
Abstract
Vitamin D is a key hormone involved in the regulation of calcium/phosphorous balance and recently it has been implicated in the pathogenesis of sub-inflammation, insulin resistance and obesity. The two main forms of vitamin D are cholecalciferol (Vitamin D3) and ergocalciferol (Vitamin D2): the active form (1,25-dihydroxyvitamin D) is the result of two hydroxylations that take place in liver, kidney, pancreas and immune cells. Vitamin D increases the production of some anti-inflammatory cytokines and reduces the release of some pro-inflammatory cytokines. Low levels of Vitamin D are also associated with an up-regulation of TLRs expression and a pro-inflammatory state. Regardless of the effect on inflammation, Vitamin D seems to directly increase insulin sensitivity and secretion, through different mechanisms. Considering the importance of low grade chronic inflammation in metabolic syndrome, obesity and diabetes, many authors hypothesized the involvement of this nutrient/hormone in the pathogenesis of these diseases. Vitamin D status could alter the balance between pro and anti-inflammatory cytokines and thus affect insulin action, lipid metabolism and adipose tissue function and structure. Numerous studies have shown that Vitamin D concentrations are inversely associated with pro-inflammatory markers, insulin resistance, glucose intolerance and obesity. Interestingly, some longitudinal trials suggested also an inverse association between vitamin D status and incident type 2 diabetes mellitus. However, vitamin D supplementation in humans showed controversial effects: with some studies demonstrating improvements in insulin sensitivity, glucose and lipid metabolism while others showing no beneficial effect on glycemic control and on inflammation. In conclusion, although the evidences of a significant role of Vitamin D on inflammation, insulin resistance and insulin secretion in the pathogenesis of obesity, metabolic syndrome and type 2 diabetes, its potential function in treatment and prevention of type 2 diabetes mellitus is unclear. Encouraging results have emerged from Vitamin D supplementation trials on patients at risk of developing diabetes and further studies are needed to fully explore and understand its clinical applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Vitamin D plays a key role in the regulation of mineral homeostasis, being mainly involved in bone and calcium/phosphorus balance [1]. However, over the last several years, a potential extra skeletal role of vitamin D has begun to emerge [1, 2].
In particular, many studies have revealed profound immunomodulatory effects of vitamin D and its derivatives [3] and that vitamin D deficiency may be associated with sub-inflammatory state, insulin resistance and obesity/ metabolic syndrome. Obesity, metabolic syndrome, essential hypertension and type 2 diabetes are all insulin-resistant states, in which different hormones and cytokines are implicated. However, all these pathologic states are characterized by a condition of chronic sub-inflammation. Considering the importance of sub-inflammation in the development of insulin resistance and obesity/metabolic syndrome, it has been speculated that Vitamin D deficiency could play a contributory role in it [4].
The focus of this review is to explore the pathophysiology of the mechanistic relationship between vitamin D, bone metabolism, inflammation and insulin resistance, in humans.
2 Vitamin D: Metabolism, actions
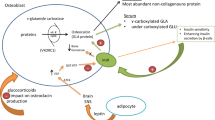
Cholecalciferol (Vitamin D3) and ergocalciferol (Vitamin D2) are the two main forms of vitamin D. The first one is mainly endogenous and produced in the skin, as a result of sun exposure; only a minor portion, instead, is exogenous and extracted from food such as fatty fish, egg yolk, mushrooms and yeast. Dairy products with nutritional supplements and nutritional supplements by themselves usually contain vitamin D2, that is the vegetal form of vitamin D [5].
Vitamin D3 is a lipophilic precursor of the major circulating form: 25(OH) Vitamin D. During sun exposure 7-dehydrocholesterol is converted to pre-vitamin D3 and then to vitamin D3 in a non-enzymatic reaction that takes place in the skin. The excess of sunlight causes degradation of pre-vitamin D3 and vitamin D3 into inactive form. Through the binding to the vitamin D-binding protein, Vitamin D3 is then transported in the bloodstream from the skin to the liver, where it is converted by vitamin 25-hydroxylase to the circulating form, 25(OH)Vitamin D [6].
