
Abstract
The mechanism and regioselectivity of [3 + 2] cycloaddition (32CA) reactions of N-methyl-1-phenylmethanimine oxide nitrone 1 and bicyclopropylidene 2 are analyzed using molecular electron density theory (MEDT) at the B3LYP/6–311 + + G(d,p) level. A study of the electron localisation function (ELF) predicts the zwitter-ionic nature of the nitrone, allowing its participation in zw-type 32CA reactions with a high energy barrier that must be surmounted by suitable electrophilic–nucleophilic interactions. The global electronic flux from the strong nucleophilic bicyclopropylidene 2 to the electrophilic nitrone 1 is predicted by an analysis of the CDFT indices. In this 32CA reaction, no new covalent bonds are generated at the TSs, and the mechanism is one-step and kinetically controlled with low asynchronicity in bond formation. The Gibbs free energy of this 32CA reaction in the gas phase is −9.88 and −15.01 kcal.mol−1 for exo and endo path, respectively. The increased thermodynamic stability of the cycloadducts 4 favors the endo regiochemical route. The ELF topological examination at the transition states is in agreement with the predictions of bonding evolution theory (BET) for the endo and exo routes, which point to a one-step process including early transition states.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The study of heterocycle synthesis is common in the field of organic chemistry and requires a particular synthetic strategy. Heterocycles play a crucial role in the synthesis of vital biological molecules, such as vitamin B6 [1]. The [3 + 2] cycloaddition (32CA) reaction is a very common class of reactions that have been the subject of extensive research, most notably by Huisgen [1], who performed a comprehensive analysis of the likely interactions between dipole and dipolarophile, thereby facilitating a better understanding of these reactions [2]. A few years after the structure of diazoacetic acid esters was determined by the reaction of diazomethane with acrylic esters [2], the 32CA reactions were discovered. They have been the subject of numerous reviews because the research in this field is so intriguing. Particularly, the 32CA reactions are an efficient method to produce 5-membered heterocycles [3,4,5,6,7]. In recent decades, the importance of the function of heterocyclic compounds has increased, leading to an astounding number of novel classes of compounds containing at least one heterocycle in their structure. Heterocycles are significant not only for their abundance and exceptional diversity, but also for their use in biological, medical, and therapeutic disciplines (vitamins, hormones, antibiotics, etc.), as well as in industrial and technical fields (such as corrosion inhibitors, dyes, stabilizing agents, insecticides, herbicides) [8,9,10,11]. Although the usefulness of 32CA reactions in organic synthesis is no longer debatable, research in this field focuses on improving the chemical reactions that generate regio- and stereochemically controlled products. Using a chiral Lewis acid catalyst appears to be the most efficient and cost-effective method for lowering the activation energy of a specific 32CA reaction in order to generate a high yield of the primary product. Numerous experimental and theoretical investigations on this topic have been published [12,13,14,15].
In the last two decades, computational chemistry has become an important instrument for analyzing experimentally reported reactivity and selectivity results by constructing a comprehensive picture of how chemical events occur [16,17,18,19,20,21,22,23,24]. In 2016, Domingo [25] proposed the molecular electron density theory (MEDT), recognizing the critical role of electron density changes in molecular reactivity. Despite the increasing number of modern applications of computational science in chemistry, the fundamental theories of organic chemistry had remained stagnant for the previous 40 years. In the past 7 years, MEDT has effectively analyzed the experimental results of numerous 32CA reactions [26,27,28,29]. Recently, MEDT has been utilized to evaluate the experimental outcomes of strain-promoted and catalyzed 32CA reactions and the reported chemo-, regio-, and stereoselective production of spiroisoxazolines [30, 31]. Based on their electronic structure, the three-atom components (TACs) that play a role in 32CA reactions are classified as pseudodiradical, pseudo(mono)radical, carbenoid, and zwitter-ionic [32]. This enables them to take part in the pdr-type, pmr-type, cb-type, and zw-type 32CA reactions [32,33,34]. 32CA reactions with pdr-type 32CA have a lower energy barrier and are simpler to perform than 32CA reactions with zw-type 32CA, which require electrophilic–nucleophilic contact to overcome a higher energy barrier [35].
The 32CA reactions of N-methyl-1-phenylmethanimine oxide nitrone 1 and bicyclopropylidene 2 under solvent-free conditions and in benzene and toluene solvents are investigated in detail. Brandi [36] and co-workers conducted experiments on this reaction in 1994, in which the 32CA reaction of N-methyl-1-phenylmethanimine oxide nitrone 1 and bicyclopropylidene 2 (Scheme 1) afforded the endo- diastereoselective adduct 4 more than exo- diastereoselective 3. The synthesis was performed using organic solvents such as benzene and toluene, furnishing exclusive endo- diastereoselectivity in each case. In this research paper, the MEDT investigation is presented in the “The ELF analysis at the ground state of the reactants N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2,” the “Analysis of the CDFT indices at the ground state of the reagents N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2,” the “Analysis of the potential energy surface (PES) of the 32CA reaction of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2,” the “Topological analysis of the ELF at the TSs,” the “Mechanistic implications along the stereoisomeric channels of 32CA reaction N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2 from bonding evolution theory (BET) study,” and the “Topological analysis of the AIM at the TSs involved in the 32CA reactions” sections.

