
Abstract
The [3+2] cycloaddition (32CA) reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 has been investigated using molecular electron density theory (MEDT) at the MPWB95/6-311++G(d,p) computational level. This 32CA reaction undergoes two stereo- and stereoisomeric reaction paths to form two different products 3 and 4. An electron localization function (ELF) study predicts that the N-methyl-C-(4-hydroxylphenyl) nitrone 1 has a zwitterionic character and it takes place through a one-step mechanism, with activation enthalpies in between 17.48 and 23.41 kJ mol−1 in the gas phase. The CDFT indices are used to forecast the global electron density flux from the strong nucleophilic N-methyl-C-(4-hydroxylphenyl) nitrone 1 to the electrophilic maleic anhydride 2. These exergonic 32CA reactions have negative Gibbs free energy along the endo and exo stereochemical routes. The endo stereochemical process is favored over the exo stereochemical pathway due to the increased thermodynamic stability of the cycloadduct. Bonding evolution theory (BET) predictions for the endo and exo routes indicate a one-step process with early transition states, which is consistent with the ELF topological investigation at the transition states.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The study of heterocycle synthesis is quite common in the area of organic chemistry and necessitates a specific synthetic approach. Heterocycles are important parts of the process of making important biological molecules, such as vitamin B6 [1]. The [3+2] cycloaddition (32CA) reaction is a very common class of reactions that have been the subject of various research, most notably by Huisgen, who did a thorough analysis of the probable interactions between dipole and dipolarophile, which enabled a better understanding of these reactions [2, 3]. The 32CA reactions were identified a few years after the structure of diazoacetic acid esters was established by the reaction of diazomethane with acrylic esters [3]. They have been the subject of multiple reviews since research in this sector is quite interesting. The 32CA reactions, in particular, are an effective way of producing 5-membered heterocycles [4,5,6,7,8]. In recent decades, the function of heterocyclic compounds has grown in importance, resulting in an astounding number of new classes of compounds with at least one heterocycle in their structure. Heterocycles are significant not only for their richness and exceptional variety, but also for their use in biological, medical, and therapeutic disciplines (vitamins, hormones, antibiotics, and so on), as well as in the industrial and technical sectors (for example, corrosion inhibitors, dyes, stabilizing agents, insecticides, herbicides, etc.) [9,10,11,12]. Although the utility of 32CA reactions in organic synthesis is no longer debatable, research in this area is focused on improving the chemical reactions to produce regulated regio- and stereochemical products. To accomplish this, it seems that the use of a chiral Lewis acid catalyst is the most efficient and cost-effective technique for lowering the activation energy of a particular 32CA reaction in order to generate the primary product at a high yield. Numerous experimental and theoretical studies on this subject are available in the literature [13,14,15].
Computational chemistry has become an important tool for analyzing experimentally reported reactivity and selectivity results by building a detailed picture of how chemical events work over the last 2 decades [16]. Despite the growing number of modern applications of computational science in chemistry, the fundamental theories of organic chemistry had remained stagnant for the last 40 years, until Domingo proposed the Molecular Electron Density Theory (MEDT) in 2016, recognizing the critical role of electron density changes in molecular reactivity [17,18,19]. MEDT has successfully analyzed the experimental results of many 32CA reactions during the previous 4 years [5, 15, 18, 20,21,22,23]. MEDT has recently been used to analyze the experimental results of strain-promoted and catalyzed 32CA reactions and the reported chemo-, regio-, and stereoselective production of spiroisoxazolines [5, 15, 22, 24,25,26,27]. The three-atom components (TACs) that play a role in the 32CA reactions are classified into pseudodiradical, pseudo(mono)radical, carbenoid, and zwitterionic types based on their electronic structure. This allows them to participate in the pdr-type, pmr-type, cb-type, and zw-types of the 32CA reaction [18, 20]. 32CA reactions with pdr-type 32CA have a lower energy barrier and are easier to do than 32CA reactions with zw-type 32CA, which have a higher energy barrier that needs to be broken down by electrophilic–nucleophilic contact [26].