To exert its biological function, interacting with the vitamin D nuclear receptor, the vitamin D’s circulating form must be subjected to a second hydroxylation reaction [7]. This reaction takes place mainly in the kidney, being carried out by a single enzyme (1 α-hydroxylase), leading to the most active vitamin D, known as 1,25 (OH)2 Vitamin D. The activating process could also take place in other cells that contains this enzyme, such as immune cells [8] or pancreatic cells [9].
Vitamin D 24-hydroxylase is in charge for the inactivation process. This event is regulated by the concentration of the active form of Vitamin D, that could induce the expression of 24-hydroxylase and convert 25(OH) vitamin D and 1,25(OH)2 vitamin D into less-active metabolites (e.g. 24,25(OH)2 vitamin D and 1,24,25(OH)3 Vitamin D) which are in turn further catabolized into inactive calcitroic acid [10].
The 1,25(OH)2 Vitamin D is the main active form and has about 1000-fold higher affinity than 25(OH) Vitamin D for the vitamin D receptor (VDR). All Vitamin D metabolites circulate in the bloodstream bound to the vitamin Dbinding protein [11]. 1,25 (OH)2 Vitamin D and 25(OH)Vitamin D are predominantly stocked in adipose and muscle tissue [12]. In particular, adipose tissue is generally considered the reservoir for Vitamin D in human subjects and rats [13–17] Interestingly, visceral fat was found to contain 20% more of this nutrient than subcutaneous fat [18].
The 25(OH) Vitamin D has a half-life between 15 to 50 days and circulates in plasma in nanomolar concentrations [12]. Vitamin D status is usually determined by serum concentrations of 25(OH) Vitamin D: a 25(OH) Vitamin D concentrations between 20 and 30 ng/mL defines insufficiency and a 25-OH Vitamin D concentration less than 20 ng/mL defines deficiency [19]. Vitamin D deficiency is considered a pandemic problem because most part of the population, even among people living in tropical areas, has this nutritional deficiency that could lead to both skeletal and extra-skeletal problems [20].
In the 2011, the Institute of Medicine set the Vitamin D Recommended Dietary Allowance (RDA, the level of intake of a nutrient judged adequate to meet the known nutrient needs of nearly all healthy people), at 600 IU/d for persons aged 1–70 years and 800 IU/d for persons aged 71 years and older [5].
3 Sub-inflammation and insulin resistance
Insulin resistance is defined as a reduced ability of insulin to exert its metabolic and biological actions at the whole-body level, where insulin in concert with glucagon, somatostatin and a number of extra-pancreatic hormones plays a key-role in the regulation of glucose homeostasis [21–23]. This condition is characterized by a decreased signaling of insulin receptors on insulin sensitive cells. In the natural history of insulin resistance/type 2 diabetes mellitus (T2DM), initially, an insulin resistant subject compensates with a greater production of insulin (compensating hyperinsulinism with normoglycemia) but, with time, beta-cell insulin secretion is reduced, leading to glucose intolerance and clinically overt type 2 diabetes mellitus [24].
This sequence of molecular and biological events occurs in humans [25] non-humans primates [26–30] and rodent models of T2DM and insulin resistance [31–33].
Insulin resistance is one of the hallmarks of type 2 diabetic patients, but it is also recognizable in more than 50% of patients without diabetes who suffered a cardiovascular event. The presence of insulin resistance increases the risk of vascular disease, also because of its association with hypertension, hyperglycemia, hyperinsulinemia, dyslipidemia, endothelial dysfunction, hypercoagulability, sub-inflammation and increased platelet reactivity [34–38].
The mechanistic link between insulin resistance, diabetes and cardiovascular diseases is not fully understood, but it is known that visceral obesity is associated with further increased cardiovascular morbidity and mortality [35–39].