32CA reaction of N-methyl-1-phenylmethanimine oxide nitrone 1 and bicyclopropylidene 2 leading to cycloadducts 3 and 4
(I) In the “The ELF analysis at the ground state of the reactants N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2” section, the electron localization function (ELF) at the ground state structures of reagents N-methyl-1-phenylmethanimine oxide nitrone 1 and bicyclopropylidene 2 is topologically analyzed so that they can be represented in terms of their electronic structure and then assessed for their reactivity in 32CA reactions. (II) In the “Analysis of the CDFT indices at the ground state of the reagents N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2” section, the reactivity indices characterized by the conceptual density functional theory [37, 38] (CDFT) are examined to comprehend the polarity of the 32CA reactions. (III) In the “Analysis of the potential energy surface (PES) of the 32CA reaction of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2” section, the potential energy surfaces (PES) along the possible stereoisomeric channels of the 32CA reactions are investigated for energy profile prediction, and the global electron density transfer [39] (GEDT) at the transition states (TSs) is determined. The “Topological analysis of the ELF at the TSs” section evaluates the ELF of the identified TSs. (V) In the “Mechanistic implications along the stereoisomeric channels of 32CA reaction N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2 from bonding evolution theory (BET) study” section, the combination of ELF and Thom’s catastrophe theory [40], namely, the Krokidis’ bonding evolution theory [41] (BET) is used to structure the process of electron density changes along stereoisomeric routes. (VI) In the “Topological analysis of the AIM at the TSs involved in the 32CA reactions” section, atoms-in-molecules [42] (AIM) topological analyses were performed at the TSs to examine their electronic structures and characterize the atomic interactions.
Computational methods
The Berny analytical gradient optimization method was used to optimize the stationary locations along the potential energy surface of the 32CA reactions at the B3LYP/6–311 + + G (d, p) level of theory [43, 44]. In the investigation of several recent 32CA reactions, the B3LYP functional has been validated as a reliable and accurate method [13, 45, 46]. At the optimized TSs, frequency calculations revealed the existence of one imaginary frequency, whereas the absence of an imaginary frequency at the local minimum was confirmed. Using the Gonzales-Schlegal integration technique, IRC calculations were performed to validate the minimal energy reaction pathway [47, 48]. The established TSs are the link between the reactants and the products. Using a polarizable continuum model (PCM) and the self-consistent reaction field (SCRF) method, the solvent effects in toluene and benzene were analyzed [49,50,51]. The CDFT [38] indices are computed for the reagents in the gas phase. Using the formula ω = (µ2/2η), determine the global electrophilicity index (ω). The chemical hardness (η) and electronic chemical potential (µ) can be approximated using the one-electron energies of the Highest Occupied Molecular Orbital (εHOMO) and Lowest Unoccupied Molecular Orbital (εLUMO), as η ≈ εLUMO-εHOMO and µ ≈ (εHOMO + εLUMO)/2, respectively. Using the HOMO energies within the Kohn–Sham scheme, the relative nucleophilicity N index is calculated. N = EHOMO(Nu)-EHOMO(TCE) is the formula for this quantity. TCE (tetracyanoethylene) was chosen as a reference due to its low HOMO energy.
Natural population analysis (NPA) was utilized to develop the global electron density theory (GEDT) [39] at the transition states (TSs) of every reacting framework.
While q represents atomic charges, the sum of charges on all atoms in the framework under consideration represents the GEDT, with a positive GEDT signifying global electronic flux from one framework to the other.
At the reagents, ELF topological analyses were conducted using Multiwfn programs Version 3.9 [52]. The UCSF Chimera software [53] was utilized to depict the ELF localization domains. Multiwfn programs Version 3.9 [52] is used to calculate TSs, IRC points, and Quantum Theory of Atoms-in-Molecules (QTAIM) parameters [42]. All calculations were conducted using the software Gaussian 16 [54].