The MEDT study is presented in five Sects. 3.1 to 3.5, in this research work.
(I) in Sect. 3.1, the electron localization function (ELF) at the ground state structures of reagents N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 is topologically analyzed so that they can be represented in terms of their electronic structure and subsequently evaluated for their reactivity in 32CA reactions [28, 29]. (II) In Sect. 3.2, reactivity indices described within the conceptual density functional theory (CDFT) are examined in order to understand the polarity of the 32CA reactions [30, 31]. (III) In Sect. 3.3., the potential energy surfaces (PES) along the possible stereoisomeric channels of the 32CA reactions are investigated in predicting the energy profiles, and the global electron density transfer (GEDT) at the transition states (TSs) is determined [32, 33]. (IV) Sect. 3.4 analyzes the ELF of the discovered TSs. (V) In Sect. 3.5, the combination of ELF and Thom’s disaster, as well as Krokidis’ proposed bonding evolution theory (BET), is used to structure the process for electron density changes along stereoisomeric routes [28, 29, 34, 35].
2 Computational methods
Berny analytical gradient optimization was used to optimize the stationary points along the potential energy surface of the 32CA reactions at the MPWB95/6-311++G (d,p) level [36, 37]. The use of the MPWB95 functional as a reliable and accurate approach has been shown in the investigation of various recent 32CA reactions [6, 7, 38].
Calculations of the imaginary frequencies at the optimal TSs revealed the existence of one imaginary frequency, whereas the absence of imaginary frequencies was proven for the local minimum. Calculations of the intrinsic reaction coordinate (IRC) using the Gonzalez–Schlegel integration technique were used to validate the lowest energy reaction pathway between reactants and products through the identified TSs [39, 40].
The effects of the solvent on toluene were investigated using a polarizable continuum model (PCM) and the self-consistent reaction field (SCRF) method [41,42,43,44,45]. The CDFT indies are determined using the equation discussed in Reference [30, 31]. At the TSs of each responding framework, natural population analysis (NPA) was used to come up with the global electron density theory (GEDT):
where q signifies atomic charges, the total of charges on all atoms in the studied framework indicates the GEDT, and the positive sign of GEDT indicates global electronic flux from that framework to another [33].
Multiwfn software [46] is used to perform the topological analysis of the ELF at the reagents, TSs, and IRC sites, as well as the computation of the surfaces are shown using UCSF Chimera software [47]. All computations were performed using the Gaussian 16 software [48].
3 Results and discussion
3.1 Analysis of the ELF topology of the N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2
A good connection has been established between the electronic structure of three atom compounds (TACs) and their reactivity in 32CA reactions by using the MEDT theory [17, 18, 20].
The ELF of the ground state structures of chemicals 1 and 2 is investigated in this study to characterize their electronic structures and reactivity in 32CA reactions. The ELF valence basin populations of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 are listed in Table 1, along with their ELF localization.
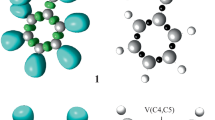
The domains are shown in Fig. 1. The Lewis structures of the reagents are provided based on their ELF valence populations and are shown in Fig. 2.

MPWB95/6-311++G(d,p) ELF localization domains represented at an isosurface value of ELF = 0.82 of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2. Green colored ones are the disynaptic basins, blue color represents the protonated basins, red color is used to represent the monosynaptic basins. The attractor positions are represented as black spheres

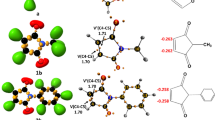
MPWB95/6-311++G(d,p) calculated natural atomic charges, in average number of electrons e, of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2. Positive charges are colored in blue and negative charges in red
The ELF of N-methyl-C-(4-hydroxylphenyl) nitrone 1 includes two monosynaptic basins, V (O1) and Vʹ (O1), that integrate 5.91 e, two disynaptic basin, V (C3, N2) and Vʹ (N2, C3) that integrate 3.75 e, and one disynaptic basin, V (N2, O1), that integrates 1.48 e, which corresponds to the non-bonding electron density on O1 oxygen, the underpopulated C3–N2 double bond, and the underpopulated N2–O1 single bond, respectively.