Sub-inflammation is an important factor involved in the pathogenesis of insulin resistance. Pro-inflammatory cytokines and acute phase proteins can affect negatively several signaling and metabolic pathways which are activated by the insulin receptor signaling machinery, the functions of lipoproteins lipase and adipose tissue function and structure [40].
Also, the metabolic syndrome, is classically characterized as a chronic state of low-grade inflammation (sub-inflammation), identifiable by slightly elevated levels of adipocyte-released Tumor necrosis factor α (TNF- α) and interleukin 6 (IL-6) [41]. Inflammation occurs primarily in adipose tissue, but also in other tissues with important metabolic activity, such as liver, pancreatic islets and hypothalamus [42].
Adipose tissue is also an endocrine organ since it secretes a variety of cytokines (TNF-α, IL-6, interleukin 1 β (IL1β)), predominantly produced by local macrophages, as well as leptin and adiponectin [43]. Interestingly leptin has also been implicated in the development of cardiac hypertrophy in obese patients [44].
A cause for cytokines overproduction in obesity and type 2 diabetes is the alteration of the tissue inhibitor of metalloproteases 3 - TNF-α converting enzyme (TIMP3-TACE) dyad in human skeletal muscle, where an elevated TACE activity and a diminished TIMP3 protein levels are present, leading to an augmented release of TNF-α. The TIMP3–TACE dyad is involved in the regulation of TNF-α shedding from pro-TNF-α as well as IL-6 release [39, 45, 46].
This abnormal release of adipokines may help explaining some features of the metabolic syndrome. Inflammation is linked to metabolic abnormalities such as increased triglycerides, insulin, glucose, reduced high density lipoproteins (HDL) levels, sympathetic over activity, endothelial dysfunction and oxidative stress. All these alterations are associated with the development of T2DM, arterial hypertension, platelet increased aggregability and accelerated atherosclerosis. [38, 47–50].
The effect of TNF-α and other pro-inflammatory cytokines on peripheral insulin sensivity occurs through the inhibition of insulin-mediated tyrosine phosphorylation of insulin receptor and insulin receptor substrate (IRS)-1, leading to defective activation of downstream insulin signaling (for example to phosphatidylinositol-3 (PI3)-kinase and translocation of GLUT4 to the cell surface) [51, 52].
Molecular and cell triggers have been recognized in metabolic sub-inflammation: 1) endoplasmic reticulum stress (ERS), 2) activation of toll-like receptor (TLR) 4 and 3) stimulation of protein kinase R (PKR).
The endoplasmic reticulum, in the presence of lipid overload becomes unable to correctly synthetize and fold proteins; long-chain saturated fatty acids can activate TLR4, leading to insulin resistance and inflammatory gene transcription; finally, nutrient overload can stimulate PKR, inducing inflammatory signaling (through c-Jun N terminal kinase (JNK) and IkB kinase (IKK)). These three signaling pathways are connected and can target the insulin signaling pathway negatively. Furthermore, dietary fats can promote changes to gut microbiota which through the activation of TLR4 and the alteration of ERS, contribute to metabolic inflammation [37, 42, 53].
Angiotensin II, cytokines, such as TNF- α, IL-6 and Monocyte chemotactic protein 1 (MCP1), and other inflammatory factors produced in the context of metabolic inflammation can activate intracellular stress kinases (protein-kinases), such as JNK, IKK, S6 kinase (S6 K) and protein kinase C (PKC), promoting the inhibitory serine phosphorylation of the insulin receptor substrates, IRS1 and IRS2, thus providing the molecular basis for insulin resistance [31, 37, 51, 52, 54–57].
4 Vitamin D and inflammation
An interesting as well as unexpected role of Vitamin D in the modulation of immune function and inflammatory processes has emerged from cellular studies [58]. Vitamin D inhibits adipose tissue inflammation both in vitro and in vivo, acting upon preadipocytes, adipocytes as well as on leucocyte infiltration [59, 60].
Vitamin D seemingly exerts broad regulatory effects on adaptive and innate immune cell system [61]. The conversion of 25(OH)Vitamin D to its active form 1,25(OH)2 Vitamin D can occur inside immune system cells that possess the 1 α-hydroxylase enzyme: dendritic cells, macrophages, T and B cells [62].