Results and discussion
The ELF analysis at the ground state of the reactants N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2
The electron localization function makes it possible to get a quantitative picture of the electronic structure of a molecular system. This makes it possible to find a fairly good link between the electronic structure and the molecular reactivity of the three atom components (TACs) in MEDT. Silvi and Savin [55] further illustrated the ELF concept developed by Becke and Edgebombe [56] to characterize distinct regions in a chemical system, namely, the atomic nuclei, bonding and non-bonding regions, associated with the core, monosynaptic and disynaptic basins. A pseudoradical atomic center has a monosynaptic basin that adds up to less than 1 e. A carbenoid atomic center has a monosynaptic basin that adds up to 2 e. Standard classification divides TACs into three types: pseudodiradical type, pseudo(mono)radical type, and carbenoid type. The pseudodiradical type has two pseudoradical centers, the pseudo(mono)radical type has one pseudoradical center, and the carbenoid type has one carbenoid center. Since the molecular reactivity of TACs in 32CA reactions is related to their electronic structure, the most reactive TACs in the series are the pseudodiradical ones with very low energy barriers. The pseudo (mono) radical TACs and carbenoid TACs have the same level of reactivity, but the zwitter-ionic TACs have the highest activation energy barrier.
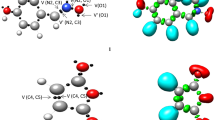
Figure 1 shows the most important valence basin populations, the ELF localization regions, and the positions of the basin attractor for the N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2. The ELF analysis of nitrone 1 shows that V(O1) and V′(O1) monosynaptic basins with an integration of 5.91 e are connected to the nonbonding electron density on O1 oxygen (see Table 1). The disynaptic basins V(C3,N2) and V(N2,O1) with an integration of 3.81 e and 1.39 e are connected to the C3–N2 and N2–O1 bonding regions. Since nitrone 1 does not have a pseudoradical or carbenoid center, it can be classified as a zwitter-ionic TAC that needs the right electrophilic-nucleophilic interactions to break through a high-energy barrier.

B3LYP/6–311 + + G(d,p) ELF localization domains represented at an isosurface value of ELF = 0.80 of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2. Blue color represents the protonated basins, green colored ones are the disynaptic basins, and red color is used to represent the monosynaptic basins. The attractor positions are represented as black spheres
ELF studies of bicyclopropylidene 2 show that there are two disynaptic basins V(C4, C5) and V′(C4, C5) that add up to a total population of 3.84 e. These basins are linked to the double bond between C4 and C5. Figure 2 shows the NBO charges and Lewis-like structures of the reactants. O1 has a charge of −0.529 e, while N2 has a charge of 0.079 e and C3 has the value of 0.020 e. Note that the charge distribution of nitrone 1 does not match the Lewis resonance structure pattern. This is because the term “zwitter-ionic,” which is used in TAC classification, does not mean that the molecule has a dipolar electronic structure. Instead, it means that the molecule has a high overall electron density. The C4 and C5 carbons of bicyclopropylidene 2 show the negative charge of −0.297 e.

B3LYP/6–311 + + G(d,p) calculated natural atomic charges, in average number of electrons e, of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2. Positive charges are colored in blue and negative charges in red
Analysis of the CDFT indices at the ground state of the reagents N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2
For CDFT calculations, the B3LYP/6-31G(d) computational level is used to describe how the reagents behave on the usual nucleophilicity [37] and electrophilicity [57] scales. The CDFT analysis gives predict how the electronic properties of the reacting partners will change and, based on that, figure out how low polar the 32CA reaction is. Table 2 shows the reagents’ estimated B3LYP/6-31G(d) CDFT indices. N-methyl-1-phenylmethanimine oxide nitrone 1 has a lower electronic chemical potential (−3.41) than bicyclopropylidene 2 (−3.07 eV). Since there is only a small difference between the values of these two reagents, this points to a non-polar reaction. Nitrone 1 is a moderate electrophile, with an electrophilicity of 1.39 eV, while bicyclopropylidene 2 are marginal electrophile, with an electrophilicity of 0.69 eV. On the standard scale for nucleophilicity, nitrone 1 is a strong nucleophile (N = 3.99 eV) and bicyclopropylidene 2 is moderate nucleophile (N = 2.98 eV) within the standard nucleophilicity scale [37, 57].
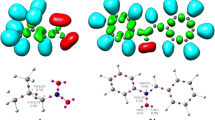
Domingo [58] introduced the local reactivity electrophilic indices \({P}_{k}^{+}\) and \({P}_{k}^{-}\) in 2013 for electrophilic and nucleophilic attacks, respectively. Parr functions are derived from the Mulliken atomic spin density (MASD) to predict the local reactivity at the reacting counterparts. Figure 3 depicts the 3D representations of the MASD of the radical cation of N-methyl-1-phenylmethanimine oxide 1•+ and the radical anion of bicyclopropylidene 2•‾, as well as the nucleophilic \({P}_{k}^{-}\) Parr functions of N-methyl-1-phenylmethanimine oxide 1•+ and the electrophilic \({P}_{k}^{+}\) Parr functions of bicyclopropylidene 2•‾. O1 oxygen of nitrone has a nucleophilic \({P}_{k}^{-}\) Parr function of 0.189, whereas C3 has a nucleophilic \({P}_{k}^{-}\) Parr function of 0.026, indicating that O1 is the nucleophilic center. Due to the symmetry of bicyclopropylidene 2, the double bond carbon atoms, C4 and C5, of bicyclopropylidene have same electrophilic \({P}_{k}^{+}\) Parr functions.