The ELF of maleic anhydride 2 reveals two disynaptic basins for the C4–C5 bonding region, with a total integrating population of 3.28 e, corresponds to the underpopulated C4–C5 double bond.
After analyzing the reagents’ bonding patterns, the atomic charge distributions of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 were determined by NPA (Fig. 2) [49, 50]. Oxygen atom O1 of N-methyl-C-(4-hydroxylphenyl) nitrone 1 is negatively charged (-0.565 e), and N2 nitrogen is positively charged (0.093 e), while C3 carbon atom shows charge of 0.021 e; this shows charge separation in the nitrone, although it is not consistent with the charges predicted by Lewis’s bonding model. Although the N-methyl-C-(4-hydroxylphenyl) nitrone 1 is classed as a zwitterionic TAC based on ELF analysis, this term does not refer to the nitrones’ dipolar electronic structure. Instead of that, it denotes the specific bonding pattern (in the absence of charges) of Huisgen’s resonance Lewis structure for "1,3-dipoles" [51]. Carbon atoms C4 and C5 in maleic anhydride 2 have negative charge values of − 0.239 and − 0.239, respectively.
3.2 Analysis of the CDFT indices of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2
The concept of "Conceptual DFT," which began with Parr’s pioneering work, has been used in a number of investigations to determine the chemical reactivity of structures involved in 32CA reactions [30]. The CDFT indices, which are specified inside the conceptual DFT, have a well-established literature and [31, 52,53,54,55] give a first understanding of molecular reactivity by addressing the chemical behavior of the reactants [31]. Domingo developed standard scales for nucleophilicity and electrophilicity indices at the B3LYP/6-31G(d) level, which was used for the CDFT study in this paper [56, 57]. Consequently, the CDFT indices, electronic chemical potential, μ, chemical hardness, ɳ, electrophilicity, ω, and nucleophilicity, N, at the ground state of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 are listed in Table 2 [58,59,60,61].
The electronic chemical potential of N-methyl-C-(4-hydroxylphenyl) nitrone 1 = − 3.61 eV is greater than that of maleic anhydride 2 = − 6.25 eV (1), suggesting that during the 32CA reaction, the electron density will flux from N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 [31, 58].
The electrophilicity ω index and the nucleophilicity N index of N-methyl-C-(4-hydroxylphenyl) nitrone 1 are 1.08 and 2.88 eV, respectively; according to the scales used, it is a moderate electrophile and a strong nucleophile, respectively. [56, 57, 60, 61]. The maleic anhydride 2 with electrophilicty indices ω = 2.61 eV is classified as strong electrophiles and with nucleophilicity indices N = 0.50 eV as weak nucleophiles. Consequently, maleic anhydride 2 will behave as an electrophile in these zw-type 32CA reactions, whereas N-methyl-C-(4-hydroxylphenyl) nitrone 1 will behave as a nucleophile, in accordance with their electronic chemical potentials μ.
3.3 Analysis of the energy profile associated with the 32CA reactions of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2
The analysis of the energy profile may provide some interesting results.
(i) The 32CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 shows that the reaction has negative free energy ranging from − 29.92 to − 58.79 kJ mol−1, confirming kinetic control and hence irreversibility.
(ii) The activation enthalpy of TS-ex is 5.12, 7.66, 1.42, 10.64, and 10.99 kJ mol−1 lower than that of TS-en in the gas phase, toluene, benzene, THF, and DCM, respectively [62] (Scheme 1).

Studied stereoisomeric paths for the 32CA reactions of N-methyl-C-(4-hydroxylphenyl) nitrone 1 with maleic anhydride 2
(iii) TS-ex has an activation enthalpy of 17.48 kJ mol−1 in the gas phase, which increases to 28.19 kJ mol−1 in DCM, 27.84 kJ mol−1 in THF, 23.33 kJ mol−1 in benzene, and 23.59 kJ mol−1 in toluene, indicating a 10.71 kJ mol−1 increase from gas phase to DCM. Thus, the reaction is energetically possible in low-polar solvents.
