
Abstract
Three new CoII coordination polymers, namely [Co(DNBA)2(pbdmbm)] (1), [Co2(H2O)2(DNBA)2(ebdmbm)2] (2) and [Co2(DNBA)2(pbbm)2] (3) have been obtained by hydrothermal reactions of CoII with flexible bis(benzimidazole) ligands [1,1′-(1,3-propanediyl)bis(5,6-dimethylbenzimidazole) (pbdmbm), 1,1′-(1,2-ethanediyl)bis(5,6-dimethylbenzimidazole) (ebdmbm), 1,1′-(1,3-propanediyl)bis(benzimidazole) (pbbm)] plus 3,5-dinitrobenzoic acid (HDNBA). The complexes have been characterized by single crystal X-ray diffraction, elemental analyses, IR and TG. Complexes 1 and 3 exhibit one-dimensional chains composed of CoII centers bridged by flexible bis(benzimidazole) ligands. Complex 2 is a three-dimensional NaCl-type supramolecular framework constructed from binuclear units, which are formed by two CoII centers and two ebdmbm ligands. The spacer length and substituents on the bis(benzimidazole) ligands are crucial for the construction of these structures. The photoluminescence properties of the complexes and the cyclic voltammetry behavior of complex 1 are described.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, the design and construction of coordination polymers with novel structures have attracted great attention due to their potential applications in magnetism, catalysis, gas storage and nonlinear optics [1–5]. Thus, a large number of coordination polymers with various structures have been prepared through rational selection of the metal and ligands, as well as the synthesis conditions [6–10]. Multidentate N-heterocyclic ligands, especially those containing flexible spacers, have been widely used in these systems, and many complexes with novel architectures have been obtained [11–14]. Some complexes with flexible N-heterocyclic ligands such as 1,3-bis(4-pyridyl)-propane (bpp), 1,4-bis(imdazol-1-ylmethyl)benzene (bix), 1,3,5-tri(imidazol-1-ylmethyl)benzene (tib), and 1,1′-(1,4-butanediyl)bis-1H-benzotriazole (bbbt) have been reported by Lang, Bu, Ma and our group [15–19].
To expand our previous work, flexible bis(benzimidazole)-based ligands have been used here [pbdmbm = 1,1′-(1,3-propanediyl)bis(5,6-dimethylbenzimidazole), ebdmbm = 1,1′-(1,2-ethanediyl)bis(5,6-dimethylbenzimidazole), pbbm = 1,1′-(1,3-propanediyl)bis(benzimidazole)] (Fig. 1) as neutral organic linkers to systematically investigate the influence of substituents and spacer length on the structures of their complexes. Complexes based on pbdmbm and ebdmbm ligands have rarely been reported up to now [20].

The ligands used in this paper
In this study, we describe three new CoII coordination polymers [Co(DNBA)2(pbdmbm)] (1), [Co2(H2O)2(DNBA)2(ebdmbm)2] (2), and [Co2(DNBA)2(pbbm)2] (3) using DNBA (3,5-dinitrobenzoic acid) as counter-ion. Complexes 1 and 3 exhibit one-dimensional chain structures, while complex 2 is binuclear. In addition, the fluorescence properties and electrochemical behaviors of the complexes are described.
Experimental
All chemicals were used as supplied from commercial sources without further purification. The heterocyclic ligands were synthesized by the literature method [21] and characterized by FT/IR spectra. FT/IR spectra (KBr pellets) were taken on a Magna 560 spectrometer. Elemental analyses were obtained on a Perkin–Elmer 240CHN analyzer. Thermogravimetric analysis was carried out with a Pyris Diamond TG-DTA instrument. Luminescence spectra were measured on a Hitachi F-4500 fluorescence spectrophotometer. Electrochemical experiments were carried out using a CHI 440 Electrochemical quartz crystal microbalance.