3D representations of the MASD of the radical anion of bicyclopropylidene 2 and the radical cation of N-methyl-1-phenylmethanimine oxide 1, in addition to the electrophilic \({P}_{k}^{+}\) Parr functions of bicyclopropylidene 2, and the nucleophilic \({P}_{k}^{-}\) Parr functions of nitrone 1 in gas phase
Analysis of the potential energy surface (PES) of the 32CA reaction of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2
The 32CA reaction of nitrone 1 and bicyclopropylidene 2 can take place along two feasible stereoisomeric reaction paths. Search for the stationary points along these two reaction paths allowed locating the reagents, products and only one TS along each reaction path, namely, TS-ex and TS-en leading to cycloadducts 3 and 4 respectively, implying one-step mechanism for this 32CA reaction (Scheme 2).

Studied stereoisomeric paths for the 32CA reactions of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2
The relative electronic energies of the TSs and the products in gas phase, benzene and toluene along with the relative enthalpies, entropies, and free energies in the solvents are given in Table 3 and Table 4.
The energy profile study could show some interesting things. (i)The 32CA reaction of nitrone 1 and bicyclopropylidene 2 is exergonic with negative reaction free energies in the benzene and toluene suggesting kinetic control in the generation of the cycloadducts. (ii) The 32CA reaction of N-methyl-1-phenylmethanimine oxide nitrone 1 and bicyclopropylidene 2 shows that the reaction has negative free energy with a value of −8.13 and −8.05 kcal.mol−1 in benzene and toluene, respectively, for exo path, see Table 4. However, the free energy decreases to −12.82 and −12.73 kcal.mol−1 in benzene and toluene, respectively, for endo path, proving kinetic control and, therefore, irreversibility. This is in complete agreement with the experimental outcome showing the generation of cycloadducts 3 and 4. (iii) TS-ex has an activation enthalpy of 20.02 kcal.mol−1 in benzene, which increases to 20.10 kcal.mol−1 in toluene. Similar trend was found for TS-en that is the activation enthalpy increases from 19.71 kcal.mol−1 in benzene to 19.81 kcal.mol−1 in toluene. Table 4 shows the GEDT at the TSs, which was used to figure out their orientation. The found TSs have a GEDT of 0.010 and 0.013 e, which is typical of a null electron density flux [59] (NEDF). This suggests that the 32CA reaction is non-polar.
Figure 4 shows the TS-en and TS-ex optimized geometry in the gas phase. The analysis of the geometrical data shows that the bond distance of C3-C4 is greater than for O1–C5 in the gas phase with a value of 2.278 and 2.093 Å, respectively, at TS-ex, and 2.275 and 2.108 Å at TS-en. This shows that TS-ex is slightly more asynchronous relative to TS-en. The inclusion of solvents shows minimal changes in the bond distance between C3-C4 and O1-C5.

B3LYP/6–311 + + G(d,p) optimized geometry of TSs in the gas phase (black), benzene (red), and toluene (blue)
Topological analysis of the ELF at the TSs
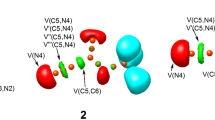
The electronic structures of optimized TSs associated with the 32CA reaction of N-methyl-1-phenylmethanimine oxide 1 with bicyclopropylidene 2 are characterized topologically. Figure 5 shows the ELF localization regions and basin attractor sites for gas phase TSs that are involved in the 32CA reaction. The ELF of TS-ex shows that there are V(O1) and V′(O1) monosynaptic basins with a total population of 5.78 e, while the ELF of TS-en shows that there are V(O1) and V′(O1) monosynaptic basins with a total population of 5.82 e related to the non-bonding electron density on O1 oxygen. V(C3, N2) and V′(C3, N2) disynaptic basins in the ELF of TS-ex and TS-en integrate a total population of 2.53 e and 2.93, respectively, associated with the C3-N2 bonding region, and the V(N2) monosynaptic basin integrates 1.29 e and 1.41 e for TS-ex and TS-en associated with the non-bonding electron density at N2 nitrogen. Note that the number of electrons in the C3–N2 bonding region has dropped from 3.82 e at N-methyl-1-phenylmethanimine oxide 1 to 2.53 e at TS-ex and 2.93 e at TS-en. This shows that the C3–N2 double bond broke at the TSs to create the non-bonding electron density at N2 nitrogen. The population in the V (O1,N2) disynaptic basin goes down from 1.39 e at N-methyl-1-phenylmethanimine oxide 1 to 1.22 e at TS-ex and 1.21 e at TS-en. So, most of the electron density in the V (N2) monosynaptic basin comes from the N2-C3 bonding region. The ELF of TS-ex and TS-en shows that there is a monosynaptic basin at V(C3) that integrates 0.39 and 0.37 e, which is related to the formation of a pseudoradical center at C3. There is one disynaptic basin in the ELF of TS-ex and TS-en that combines 2.98 e and 2.97 e from the C4–C5 bonding region. Note that the C4–C5 bonding region depopulated from 3.84 e in the bicyclopropylidene 2 to 2.98 e and 2.97 e at the TSs. This causes the pseudoradical center to form at C4, as shown by the appearance of the monosynaptic basin V (C4), which integrates 0.60 e at TS-ex and TS-en (Table 5).