Vitamin D’s effect on dendritic cells includes increased interleukin 10 (IL-10) production (which is an anti-inflammatory cytokine) and reduced release of TNF-α, interferon γ (IFN-γ) and interleukin 12 (IL-12) (which are pro-inflammatory cytokines). With the stimulation of Vitamin D, dendritic cells acquire a peculiar immunoregulatory role as well as tolerogenic properties [3].
The effect on monocytes consist in down-regulation of the expression and production of several pro-inflammatory cytokines including TNF- α, IL-1β, IL-6 and interleukin 8 (IL-8) [63, 64].
On the other hand, on lymphocytes, the overall effect is a switch from the more inflammatory T-helper 1 (Th1)/Th17 response to the less inflammatory Th2/Treg profile [65].
The immunoregulatory effects of vitamin D have been studied in type 1 diabetes and in particular in islet transplantation. Islet transplantation is a novel and effective treatment, able to significantly change the natural history of the disease, but it is burdened by the need of an appropriate immunosuppression to avoid the risk of rejection. Traditional immunosuppressive regimens are in fact responsible for numerous side effects (risk of infections, neoplastic diseases, nephrotoxicity, now onset diabetes etc.) and this lead to development of new immunoregulatory protocols. Animal studies demonstrated that 1,25 (OH)2 Vitamin D administration with mycophenolate mofetil is able to induce tolerance after islet transplantation: it affects antigen presenting cells (APCs) and in particular dendritic cells (DCs), promoting down regulation of the expression of CD40, CD80, and CD86 costimulatory molecule, inhibits alloreactive T cells and responsiveness to alloantigens. These results suggest a potential role in transplant immunosuppression and could allow reduction of traditional immunosuppressive regimen doses [66–69].
The effect of the active form of vitamin D can occur in two ways: through the interaction with the membrane Vitamin D receptor (mVDR) or through binding with the nuclear Vitamin D receptor (nVDR), that has high affinity and avidity for the active metabolite and regulate gene expression [70]. There are more than a thousand genes that are directly or indirectly regulated by (OH)2 Vitamin D and involved in various physiological processes such as cell proliferation, differentiation, apoptosis and angiogenesis [71]. The nVDR is found in multiple cells of the immune system such as human T-reg cells, neutrophils, dendritic cells, B cells and macrophages [70]. Moreover, numerous studies demonstrated the expression of VDRs in pancreatic β-cells and in all insulin-responsive tissues [2, 72–74].
Vitamin D can affect inflammation also through the mediation of TLRs, that are trans-membrane receptors located on monocytes and macrophages, implicated in the innate immune response to pathogens [75, 76]. In fact, it has been reported that elevated vitamin D levels during the summer coincided with decreased monocyte expression of TLR-2 and TLR-4; decreased vitamin D levels, on the opposite, were associated with an increased expression of TLRs [77]. In obese subjects a reduced degree of plasmatic Vitamin D seasonal variations has been observed: this may contribute to an up-regulation of TLRs expression and consequent increased level of inflammation [78].
5 Vitamin D, insulin sensitivity and insulin secretion
Vitamin D, besides its effect on inflammation and immune system, also seems to directly influence insulin sensitivity/resistance.
In fact, several studies have shown an increase in insulin sensitivity which could be mediated through the interaction with VDRs in skeletal muscles [79], the stimulation of expression of insulin receptors on target tissues [80] and, finally, through the activation of peroxisome proliferator activator receptorδ (PPAR δ) [81], that is a transcription factor involved in metabolism and mobilization of fatty acids in adipose tissue and skeletal muscle [82].
Other proposed mechanisms for increasing insulin sensitivity are the inhibition of the renin-angiotensin-aldosterone system (RAAS), which is a well-known inhibitor of insulin action in peripheral tissues, and the regulation of cellular calcium concentration in skeletal muscle cells, that might enhance glucose transport through the membrane via the recruitment of Glucose transporter type 4 (GLUT4). [83–85].