The GEDT at the TSs was determined to determine their polarity and is presented in Table 3. The found TSs have a GEDT between 0.18 and 0.35 e, which is typical of a forward electron density flux [63] (FEDF), suggesting the 32CA reaction is polar.
Figure 3 shows the gas phase geometries of TS-ex and TS-en. At TS-ex, the distance between C3 and C4 interacting centers is 0.599 Å times greater than the distance between C5 and O1 interacting centers, whereas in TS-en, the distance between the C3 and C4 interacting centers is 0.317 more than the distance between the C5 and O1 interacting centers, showing that TS-ex has a greater degree of asynchronicity than TS-en. The inclusion of the solvents has no effect on the distance between the C3 and C4 interacting centers, which is 2.369 Å in TS-ex and 2.173 Å in TS-en, and also the distance between the C5 and O1 interacting centers, which is 1.770 Å in TS-ex and 1.856 Å in TS-en.

MPWB95/6-311++G(d,p) optimized gas phase TSs
3.4 Topological analysis of the ELF at the TSs
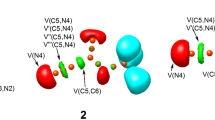
The topological examination of the ELFs at the TSs enables the determination of their electronic structure and the extent to which bonds are formed. The ELF localization domains and basin attractor sites for gas phase TSs associated with the 32CA reaction are given in Fig. 4. The ELF of TS-ex indicates the presence of V(O1) and V′(O1) monosynaptic basins with a total population of 5.76 e, while the ELF of TS-en shows the presence of V(O1) and V′(O1) with a total population of 5.73 e related with the non-bonding electron density on O1 oxygen. The ELF of TS-ex and TS-en shows the presence of V(C3, N2) and V′(C3, N2) disynaptic basins integrating a total population of 5.16 e associated with the C3–N2 bonding region, as well as the V(N2) monosynaptic basin integrating 1.45 e and 1.36 e associated with the non-bonding electron density at N2 nitrogen at TS-ex and TS-en, respectively. Note that the C3–N2 bonding region has been depopulated from 3.75 e at N-methyl-C-(4-hydroxylphenyl) nitrone 1 to 2.63 and 2.53 e at TS-ex and TS-en, respectively, indicating the rupture of the C3–N2 double bond at the TSs to create the non-bonding electron density at N2 nitrogen. The V (N2, O1) disynaptic basin depopulates from 1.48 e at N-methyl-C-(4-hydroxylphenyl) nitrone 1 to 1.20 and 1.23 e, respectively, at TS-ex and TS-en. Thus, the electron density in the V (N2) monosynaptic basin is mostly derived from the C3–N2 bonding region. The ELF of TS-ex and TS-en demonstrates the existence of a V (C3) monosynaptic basin integrating 0.39 and 0.23 e, respectively, which is related to the creation of a pseudoradical center at C3. There is one disynaptic basin integrating 2.65 e and 2.77 e in the ELF of TS-ex and TS-en that are related to the C4–C5 bonding region. Note, the C4–C5 bonding region depopulates from 3.28 e in maleic anhydride 2 to 2.65 e and 2.77 e at the TSs, resulting in the formation of the pseudoradical center at C4, as shown by the appearance of the monosynaptic basin V (C4) integrating 0.13 e and 0.40 e at TS-ex and TS-en, respectively (Table 4).

MPWB95/6-311G++(d,p) ELF localization domains and the basin attractor positions of gas phase TSs TS-ex and TS-en. Protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green and the attractor positions in magenta color (Iso value = 0.83)
3.5 Mechanistic implications along the stereoisomeric channels of 32CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 from bonding evolution theory (BET) study
Krokoidis developed the bonding evolution theory (BET) by examining consecutive bonding alterations using the ELF topological analysis and the Thoms catastrophe theory, gives mechanistic implications along a reaction route [28, 29, 34, 35]. For the 32CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2, comprehensive BET research has been conducted to determine the bonding pattern along the exo and endo stereoisomeric routes.