B3LYP/6-311G + + (d,p) ELF localization domains and the basin attractor positions of TS-ex and TS-en in the gas phase. Protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green and the attractor positions in black color
Mechanistic implications along the stereoisomeric channels of 32CA reaction N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2 from bonding evolution theory (BET) study
Krokoidis [41] came up with the bonding evolution theory (BET) by looking at consecutive changes in bonding using the ELF topological analysis and the Thom’s catastrophe theory [40]. This gives mechanistic implications along a reaction route. For the 32CA reaction of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2, numbers of BET study have been done to figure out the bonding pattern along the exo and endo stereoisomeric routes.
Tables 6 and 7 show the ELF basin populations at the sites where N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2 react during the 32CA reaction along the stereoisomeric route in TS-ex and TS-en. From the analysis of ELF basins, seven topological phases were found for exo and endo paths.
The pattern of bonds in the ELF structure of the starting point S1 is similar as that of the individual compounds (see Table 1). At S2, 0.92 e for exo path and 0.96 e for endo path are integrated by the monosynaptic basin V (N2) associated with the N2 nitrogen lone pair. This is accomplished by obtaining electron density from the C3–N2 bonding region. At S3, pseudoradical centers form at C3 and C4, which leads to the formation of monosynaptic basins V(C3) and V(C4). Phase IV, which concern to the transition structure TS-ex and TS-en, the single bonds O1–C5 and C3–C4 have not yet been formed. In phase V, pseudoradical centers form at C5, which leads to the formation of monosynaptic basins V(C5). In phase VI, the first single bonds are formed between C3 and C4. In addition, in the same phase as IRC point S5, a pseudoradical centers form at O1 for exo path; however, this pseudoradical centers was not found for endo path. In phase VII, initial O1–C5 single bond formation begins, as shown by the development of disynaptic basin V (C3, C4). These results show that more advanced (C3–C5) and (O1–C4) bonds are likely to form along the endo reaction route.
Topological analysis of the AIM at the TSs involved in the 32CA reactions
Bader and Coworkers [42, 60] found that the electron density buildup and the Laplacian of electron density \({\nabla }^{2}\rho ({r}_{c})\) at the bond critical points (BCPs) can be used to describe the covalent and non-covalent interactions between atomic pairs connected by a bond path. At the TSs, the BCPs, CP1 and CP2, are linked to the formation of N3-C4 and N1-C5 links. Table 8 shows the total electron density,\(\rho (a.u.)\), and the Laplacian of electron density, \({\nabla }^{2}\rho ({r}_{c})\), at CP1 and CP2. In each case, the total electron density is less than 0.1 a.u. and the Laplacian of electron density is positive. This is consistent with the ELF topological analysis and the fact that the distance between forming bonds is more than 2.0 Ǻ.
Conclusion
The reaction mechanism of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2 was studied at the B3LYP/6–311 + + G(d,p) level of theory using MEDT. The ELF topological investigation of the ground state structures classified N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2 as a zwitter-ionic TAC, indicating its participation in zw-type 32CA reactions that require suitable electrophilic-nucleophilic interactions.
By comparing the electronic chemical potentials and nucleophilic strengths of N-methyl-1-phenylmethanimine oxide 1 and the bicyclopropylidene 2, the global electron flow was predicted. This prediction was subsequently validated through GEDT calculations at the TSs, indicating that the 32CA reactions possessed negative free energy and were exergonic. This is in complete agreement with the experimental outcomes by Brandi and co-workers [36].
BET analysis along the regioisomeric reaction channels allows for some significant mechanistic results. Along both reaction pathways, The C = N bonding region of N-methyl-1-phenylmethanimine oxide 1 and the double bond bonding region of the bicyclopropylidene 2 are depopulated in the first four phases to form pseudoradical centers at C3, C4, and C5 carbons and lone pair electron density at N2 nitrogen. ELF studies at the TSs further showed early TSs along the exo and endo pathways before the formation of a new single covalent bond began. These findings provide a more profound comprehension of the mechanism of this type of reaction, which is valuable for designing more efficient and selective synthetic processes in organic chemistry.
Availability of data and materials
All datasets generated and/or analyzed during this study are available from the corresponding author on reasonable request.