In addition, vitamin D could stimulate insulin release through the regulation of beta cell intracytoplasmic calcium concentration and stimulation of exocytosis mechanism. The interaction between 1,25 (OH)2 Vitamin D and nVDR lead to the transcription of insulin, cellular structure and growth genes. Two pathways have been recognized: the first includes the activation of protein kinases A (PKA) that phosphorylates voltage-dependent calcium channels and proteins involved in exocytosis; the second, instead, includes the activation of phospholipase C (PLC), synthesis of inositol 1,4,5-trisphosphate (IP3), that causes the release of calcium from the endoplasmic reticulum, and diacylglycerol (DAG), that activates protein kinase C (PKC), in turn responsible for the activation of voltage-dependent calcium Channels and the KATP channels, but also the mobilization of insulin secretory vesicles. All these steps lead to the depolarization of the cytoplasmic membrane, opening of calcium channels, increasing of intracellular calcium content and subsequent insulin secretion. [86].
However, in neoplastic β-cell lines, Vitamin D has a pro-apoptotic and anti-insulinogenic effect. In fact, some cellular studies demonstrated that calcitriol inhibits growing, induce apoptosis, decrease cell viability, gene expression, insulin content and secretion in murine insulinoma β-cells [87, 88].
6 Interaction between vitamin D, inflammation and insulin resistance
A number of basic experimental, epidemiological and clinical studies support the role of low-grade inflammation in the development of metabolic diseases, insulin resistance and type 2 diabetes [12].
As discussed above, several studies have demonstrated that vitamin D acts as a negative modulator of TNF-α and IL-6 release [54, 55, 89], decreasing TNF-α, IL-6, and C-reactive protein (CRP) both in vivo as well as in vitro [90, 91] and thus acting on adipose tissue and immune system [56, 57].
The importance of inflammation in the pathogenesis of metabolic syndrome, the evidence of the anti-inflammatory activity and the direct effects on insulin sensitivity of Vitamin D, suggest a possible role for this hormone in insulin resistant clinical conditions.
In recent years, several cross-sectional, cohort, longitudinal as well as interventional clinical studies, have investigated the relationship between Vitamin D, inflammation and insulin resistance.
Many cross-sectional and cohort studies showed an association of 25(OH) Vitamin D concentrations with inflammatory status, glucose intolerance, insulin resistance, metabolic syndrome and the risk of type 2 diabetes mellitus [92–106].
These association seem to be more relevant in patient with Vitamin D deficiency than in patients with normal level: in a large Canadian cohort study on non-diabetic adults it was demonstrated that vitamin D status was inversely associated with insulin responsiveness. Furthermore, insulin response correlated with 25(OH) Vitamin D level after adjusting for BMI, waist circumference, weight, age and sex, in patients with a 25 (OH) Vitamin D baseline level from 40 to 90 nmol/L [107].
In addition, Vitamin D plasma levels were found to be inversely correlated to the classical parameters of obesity such as BMI, fat mass and waist circumference [108, 109]. Interestingly, it has been demonstrated that serum 25(OH) Vitamin D is significantly lower in obese individuals as compared to lean ones [110]. The prevalence of Vitamin D deficiency is respectively a 25% and 35% higher in overweight and obese than lean subjects [111]. Some authors, considering the fat-soluble nature of this nutrient and the adipose tissue as its main storage place, have suggested that vitamin D and its metabolites could be sequestered in the excess fat mass which is present in obese persons [112–114]. Another hypothesis is that individuals with a higher BMI might have a higher volume in which 25-OH Vitamin D is diluted and would require greater Vitamin D dietary intake as compared to lean individuals to achieve a normal Vitamin D level [115].
Furthermore, vitamin D seem to be a predictive factor for death by cardiovascular disease in diabetic patients [116, 117].
Type 2 diabetic patients showed a prevalent impairment of Ventricular diastolic filling but a similar ventricular systolic performance, if compared to type 1 diabetic patients.