Synthesis of [Co(DNBA)2(pbdmbm)] (1)
A mixture of CoCl2·6H2O (0.0237 g, 0.1 mmol), HDNBA (0.0424 g, 0.2 mmol), pbdmbm (0.0334 g, 0.1 mmol), and H2O (10 mL) was sealed in a 25-mL Teflon reactor at 150 °C for 3 days. After slow cooling to room temperature, purple block crystals of 1 were obtained. Yield: 30% based on CoII . Calc. for C35H30CoN8O12: C, 51.7; H, 3.7; N, 13.8. Found: C, 51.5; H, 3.6; N, 13.4%. IR (KBr, cm−1): 3,449m, 2,950m, 2,362s, 2,327s, 1,625w, 1,539m, 1,454m, 1,342m, 1,224m, 1,085m, 992m, 844m, 782m, 747s, 649m, 565s.
Synthesis of [Co2(H2O)2(DNBA)2(ebdmbm)2] (2)
Complex 2 was prepared in the same manner as for 1 but using ebdmbm (0.0323 g, 0.1 mmol) in place of pbdmbm. Yield: 40% based on CoII. Calc. for C68H60Co2N16O26: C, 49.9; H, 3.7; N, 13.7. Found: C, 50.0; H, 3.6; N, 13.7%. IR (KBr, cm−1): 3,560m, 3,460s, 3,401s, 3,092s, 2,960m, 2,368s, 1,634m, 1,590m, 1,546s, 1,484m, 1,453s, 1,414m, 1,344s, 1,274m, 1,224s, 1,070s, 1,000s, 915s, 854s, 782s, 726s, 626s.
Synthesis of [Co2(DNBA)2(pbbm)2] (3)
The synthesis complex 3 was similar to that of 1, except that pbbm (0.0278 g, 0.1 mmol) was used instead of pbdmbm. Yield: 35% based on CoII . Calc. for C62H44Co2N16O24: C, 49.2; H, 2.9; N, 14.8. Found: C, 49.2; H, 2.9; N, 14.7%. IR (KBr, cm−1): 3,449m, 3,338m, 3,085m, 2,362s, 2,327s, 1,637s, 1,541m, 1,458m, 1,394s, 1,348m, 1,288m, 1,259m, 1,190s, 1,070s, 1,007w, 930s, 890m, 719s.
Preparation of compound 1 bulk-modified carbon paste electrode (1-CPE)
The 1-CPE was obtained as follows: complex 1 (0.034 g) and graphite powder (0.5 g) were ground together with an agate mortar and pestle for approximately 30 min to achieve an even, dry mixture. Paraffin oil (1.1 mL) was added, and the mixture was stirred with a glass rod; then the homogenized mixture was used to pack 3-mm inner diameter glass tubes to a length of 0.6 cm. The electrical contact was established with a copper stick, and the surface of the 1-CPE was wiped with weighing paper.
X-Ray crystallographic study
Diffraction data for the complexes 1–3 were collected on a Bruker Smart 1000 CCD area detector diffractometer (Mo Kα radiation, graphite monochromator, λ = 0.71073 Å for 1, 3 and λ = 0.71069 Å for 2). The crystal structures were solved by direct method and refined on F 2 by full-matrix least-squares techniques with the SHELXL-97 software package [22]. All non-hydrogen atoms were refined anisotropically, and hydrogen atoms of the ligands were placed in geometrically idealized positions and refined isotropically. The crystallographic information is summarized in Table 1. Selected bond lengths and angles for complexes 1–3 are listed in Table 2.
Results and discussion
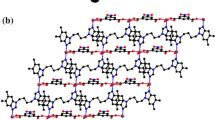
Complex 1 exhibits a 1D chain structure based on the building blocks of one CoII atom, two DNBA anions and one pbdmbm ligand. As depicted in Fig. 2, each Co atom is tetra-coordinated by two oxygen atoms (O2, O8) from two anionic DNBA ligands and two nitrogen atoms (N1, N4) from two pbdmbm ligands, forming a distorted tetrahedral geometry [CoN2O2]. Each DNBA is monodentate. The adjacent [CoN2O2] geometries are linked into a 1D chain by pbdmbm ligands, with a Co···Co distance of 10.2178(20) Å (Fig. 3). In complex 1, pbdmbm adopts a trans–trans conformation, and the dihedral angle between the imidazole rings is 70.87°. Two adjacent chains are expanded to a 1D supramolecular double chain via π–π stacking interactions, and the face-to-face distances between DNBA and the benzene ring of the pbdmbm ligand are 3.5503(4) Å and 3.5761(4) Å (Fig. S4).

