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Geng, Jc., Qin, L., He, Ch. et al. Synthesis, crystal structures and catalytic properties of copper(II) and cobalt(II) coordination polymers based on a flexible benzimidazole ligand. Transition Met Chem 37, 579–585 (2012). https://doi.org/10.1007/s11243-012-9624-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-012-9624-1