In type 1 diabetic patients an alteration of high-energy phosphates (HEPs) metabolism may contribute to Left ventricular dysfunction. [118, 119].
In diabetic patients, the effect of vitamin D on cardiac diastolic or systolic function have not been studied while some animal models suggested some effect of vitamin D supplementation on cardiac metabolism. 1,25(OH)2 Vitamin D treated rats had lower body weight, smaller left ventricular end-diastolic diameter, shorter QT interval, higher cardiac PPAR-α and PPAR-δ protein expressions, but lower cardiac PPAR-γ protein levels and inflammatory cytokines (TNF-α and IL-6) than non-treated diabetic rats. [120].
Some beneficial effects of Vitamin D administration on arterial pressure and stiffness have also been reported. [121–125].
An inverse association between vitamin D status and incident type 2 diabetes mellitus was also demonstrated by longitudinal studies [6]. Furthermore, two meta-analyses [126, 127] of longitudinal observational studies, have confirmed these results.
Nevertheless, confounding factors that may influence type 2 diabetes incidence but also vitamin D status, such as age, diet and lifestyle, may have influenced the results of these studies and therefore causality cannot be established with certainty. This is a major limitation of these studies [6, 128, 129].
7 Vitamin D supplementation clinical trials
Whereas animal studies have found that vitamin D supplementation in deficient animals can improve insulin sensitivity and inflammation, these findings have not been unequivocally reproduced in humans.
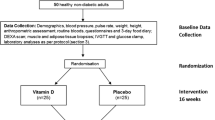
In Tables 1 and 2 we have listed the main prospective and interventional clinical trials with Vitamin D.
No significant effects on inflammation status have been observed in clinical trials after supplementation with vitamin D [12, 96, 128, 130, 131].
Clinical trials in humans affected by type 2 diabetes mellitus produced controversial results. In some of them there were improvements in glycaemia, insulin sensitivity, lipid profile and endothelial function [121, 132]. Other studies however, demonstrated no significant effects on glycaemia, glucose metabolism, insulin sensitivity, as well as no reduction in diabetes incidence [122, 123, 133–137]. To add further fuel to this controversy some others showed no significant changes in insulin sensitivity and inflammatory markers, although there was a minimal improvement of insulin secretion [138].
Controversial effects were observed also in interventional trials on healthy subjects.
In particular, a neutral effect of vitamin D supplementation was seen in two large clinical trials: Avenell and colleagues demonstrated no changes in the incidence of diabetes and no beneficial effect on glucose metabolism in a group of 5292 participants aged ≥70 years, followed for 24–62 months. This was a blinded randomized trial lead in UK from 1999 to 2002; the patients were randomized to take oral 800 IU (20 μg) daily vitamin D 3 and 1000 mg calcium or placebo. [134] In USA, De Boer and colleagues in a double blinded clinical trial with a median follow-up time of 7 years, observed no reduction in the development of diabetes after administration of Calcium plus vitamin D3 supplementation (1000 mg elemental calcium plus 400 IU of vitamin D3 daily) in a group of 33,951 women without self-reported diabetes at baseline clinical trial. 2291 of these women had a new diagnosis of diabetes. [135].
A positive effect of Vitamin D supplementation, instead, was described by Tepper et al. in a double blind randomized controlled trial lead in Israel on 130 men aged 20–65 years without diabetes with serum 25-hydroxyvitamin D levels <50 nmol/l. It was observed that insulin levels and HOMA-IR values remained steady during the study period in the treatment group but increased in the control group. The patients were treated with 100,000 IU vitamin D bimonthly or placebo and revaluated at 6 and 12 months of follow up. [139].
Von Hurst and colleagues also find positive results following for 6 months a group of 81 subjects in a randomized controlled, double-blind intervention. 42 patients were given 100 microg (4000 IU) of vitamin D (3) and 39 were given placebo. This study demonstrated that after vitamin D supplementation there was a significant decrease in fasting insulin and a significant improvement in insulin sensitivity and resistance. [140].