The coordination environment of CoII in complex 1, showing a distorted tetrahedral geometry. The hydrogen atoms are omitted for clarity

1D chain connected by pbdmbm ligands in complex 1
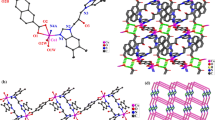
Single crystal X-ray diffraction reveals that complex 2 is a three-dimensional supramolecular framework constructed from binuclear structures. Each CoII atom is coordinated by two nitrogen atoms (N1, N4) from two ebdmbm ligands and two oxygen atoms (O1, O7) from two anionic DNBA ligands, giving a distorted tetrahedral geometry [CoN2O2] (Fig. 4). Each DNBA ligand is monodentate, similar to complex 1. The neutral ebdmbm ligand in complex 2 has a shorter linker than pbdmbm. The ebdmbm ligands adopt trans conformation linking two adjacent [CoN2O2] geometries to form a binuclear structure, with a Co···Co distance of 7.3463(20) Å. The dihedral angle between the imidazole rings is 68.93°. There are three kinds of π–π stacking interactions in 2. Two adjacent binuclear units are connected into two-dimensional supramolecular layers by π–π stacking interactions between the aromatic rings from two parallel DNBA ligands with face-to-face distances of 3.6364(11) and 3.7437(10) Å (Fig. S5). The neighboring layers are further extended into a 3D supramolecular framework through π–π stacking interactions between the aromatic rings of the ebdmbm ligands with a face-to-face distance of 3.8165(12) Å. Finally, the structure of complex 2 can be described as a NaCl-type supramolecular framework if each binuclear unit is considered as a node (Fig. S6).

The coordination environment of CoII in complex 2, showing a distorted tetrahedral geometry. The hydrogen atoms are omitted for clarity
Complex 3 is a 1D coordination polymer, which is further extended into a 1D double chain by π–π stacking interactions. Each CoII center is tetrahedrally coordinated by two nitrogen atoms (N3, N5) from two pbbm ligands, two oxygen atoms (O3, O5) of two anionic monodentate DNBA ligands forming [CoN2O2] units (Fig. 5). Compared with the pbdmbm ligand in complex 1, pbbm has no substituents in the aromatic rings and bridges two adjacent [CoN2O2] units to build a 1D zigzag chain with a Co···Co distance of 9.3955(10) Å, which is 0.9 Å shorter than complex 1, as shown in Fig. 6. The pbbm ligands in 3 also show trans–trans conformation, and the dihedral angle between the benzimidazole rings is 74.45°. Two adjacent chains are expanded to a 1D supramolecular double chain by π–π stacking interactions, and the face-to-face distance between DNBA ligands is 3.6683(5) Å (Fig. S7).

The coordination environment of CoII in complex 3, showing a distorted tetrahedral geometry. The hydrogen atoms are omitted for clarity

1D zigzag chain connected by pbbm ligands in complex 3
Although these complexes are all constructed from HDNBA and different bis(benzimidazole) derivatives, and synthesized under similar conditions, they show different structures. As shown in Fig. S8, the pbdmbm ligand of complex 1 adopts trans–trans conformation with an N···N distance of 4.9110(97) Å. The angles between the adjacent –CH2– spacers are 114.37°, 113.99° and 114.17°. In complex 3, the pbbm ligands also display trans–trans conformation with spacer angles of 111.32°, 111.60° and 112.24°, leading to an N···N distance of 4.3840(25) Å, a little shorter than in complex 1, which may be attributed to the steric hindrance of the methyl substituents of pbdmbm. In complex 2, we choose ebdmbm as ligand with an N···N distance of 3.1249(23) Å, much shorter than the ligands used in 1 and 3 because of the two –CH2– spacers. Ebdmbm shows trans conformation, and the angles of the adjacent –CH2– spacers are 113.94° and 114.64°. Hence, variations in spacer length and substituents exert influence on the final structures of the complexes (Fig. S9). Both complexes 1 and 3 exhibit 1D chains with different shapes, which are expanded into 1D double chains through π–π stacking interactions, whereas complex 2 displays a binuclear structure with a NaCl-type supramolecular framework involving π–π stacking interactions.