References
Huisgen R (1963) 1, 3‐dipolar cycloadditions. Past and future. Angewandte Chemie Int Ed English 2(10):565–598. https://doi.org/10.1002/anie.196305651
Su YL, Dong K, Zheng H, Doyle MP (2021) Generation of diazomethyl radicals by hydrogen atom abstraction and their cycloaddition with alkenes. Angew Chem 133(34):18632–18636. https://doi.org/10.1002/ange.202105472
Mohammad-Salim H, Hassan R, Abdallah HH, Oftadeh M (2020) Theoretical study on the mechanism of [3+ 2] cycloaddition reactions between α, β-unsaturated selenoaldehyde with nitrone and with nitrile oxide. J Mexican Chem Soc 64(2):147–164. https://doi.org/10.29356/jmcs.v64i2.1111
Abbiche K, Mohammad-Salim H, Salah M, Mazoir N, Zeroual A, El Alaoui El Abdallaoui H, El Hammadi A, Hilali M, AbdallahH, Hochlaf M (2020) Insights into the mechanism and regiochemistry of the 1, 3-dipolar cycloaddition reaction between benzaldehyde and diazomethane. Theor Chem Acc 139:1–12. https://doi.org/10.1007/s00214-020-02662-4
Domingo LR, Acharjee N, Mohammad-Salim HA (2020) Understanding the reactivity of trimethylsilyldiazoalkanes participating in [3+ 2] cycloaddition reactions towards diethylfumarate with a molecular electron density theory perspective. Organics 1(1):3–18. https://doi.org/10.3390/org1010002
Mohammad-Salim HA, Basheer HA, Abdallah HH, Zeroual A, Jamil LA (2021) A molecular electron density theory study for [3+ 2] cycloaddition reactions of N-benzylcyclohexylnitrone with methyl-3-butenoate. New J Chem 45(1):262–267. https://doi.org/10.1039/D0NJ04049E
Mohammad-Salim HA, Acharjee N, Abdallah HH (2021) Insights into the mechanism and regioselectivity of the [3+ 2] cycloaddition reactions of cyclic nitrone to nitrile functions with a molecular electron density theory perspective. Theoret Chem Acc 140:1–14. https://doi.org/10.1007/s00214-020-02703-y
Padwa A, Pearson WH (2003) Synthetic applications of 1, 3-dipolar cycloaddition chemistry toward heterocycles and natural products Vol. 59. John Wiley & Sons
Eicher TS (2013)Hauptmann, and A. Speicher, The chemistry of heterocycles: structures, reactions, synthesis, and applications. John Wiley & Sons
Majumdar KC, Chattopadhyay SK (2011) Heterocycles in natural product synthesis. John Wiley & Sons
Lednicer D (2007) The organic chemistry of drug synthesis, Volume 7. Vol. 7. John Wiley & Sons
Artemov A, Sazonova E, Zarovkina NY (2013) 1, 3-Dipolar cycloaddition reaction of nitrone η 6-(arene) chromium tricarbonyl complexes with styrene and η 6-(styrene) chromium tricarbonyl. Russ Chem Bull 62:1382–1387. https://doi.org/10.1007/s11172-013-0197-8
Mohammad-Salim HA (2021) Understanding the reactivity of C-cyclopropyl-N-methylnitrone participating in [3+ 2] cycloaddition reactions towards styrene with a molecular electron density theory perspective. J Mexican Chem Soc 65(1):129–140. https://doi.org/10.29356/jmcs.v65i1.1437
Mohammad‐Salim HA, Acharjee N, Domingo LR, Abdallah HH (2021) A molecular electron density theory study for [3+ 2] cycloaddition reactions of 1‐pyrroline‐1‐oxide with disubstituted acetylenes leading to bicyclic 4‐isoxazolines. Int J Quantum Chem 121(5):e26503. https://doi.org/10.1002/qua.26503
Acharjee N, Mohammad‐Salim HA, Chakraborty M, Rao MP, Ganesh M (2021) Unveiling the high regioselectivity and stereoselectivity within the synthesis of spirooxindolenitropyrrolidine: a molecular electron density theory perspective. J Phys Organ Chem 34(6):e4189. https://doi.org/10.1002/poc.4189
Krylov A, Windus TL, Barnes T, Marin-Rimoldi E, Nash JA, Pritchard B, Smith DG, Altarawy D, Saxe P, Clementi C (2018) Perspective: computational chemistry software and its advancement as illustrated through three grand challenge cases for molecular science. J Chem Phys 149(18):180901. https://doi.org/10.1063/1.5052551
Salim HM (2021) Theoretical study of the reaction of ketene with methanimine using DFT method. Zanco J Pure Appl Sci 33(4):1–10. https://doi.org/10.21271/ZJPAS.33.4.1
Mohammad-Salim HA (2021) Ab initio study of Diels-Alder reaction between cyclic dienes and olefin. Asian Journal of Applied Chemistry Research 9(3):10–17. https://doi.org/10.9734/ajacr/2021/v9i330214