Table 5 shows the ELF basin populations at the reacting centers for the 32CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 along the stereoisomeric route in TS-ex. The study of ELF basins enables the identification of six topological phases: I, II, III, IV, V, and VI, denoted by the beginning points P1-I, P2-I, P3-I, P4-I, p5-I and P6-I, respectively (See Table 5).
The ELF structure of the starting point P1 exhibits a pattern of bonding that is identical to that of the specific compounds (see Table 1). At P2-I, (d (C3–C4) = 2.485 Å, d (O1–C5) = 1.918 Å, the monosynaptic basin V (N2) associated with the N2 nitrogen lone pair integrates 1.03 e. This is accomplished by obtaining electron density from the C3–N2 bonding region, which has depopulated from 3.75 e at P1-I to 2.88 e at P2-I. Phase III is defined by the establishment of pseudoradical centers at C3 and C5 at P3-I, which results in the production of monosynaptic basins V(C3) and V(C5) integrating 0.39 e and 0.13 e, respectively. Note that the C3–N2 bonding region depopulates from 2.88 to 2.63 e at P2-I, and the C4–C5 bonding region depopulates from 3.12 to 2.65 e at P3-I. TS-ex in found in Phase III transition structure. Note that the formation of the O1–C5 and C3–C4 single bonds has not yet commenced at the TS-ex. In phase IV, initial C3–C4 single bond formation begins, as shown by the development of disynaptic basin V (C3, C4) integrating 1.12 e, at the C3–C4 distance of 2.236 Å, as shown by the development of disynaptic basin V (C3, C4). Also, in the same phase as characterized by IRC point P4-I, the second O1–C5 single bond formation occurs at the O1–C5 distance of 1.636 Å, as shown by the development of disynaptic basin V (O1, C5) integrating 1.00 e. Finally, the molecular geometry is relaxed at O1–C5 and C3–C4 distances of 1.418 and 1.572 Å, respectively, in the cycloadduct 3 (Scheme 2).

Sequential bonding changes and most significant valence basin populations in average number of electrons e at the representative IRC points along the exo reaction path of the 32 CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2
Scheme 3 shows the ELF basin populations at the reacting centers for the 32CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 along the endo stereoselective path, as well as the sequential bonding modifications. The identification of catastrophes using ELF basin analysis enables the characterization of six topological phases, I, II, III, IV, V, and VI, as indicated by the beginning points P1-II, P2-II, P3-II, P4-II, P5-II, and P6-II, respectively (Scheme 3). The ELF topology of the starting point P1-II, like the individual reagents, has the same bonding pattern as the individual reagents (see Table 1). (d(C3–C5) = 2.571 Å, d(O1–C4) = 2.243 Å is derived from the C3–N2 bonding region at P2-II, demonstrating a depopulation from 3.67 e at P1-II to 3.66 e at P2-II. Phase P3-II is defined by the formation of monosynaptic basins V(C3), V(C4), and V(N2) integrating 0.21 e, 0.40 e, and 1.37 e, respectively, whereas phases III are defined by the formation of pseudoradical centers at C3, C4, and N2 nitrogen. It should be noted that the C4–C5 bonding region has been depopulated from 3.18 e at P2-II to 2.76 e at P3-II, while the C3–N2 bonding region has been depopulated from 2.48 e at P3-II to 2.08 e at P4-II.

Sequential bonding changes and most significant valence basin populations in average number of electrons e at the representative IRC points along the endo reaction path of the 32 CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2
The initial O1–C4 single bond formation starts in phase IV, defined by the IRC point P4-II, at the O1–C4 distance of 1.613 Å, as shown by the establishment of disynaptic basin V (O1, C4) integrating 0.89 e. Note that at the TS.
Finally, in phase V, at P5-II, the formation of the second C3–C5 single bond began at a C3–C5 distance of 1.632 Å, as shown by the development of disynaptic basin V (C3, C5) integrating 1.71 eV. In the cycloadduct 4, the molecular geometry is completely relaxed at O1–C4 and C3–C5 distances of 1.413 and 1.540 Å, respectively. It is important to note that the creation of the second C3–C5 starts when the total integrating population of disynaptic basin V (O1, C4) reaches 1.20 e. As a result, the endo reaction route is projected to have a one-step, two-stage mechanism.