The IR spectra of complexes are shown in Figs. S1–S3. The bands at 844, 1,454, and 1,539 cm−1 for 1 (854, 1,453, and 1,546 cm−1 for 2, 890, 1,458, and 1,541 cm−1 for 3) can be attributed to ν C–N of the N-heterocyclic rings of the ligands. The bands at 2,950 cm−1 for 1, 2,960 cm−1 for 2, and 3,085 cm−1 for 3 can be assigned to the methylene ν C–C of the ligands, while bands at 1,342, and 1,625 cm−1 for 1, 1,344, and 1,634 cm−1 for 2, 1,348 and 1,637 cm−1 for 3 can be attributed to ν(–NO2) and ν(–COO) of DNBA. For complex 2, a band at 3,460 cm−1 is assigned to ν(H2O) [23].
To estimate the thermal stabilities of the complexes, thermogravimetric analyses were performed in the temperature range of 50–800 °C (Figure S10). The TG curves of complexes 1 and 3 are similar, showing only one weight loss step from 329 to 614 °C for 1 and from 302 to 619 °C for 3. The remaining weights (9.7% for 1, 10.3% for 3) correspond to CoO (calculated 9.2% for 1, 9.9% for 3). The TG curve of complex 2 exhibits two weight loss steps in the temperature range of 116–505 °C. The first weight loss of 2.5% at 116–152 °C is ascribed to the loss of lattice water molecules (calculated value 2.2%). Decomposition of the organic ligands ebdmbm and DNBA can be observed from 316 to 495 °C, and the remaining weight of 10.0% is equivalent to CoO (calculated 9.2% for 2).
The photoluminescence properties of the complexes, together with those of the bis(benzimidazole)-based ligands, were studied in the solid state at room temperature. The emission spectra of the complexes are shown in Fig. 7. Bands are observed at 465 nm for 1, 446 nm for 2, and 426 nm for 3 with excitation wavelengths of 352, 370 and 400 nm, respectively. The emission spectra of the free ligands are shown in the inset of Fig. 7. The main emission bands of the complexes are similar to those of the free ligands, which may be attributed to intraligand charge transfer [24, 25].

Emission spectra of the free ligands and complexes 1–3 in the solid state at room temperature
In order to study the redox properties of the CoII coordination polymer, complex 1 was used as a solid modifier to fabricate a bulk-modified carbon paste electrode (CPE) due to its insolubility in water and common organic solvents. The electrochemical behavior of 1-CPE was investigated in 1 M H2SO4 at room temperature. As shown in Fig. 8, in the potential range of +750 to −100 mV with scan rate of 100 mV s−1, no redox peak was observed at the bare CPE. In contrast, 1-CPE showed one quasi-reversible redox peak and the mean peak potential E 1/2 = (E pa + E pc)/2 was approximately 400 mV, which could be attributed to the CoIII/CoII redox couple [26].

Cyclic voltammograms of the bare CPE with scan rate 100 mV s−1 and 1-CPE at different scan rates (from inner to outer): 20, 40, 60, 80, 100, 120, 140, 160, 180, 200, 250, 300, 350, 400, 450 mV s−1 in 1 M H2SO4 aqueous solution in the potential range of +750 to −100 mV. The inset shows the plots of the anodic and the cathodic peak currents versus scan rates
The effect of varying scan rates on the electrochemical behavior of the 1-CPE was also investigated under the same conditions. It can be seen from Fig. 8 that the peak potentials changed gradually: the cathodic peak potentials gradually shifted to negative direction and the corresponding anodic peak potentials shifted to positive direction as the scan rate was increased from 20 to 450 mV s−1. The inset of Fig. 8 shows that the anodic and cathodic peak currents are proportional to the scan rates, suggesting that the redox process for 1-CPE is surface-controlled [27].
Conclusion
In summary, three new CoII complexes were synthesized using the flexible double benzimidazole derivative ligands pbdmbm, ebdmbm, and pbbm with HDNBA co-ligand. The different structures of the complexes imply that the ligands are important in determining the final crystal structures and properties of the CoII coordination polymers.