Although the effect of Vitamin D supplementation on diabetic patients seem to be non-significant, more interesting could be to study the effect on people at risk of developing diabetes such impaired glucose tolerance (IGT) and impaired fasting glucose (IFG).
In fact, some recent trials demonstrated that vitamin D supplementation could be useful for patients with IFG or IGT which are at high (50%) risk of type 2 diabetes: patients with IFG taking vitamin D had a lower rise in FPG at 3 years compared to those on placebo in a trial on non-diabetic adults [128]; a low calorie diet that lead to a rise in serum 15 OH Vitamin D was associated with an increase in Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) and improved lipid profile in a group of obese women [131]; vitamin D supplementation in adults at risk for type 2 diabetes lead to improved β-cell function and to a minor, but non-significant, increase in HbA1c values than in the control group [129]; finally, insulin sensitivity improved after 4 weeks of vitamin D administration in persons with IFG [141].
On the contrary, other authors found no effect of vitamin D supplementation on insulin secretion, insulin sensitivity or incident diabetes in a population affected by IGT or IFG [125, 142–144].
Two meta-analysis of randomized clinical trials concluded that at the moment there are not enough evidences to recommend supplementation with vitamin D to improve glycemic metabolism and prevent diabetes, even in high risk IGT or IFG individuals [145, 146].
8 Conclusions and perspective
Numerous studies have demonstrated that vitamin D plays a critical role in the regulation of inflammation, insulin resistance and, probably, insulin secretion. These findings suggest a potential function of vitamin D deficiency in the pathogenesis of the metabolic syndrome and type 2 diabetes, in which low-grade inflammation and alteration in insulin signaling pathways are key elements.
Many authors investigated the connection between vitamin D, inflammation and insulin resistance: numerous cross sectional, cohort and longitudinal studies showed an association between 25-OH Vitamin D levels, metabolic syndrome and diabetes. Interventional studies failed to demonstrate an unequivocal beneficial effect of vitamin D supplementation in type 2 diabetic patients, but some encouraging results have emerged from trials on patients at risk of developing diabetes (insulin resistant, IFG and IGT).
In conclusion, despite the presence of evidences of a mechanistic connection between signaling pathways of vitamin D, inflammatory cytokines (IL-6 and TNF- α) and insulin, the current available data are insufficient to demonstrate a general causal role of vitamin D deficiency in the pathogenesis of diabetes and metabolic syndrome, nor a therapeutic role for its supplementation in type 2 diabetes. We believe that long term, well designed, interventional clinical trials will be started to achieve a better understanding of the therapeutic potential of supplementation in Vitamin D deficient pre-diabetic subjects with attention to doses, duration of therapy, side effects, short term and long term results. In fact, we believe that Vitamin D deficient patients at risk of developing diabetes are the most promising target for supplementation: administration doses should be decided according to RDA, considering vitamin D status (insufficient or deficient) and age; supplementation should be long term (years); there must be a complete assessment of metabolic and cardiovascular parameters.
Interestingly also, the bone is emerging as a new important organ in the regulation of glucose metabolism in humans and it is conceivable that Vitamin D action and status in the regulation of calcium-phosphorous balance and bone metabolism might also mirror the interplay between other bone remodeling hormones such as sclerosin, osteopontin, osteoprotegerin, fractalkine and insulin in insulin resistant states and type 2 diabetes mellitus [44, 147–149].
References
Muscogiuri G, Altieri B, Annweiler C, Balercia G, Pal HB, Boucher BJ, et al. Vitamin D and chronic diseases: the current state of the art. Arch Toxicol. 2016.
Muscogiuri G, Mitri J. MathieuC, Badenhoop K, tamer G, Orio F et al. mechanisms in endocrinology: vitamin D as a potential contributor in endocrine health and disease. Eur J Endocrinol. 2014;171:101–10.
Barragan M, Good M, Kolls JK. Regulation of dendritic cell function by vitamin D. Nutrients. 2015;7:8127–51.