Mohammad-Salim HA, Abdallah HH, Maiyelvaganan KR, Prakash M, Hochlaf M (2020) Mechanistic study of the [2+ 2] cycloaddition reaction of cyclohexenone and its derivatives with vinyl acetate. Theoret Chem Acc 139:1–11. https://doi.org/10.1007/s00214-019-2542-y
Mohammad-Salim HA, Abdallah HH (2019) Theoretical study of the [4+ 2] cycloaddition reaction of trifluoroethylene with five-membered chalcogens heterocyclic compounds. ARO- Sci J Koya Univ 7(2):69–77. https://doi.org/10.14500/aro.10575
Al-Douh MH, Selim EAB, Salim HAM, Abdullah HH (2019) Molecular structure and spectroscopic studies of some diazo dyes compounds using DFT method. in 2019 First International Conference of Intelligent Computing and Engineering (ICOICE). IEEE. https://doi.org/10.1109/ICOICE48418.2019.9035176
Mohammad Salim HA, Abdallah HH (2019) Theoretical study for the [2+ 2] cycloaddition reaction mechanism of ketenes and their derivatives. Orien J Chem 35(5):1550–1556. https://doi.org/10.13005/ojc/350512
Salim HAM, Abdallah HH, Ramasami P (2018) Stereoselectivity and regioselectivity of the cycloaddition dimerization of allyl 3-(2-pyridyl) acrylate and allyl 3-(2-pyrryl) acrylate: DFT calculations. in IOP Conference Series: Materials Science and Engineering. IOP Publishing. https://doi.org/10.1088/1757-899X/454/1/012049
Salim HM, Abdallah HH, Ramasami P (2018) Mechanism and thermodynamic parameters of paternὸ-büchi reaction of benzene and furan: DFT study. in 2018 International Conference on Advanced Science and Engineering (ICOASE). IEEE. https://doi.org/10.1109/ICOASE.2018.8548917
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21(10):1319. https://doi.org/10.3390/molecules21101319
Mohammad-Salim HA (2021) A molecular electron density theory study of the [3+ 2] cycloaddition reaction of nitronic ester with methyl acrylate. Theoret Chem Acc 140(7):83. https://doi.org/10.1007/s00214-021-02789-y
Acharjee N, Mohammad-Salim HA, Chakraborty M (2021) Unveiling [3+ 2] cycloaddition reactions of benzonitrile oxide and diphenyl diazomethane to cyclopentene and norbornene: a molecular electron density theory perspective. Theoret Chem Acc 140(8):113. https://doi.org/10.1007/s00214-021-02811-3
Acharjee N, Mohammad-Salim HA, Chakraborty M (2022) Unveiling the synthesis of spirocyclic, tricyclic, and bicyclic triazolooxazines from intramolecular [3+ 2] azide-alkyne cycloadditions with a molecular electron density theory perspective. Struct Chem 33(2):555–570. https://doi.org/10.1007/s11224-021-01870-3
Mohammed Salih SA, Basheer HA, Mohammad-Salim HA (2022) Insights into the mechanism and stereoselectivity of the [3+ 2] cycloaddition reaction between N-methyl-C-(4-hydroxylphenyl) nitrone and maleic anhydride with a molecular electron density theory perspective. Theor Chem Acc 141(7):33. https://doi.org/10.1007/s00214-022-02891-9
Mohammed Salih SA, Basheer HA, Mohammad-Salim HA (2022) Unveiling [3+ 2] cycloaddition reactions of N-methyl-C-3-bromophenyl-nitrone to dimethyl maleate: molecular electron density theory perspective. J Mexican Chem Soc 66(4)560–574. https://doi.org/10.29356/jmcs.v66i4.1801
Mondal A, Mohammad-Salim HA, Acharjee N (2023) Unveiling substituent effects in [3+ 2] cycloaddition reactions of benzonitrile N-oxide and benzylideneanilines from the molecular electron density theory perspective. Sci Rad 2:75–92. https://doi.org/10.58332/scirad2023v2i1a05
Rıos-Gutiérrez M, Domingo L (2019) Unravelling the mysteries of the [3? 2] cycloaddition reactions. Eur J Org Chem 267. https://doi.org/10.1002/ejoc.201800916
Ríos-Gutiérrez M, Darù A, Tejero T, Domingo LR, Merino P (2017) A molecular electron density theory study of the [3+ 2] cycloaddition reaction of nitrones with ketenes. Org Biomol Chem 15(7):1618–1627. https://doi.org/10.1039/C6OB02768G
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the reactivity and selectivities in [3+ 2] cycloaddition reactions of C, N-dialkyl nitrones with ethylene derivatives. J Org Chem 83(4):2182–2197. https://doi.org/10.1021/acs.joc.7b03093
Domingo LR, Ríos-Gutiérrez M, Acharjee N (2019) A molecular electron density theory study of the chemoselectivity, regioselectivity, and diastereofacial selectivity in the synthesis of an anticancer spiroisoxazoline derived from α-santonin. Molecules 24(5):832. https://doi.org/10.3390/molecules24050832