A comparison of the BET values for the endo and exo reaction paths for the 32CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 enables the conclusion of many significant findings. The first phase of the exo reaction path PI results in the creation of the lone pair electron density at N2 nitrogen, as well as the pseudoradical centers at C3 and C5, which are required for the creation of the new (C3–C4) and (O1–C5) bonds. At the beginning locations, the total integrating population of the V(C3) and V(C5) basins is 0.39 e and 0.13 e, respectively, while the populations along the endo reaction routes PII of V(C3) and V(C4) basins are 0.21 e and 0.40 e, respectively. These findings point to the development of more advanced (C3–C5) and (O1–C4) bonds at the endo reaction route.
As a result, GEDT at P2-I (0.20 e) and P3-I (0.19 e) associated with the exo reaction path increased compared to P2-II (0.14 e) and P3-II (0.16 e) associated with the endo reaction path (Table 6).
4 Conclusion
The 32CA reactions of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 were investigated using MEDT at the MPWB95/6-311++G(d,p) level of theory. The ELF topological study of N-methyl-C-(4-hydroxylphenyl) nitrone 1 clearly shows that this TAC participates in zw-type 32CA. The 32CA reactions of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 take occur in a one-step mechanism. The global electronic flux from N-methyl-C-(4-hydroxylphenyl) nitrone 1 to maleic anhydride 2 is predicted, since N-methyl-C-(4-hydroxylphenyl) nitrone 1 has higher electronic chemical potential and stronger nucleophilicity compared to maleic anhydride 2. These exergonic 32CA reactions have negative Gibbs free energy along the both stereochemical routes.
The activation enthalpy for the 32CA reaction leading to the exo cycloadduct 3 is lower than that along the endo reaction path. The geometrical characteristics show that at all TSs, the production of C–O or C–C single bonds has not yet began.
The BET study of the 32CA reaction between N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 that results in the synthesis of 3 reveals the creation of non-bonding N2 electron density region at the begin of the reaction path with a GEDT of 0.09 e. As a result, a rise in GEDT results in a decrease in the energy cost along the reaction routes. The current MEDT analysis enables us to conclude that the increased acceleration for the exo stereoselective zw-type 32CA reaction of N-methyl-C-(4-hydroxylphenyl) nitrone 1 and maleic anhydride 2 passing through TS-ex is due to the high polar character of this 32CA reaction of FEDF.
References
Huisgen R (1963) 1, 3-dipolar cycloadditions. past and future. Angew Chem Int Ed Engl 2(10):565–598
Curtius T (1883) Theodor Curtius: Ueber die. Ber Dtsch Chem Ges 16:2230–2231
Su YL et al (2021) Generation of diazomethyl radicals by hydrogen atom abstraction and their cycloaddition with alkenes. Angew Chem 133(34):18632–18636
Mohammad-Salim HA (2021) Understanding the reactivity of C-cyclopropyl-N-methylnitrone participating in [3+ 2] cycloaddition reactions towards styrene with a molecular electron density theory perspective. J Mex Chem Soc 65(1):129–140
Mohammad-Salim H et al (2020) theoretical study on the mechanism of [3+ 2] cycloaddition reactions between α, β-unsaturated selenoaldehyde with nitrone and with nitrile oxide. J Mex Chem Soc 64(2):147–164
Acharjee N, Mohammad-Salim HA, Chakraborty M (2022) Unveiling the synthesis of spirocyclic, tricyclic, and bicyclic triazolooxazines from intramolecular [3+ 2] azide-alkyne cycloadditions with a molecular electron density theory perspective. Struct Chem 33:1–16
Acharjee N, Mohammad-Salim HA, Chakraborty M (2021) Unveiling [3+ 2] cycloaddition reactions of benzonitrile oxide and diphenyl diazomethane to cyclopentene and norbornene: a molecular electron density theory perspective. Theor Chem Acc 140(8):1–15