Supplementary materials
CCDC 826289, 826292, 826832 contain the supplementary crystallographic data for compounds 1–3, which offer TG, IR, tables of selected bond lengths and angles and structural figures. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223–336–033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Matsuda R, Kitaura R, Kitagawa S, Kubota Y, Belosludov RV, Kobayashi TC, Sakamoto H, Chiba T, Takata M, Kawazoe Y (2005) Nature 436:238
Millward AR, Yaghi OM (2005) J Am Chem Soc 127:17998
Morris RE, Wheatley PS (2008) Angew Chem Int Ed 47:4966
Long DL, Blake AJ, Champness NR, Wilson C, Schröder M (2001) J Am Chem Soc 123:3401
Furukawa H, Miller MA, Yaghi OM (2007) J Mater Chem 17:3197
Cui Y, Evans OR, Ngo HL, White PS, Lin W (2002) Angew Chem Int Ed 7:1159
Cui Y, Lee SJ, Lin WJ (2003) J Am Chem Soc 20:6014
Noyori R (2002) Angew Chem Int Ed 12:2008
Gutiérrez A, Perpiñán MF, Sánchez AE, Torralba MC, Torres MR, Pardo MP (2007) Inorg Chem Acta 46:604
Hu S, Zhang ZM, Meng ZS, Lin ZJ, Tong ML (2010) CrystEngComm 12:4378
Wang XL, Chen YQ, Liu GC, Zhang JX, Lin HY, Chen BK (2010) Inorg Chim Acta 363:773
Barnett SA, Champness NR (2003) Coord Chem Rev 246:145
Chen SS, Fan J, Okamura TA, Chen MS, Su Z, Sun WY, Ueyama N (2010) Cryst Growth Des 10:812
Wang XL, Chen YQ, Liu GC, Lin HY, Zheng WY, Zhang JX (2009) J Organomet Chem 694:2263
Li LL, Liu LL, Zheng AX, Chang YJ, Dai M, Ren ZG, Li HX, Lang JP (2010) Dalton Trans 39:7659
Li LL, Yuan RX, Liu LL, Ren ZG, Zheng AX, Cheng HJ, Li HX, Lang JP (2010) Cryst Growth Des 10:1929
Li ZX, Hu TL, Ma H, Zeng YF, Li CJ, Tong ML, Bu XH (2010) Cryst Growth Des 10:1138
Liu YY, Wang ZH, Yang J, Liu B, Liu YY, Ma JF (2011) CrystEngComm 13:3811
Wang XL, Zhang JX, Liu GC, Lin HY, Chen YQ, Kang ZH (2010) Inorg Chem Acta 368:207
Wang XL, Yang S, Liu GC, Zhang JX, Lin HY, Tian AX (2011) Inorg Chem Acta 375:70
Li LL, Li HX, Ren ZG, Liu D, Chen Y, Zhang Y, Lang JP (2009) Dalton Trans 40:8567
Sheldrick GM (2008) Acta Cryst A 64:112
Bellamy LJ (1958) The infrared spectra of complex molecules. Wiley, New York
Allendorf MD, Bauer CA, Bhakta RK, Houk RJT (2009) Chem Soc Rev 38:1330
Lakowicz JR (2006) Principles of fluorescence spectroscopy. Springer, New York
Mitkina TV, Zakharchuk NF, Naumov DY, Gerasko OA, Fenske D, Fedin VP (2008) Inorg Chem 47:6748
Wang XL, Mu B, Lin HY, Liu GC (2011) J Organomet Chem 696:2313
Acknowledgments
This work was supported by the NCET-09-0853, the National Natural Science Foundation of China (No. 20871022) and the Foundation of Liaoning Province (No.2009R03 and 2009402007).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11243_2011_9546_MOESM1_ESM.doc
Crystallographic data in CIF, IR spectra, TG curves and the tables with the selected bond lengths and bond angles can be found, in the online version. (DOC 6627 kb)
Rights and permissions
About this article
Cite this article
Wang, XL., Yang, S., Liu, GC. et al. Cobalt (II) coordination polymers based on 3,5-dinitrobenzoate and flexible bis(benzimidazole) derivatives bearing different spacer lengths and substituents. Transition Met Chem 36, 891–896 (2011). https://doi.org/10.1007/s11243-011-9546-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-011-9546-3