Meijere A (1994) Nitrone and nitrile oxide cycloadditions to bicyclopropylidene. Rearrangement of the isoxazolidine adducts to 3-spirocyclopropane-4-pyridone derivatives. J Chem Soc Chem Comm 1994(18):2185–2186. https://doi.org/10.1039/C39940002185
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21(6):748. https://doi.org/10.3390/molecules21060748
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103(5):1793–1874. https://doi.org/10.1021/cr990029p
Domingo LR (2014) A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4(61):32415–32428. https://doi.org/10.1039/C4RA04280H
Thom R (1977) Stabilité structurelle et morphogénèse: essai d'une théorie générale des modèles. InterÉditions
Krokidis X, Silvi B, Alikhani M (1998) Topological characterization of the isomerization mechanisms in XNO (X= H, Cl). Chem Phys Lett 292(1–2):35–45. https://doi.org/10.1016/S0009-2614(98)00650-2
Bader RF (1985) Atoms in molecules. Acc Chem Res 18(1):9–15
Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J Comput Chem 3(2):214–218. https://doi.org/10.1002/jcc.540030212
Wiberg KB (1986) Ab initio molecular orbital theory by WJ Hehre, L. Radom, P. v. R. Schleyer, and JA Pople, John Wiley, New York, 548pp. Price: $79.95 (1986). Wiley Online Library
El Idrissi M, El Ghozlani M, Eşme A, Ríos-Gutiérrez M, Ouled Aitouna A, Salah M, El Abdallaoui HEA, Zeroual A, Mazoir N, Domingo LR (2021) Mpro-SARS-CoV-2 inhibitors and various chemical reactivity of 1-bromo-and 1-chloro-4-vinylbenzene in [3+ 2] cycloaddition reactions. Organics 2(1):1–16. https://doi.org/10.3390/org2010001
Domingo LR, Ríos-Gutiérrez M, Chamorro E, Pérez P (2019) Are one-step aromatic nucleophilic substitutions of non-activated benzenes concerted processes? Org Biomol Chem 17(35):8185–8193. https://doi.org/10.1039/C9OB01589B
Hratchian HP, Schlegel HB (2004) Accurate reaction paths using a Hessian based predictor–corrector integrator. J Chem Phys 120(21):9918–9924. https://doi.org/10.1063/1.1724823
Hratchian H, Schlegel H (2005) Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J Chem Theory Comput 1(1):61–69. https://doi.org/10.1021/ct0499783
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94(7):2027–2094
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107(8):3032–3041. https://doi.org/10.1063/1.474659
Barone V, Cossi M, Tomasi J (1998) Geometry optimization of molecular structures in solution by the polarizable continuum model. J Comput Chem 19(4):404–417. https://doi.org/10.1002/(SICI)1096-987X(199803)19:4%3C404::AID-JCC3%3E3.0.CO;2-W
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592. https://doi.org/10.1002/jcc.22885
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Frisch M, Trucks G, Schlegel HB, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Petersson G, Nakatsuji H (2016) Gaussian 16, Gaussian, Inc. Wallingford, CT
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371(6499):683–686. https://doi.org/10.1038/371683a0
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92(9):5397–5403. https://doi.org/10.1063/1.458517
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58(22):4417–4423. https://doi.org/10.1016/S0040-4020(02)00410-6
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3(5):1486–1494. https://doi.org/10.1039/C2RA22886F
Domingo LR, Kula K, Ríos-Gutiérrez M (2020) Unveiling the reactivity of cyclic azomethine ylides in [3+ 2] cycloaddition reactions within the molecular electron density theory. Eur J Org Chem 2020(37):5938–5948. https://doi.org/10.1002/ejoc.202000745
Bader RF, Essén H (1984) The characterization of atomic interactions. J Chem Phys 80(5):1943–1960. https://doi.org/10.1063/1.446956
Author information
Authors and Affiliations
Contributions
HMS and JVJO performed the calculations, and planned the methodology, while HMS wrote the manuscript. All authors discussed and contributed to reviewing of the manuscript to its final version.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohammad-Salim, H.A., de Julián-Ortiz, J.V. Theoretical insight into the mechanism and selectivity of the [3 + 2] cycloaddition reaction of N-methyl-1-phenylmethanimine oxide and bicyclopropylidene from the MEDT perspective. Struct Chem 35, 541–551 (2024). https://doi.org/10.1007/s11224-023-02206-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-023-02206-z