Acharjee N, Mohammad-Salim HA, Chakraborty M (2021) Unveiling the regioselective synthesis of antiviral 5-isoxazol-5-yl-2ʹ deoxyuridines with a molecular electron density theory perspective. J Serb Chem Soc 00:106–106
Padwa A, Pearson WH (2003) Synthetic applications of 1, 3-dipolar cycloaddition chemistry toward heterocycles and natural products, vol 59. John Wiley & Sons, Hoboken, NJ
Eicher T, Hauptmann S, Speicher A (2013) The chemistry of heterocycles: structures, reactions, synthesis, and applications. John Wiley & Sons, Hoboken, NJ
Majumdar KC, Chattopadhyay SK (2011) Heterocycles in natural product synthesis. John Wiley & Sons, Hoboken, NJ
Lednicer D, Mitscher LA (1980) The organic chemistry of drug synthesis, volume 2, vol 1. John Wiley & Sons, Hoboken, NJ
Artemov A, Sazonova E, Zarovkina NY (2013) 1, 3-Dipolar cycloaddition reaction of nitrone η6-(arene) chromium tricarbonyl complexes with styrene and η6-(styrene) chromium tricarbonyl. Russ Chem Bull 62(6):1382–1387
Mohammad-Salim HA et al (2021) A molecular electron density theory study for [3+ 2] cycloaddition reactions of N-benzylcyclohexylnitrone with methyl-3-butenoate. New J Chem 45(1):262–267
Mohammad-Salim HA (2021) A molecular electron density theory study of the [3 + 2] cycloaddition reaction of nitronic ester with methyl acrylate. Theor Chem Acc 140(7):1–9
Krylov A et al (2018) Perspective: Computational chemistry software and its advancement as illustrated through three grand challenge cases for molecular science. J Chem Phys 149(18):180901
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21(10):1319
Rıos-Gutiérrez M, Domingo L (2019) Unravelling the mysteries of the [3+ 2] cycloaddition reactions. Eur J Org Chem 2019(2–3):267–282
Ríos-Gutiérrez M et al (2017) A molecular electron density theory study of the [3+ 2] cycloaddition reaction of nitrones with ketenes. Org Biomol Chem 15(7):1618–1627
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of C, N-dialkyl nitrones with ethylene derivatives. J Org Chem 83(4):2182–2197
Domingo LR, Ríos-Gutiérrez M, Pérez P (2019) Unveiling the high reactivity of cyclohexynes in [3 + 2] cycloaddition reactions through the molecular electron density theory. Org Biomol Chem 17(3):498–508
Domingo LR, Acharjee N (2020) Unravelling the strain-promoted [3+ 2] cycloaddition reactions of phenyl azide with cycloalkynes from the molecular electron density theory perspective. New J Chem 44(32):13633–13643
Domingo LR, Kula K, Ríos-Gutiérrez M (2020) Unveiling the reactivity of cyclic azomethine ylides in [3 + 2] cycloaddition reactions within the molecular electron density theory. Eur J Org Chem 2020(37):5938–5948
Domingo LR, Acharjee N (2020) Unveiling the high reactivity of strained dibenzocyclooctyne in [3+ 2] cycloaddition reactions with diazoalkanes through the molecular electron density theory. J Phys Org Chem 33(11):e4100
Domingo LR, Acharjee N (2020) A molecular electron density theory study of the Grignard reagent-mediated regioselective direct synthesis of 1, 5-disubstituted-1, 2, 3-triazoles. J Phys Org Chem 33(8):e4062
Domingo LR, Ríos-Gutiérrez M, Acharjee N (2019) A molecular electron density theory study of the chemoselectivity, regioselectivity, and diastereofacial selectivity in the synthesis of an anticancer spiroisoxazoline derived from α-santonin. Molecules 24(5):832
Abbiche K et al (2020) Insights into the mechanism and regiochemistry of the 1, 3-dipolar cycloaddition reaction between benzaldehyde and diazomethane. Theor Chem Acc 139(9):1–12
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92(9):5397–5403
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371(6499):683–686
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103(5):1793–1874
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21(6):748
Sutcliffe BT (2006) The idea of a potential energy surface. Mol Phys 104(5–7):715–722
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4(61):32415–32428
Thom R (1974) “Stabilité Structurelle et Morphogenèse (Interéditions) 1972” THOM, R., Modèles Mathématiques de la Morphogenèse (Editions 10-18)
Krokidis X, Silvi B, Alikhani M (1998) Topological characterization of the isomerization mechanisms in XNO (X = H, Cl). Chem Phys Lett 292(1–2):35–45
Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J Comput Chem 3(2):214–218
Wiberg KB (1986) Ab initio molecular orbital theory by WJ Hehre, L. Radom, P. v. R. Schleyer, and JA Pople, John Wiley, New York, 548pp. Price: $79.95 (1986). Wiley Online Library, Hoboken, NJ
Acharjee N et al (2021) Unveiling the high regioselectivity and stereoselectivity within the synthesis of spirooxindolenitropyrrolidine: a molecular electron density theory perspective. J Phys Org Chem 34(6):e4189
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74(23):4161–4163
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94(14):5523–5527
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94(7):2027–2094
Simkin BI, Sheĭkhet II (1995) Quantum chemical and statistical theory of solutions: a computational approach. Ellis Horwood, Chichester
Cossi M et al (1996) Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem Phys Lett 255(4–6):327–335
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107(8):3032–3041
Barone V, Cossi M, Tomasi J (1998) Geometry optimization of molecular structures in solution by the polarizable continuum model. J Comput Chem 19(4):404–417
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592
Pettersen EF et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Frisch M et al (2016) Gaussian 16 revision a. 03. 2016. Gaussian Inc., Wallingford, CT
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83(2):735–746
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem Rev 88(6):899–926
Huisgen R (1976) 1, 3-Dipolar cycloadditions. 76. Concerted nature of 1, 3-dipolar cycloadditions and the question of diradical intermediates. J Org Chem 41(3):403–419
Parr RG, Yang W (1995) Density-functional theory of the electronic structure of molecules. Annu Rev Phys Chem 46(1):701–728
Domingo LR et al (2016) Understanding the carbenoid-type reactivity of nitrile ylides in [3 + 2] cycloaddition reactions towards electron-deficient ethylenes: a molecular electron density theory study. Theor Chem Acc 135(7):1–12
Domingo LR, Acharjee N (2018) [3+ 2] Cycloaddition reaction of C-phenyl-N-methyl nitrone to acyclic-olefin-bearing electron-donating substituent: a molecular electron density theory study. ChemistrySelect 3(28):8373–8380
Acharjee N, Banerji A (2020) A molecular electron density theory study to understand the interplay of theory and experiment in nitrone-enone cycloaddition. J Chem Sci 132(1):1–11
Domingo LR et al (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58(22):4417–4423
Jaramillo P et al (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct (Thoechem) 865(1–3):68–72
Parr RG, Weitao Y (1994) Aspects of atoms and molecules. In: Parr RG, Weitao Y (eds) Density-functional theory of atoms and molecules. Oxford University Press, Oxford
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105(26):7512–7516
Parr R, Szentpaly L, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J Org Chem 73(12):4615–4624
Lee Y-S, Park SM, Kim BH (2009) Synthesis of 5-isoxazol-5-yl-2′-deoxyuridines exhibiting antiviral activity against HSV and several RNA viruses. Bioorg Med Chem Lett 19(4):1126–1128
Domingo LR, Ríos-Gutiérrez M, Pérez P (2020) Unveiling the Lewis acid catalyzed Diels-Alder reactions through the molecular electron density theory. Molecules 25(11):2535
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohammed Salih, S.A., Basheer, H.A. & Mohammad-Salim, H.A. Insights into the mechanism and stereoselectivity of the [3+2] cycloaddition reaction between N-methyl-C-(4-hydroxylphenyl) nitrone and maleic anhydride with a molecular electron density theory perspective. Theor Chem Acc 141, 33 (2022). https://doi.org/10.1007/s00214-022-02891-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-022-02891-9