McGill A-T, Stewart JM, Lithander FE, Strik CM, Poppitt SD. Relationships of low serum vitamin D3 with anthropometry and markers of the metabolic syndrome and diabetes in overweight and obesity. Nutr J. 2008;28:7–4.
Ross AC, Taylor CL, Yaktine AL, Del Valle HB, editors. Dietary reference intakes for calcium and vitamin D. In: Committee to review dietary reference intakes for calcium and vitamin D. Washington (DC): National Academies Press (USA); 2011.
Mitri J, Pittas AG. Vitamin D and diabetes. Endocrinol Metab Clin N Am. 2014;43:205–32.
Schuster I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim Biophys Acta. 1814;2011:186–99.
Baeke F, Gysemans C, Korf H, Mathieu C. Vitamin D insufficiency: implications for the immune system. Pediatr Nephrol. 2010;25:1597–606.
Bland R, Markovic D, Hills CE, Hughes SV, Chan SL, Squires PE, Hewison M. Expression of 25-hydroxyvitamin D3-1alphahydroxylase in pancreatic islets. J Steroid Biochem Mol Biol. 2004;89-90:121–5.
Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–F28.
Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78:1193–231.
Wamberg L, Pedersen SB, Rejnmark L, Richelsen B. Causes of vitamin D deficiency and effect of vitamin D supplementation on metabolic complications in obesity: a review. Curr Obes Rep. 2015;4:429–40.
Mawer EB, Backhouse J, Holman CA, et al. The distribution and storage of vitamin D and its metabolites in human tissues. Clin Sci. 1972;43:413–31.
Blum M, Dolnikowski G, Seyoum E, et al. Vitamin D (3) in fat tissue. Endocrine. 2008;33:90–4.
Pramyothin P, Biancuzzo RM, Lu Z, et al. Vitamin D in adipose tissue and serum 25-hydroxyvitamin D after roux-en-Y gastric bypass. Obesity (Silver Spring). 2011;19:2228–34.
Landrier JF, Marcotorchino J, Tourniaire F. Lipophilic micronutrients and adipose tissue biology. Nutrients. 2012;4:1622–49.
Marcotorchino J, Tourniaire F, Landrier JF. Vitamin D, adipose tissue, and obesity. Horm Mol Biol Clin Investig. 2013;15:123–8.
Beckman LM, Earthman CP, Thomas W, et al. Serum 25(OH) vitamin D concentration changes after roux-en-Y gastric bypass surgery. Obesity (Silver Spring). 2013;21:E599–606.
Hollick MF, Vitamin D. Deficiency. N Engl J Med. 2007;357:266–81.
Holick MF. The vitamin D deficiency pandemic and consequences for nonskeletal health: mechanisms of action. Mol Asp Med. 2008;29:361–8.
Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6
Kahn CR, White MF, Shoelson SE, Backer JM, Araki E, Cheatham B, Csermely P, Folli F, Goldstein BJ, Huertas P, et al. The insulin receptor and its substrate: molecular determinants of early events in insulin action. Recent Prog Horm Res. 1993;48:291–339.
Kahn CR, Folli F. Molecular determinants of insulin action. Horm Res. 1993;39:93–101.
DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care. 1992;15:318–68.
Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet. 1992;340:925–9.
Chavez AO, Lopez-Alvarenga JC, Tejero ME, Triplitt C, Bastarrachea RA, Sriwijitkamol A, Tantiwong P, Voruganti VS, Musi N, Comuzzie AG, DeFronzo RA, Folli F. Physiological and molecular determinants of insulin action in the baboon. Diabetes. 2008;57:899–908.
Chavez AO, Gastaldelli A, Guardado-Mendoza R, Lopez-Alvarenga JC, Leland MM, Tejero ME, Sorice G, Casiraghi F, Davalli A, Bastarrachea RA, Comuzzie AG, DeFronzo RA, Folli F. Predictive models of insulin resistance derived from simple morphometric and biochemical indices related to obesity and the metabolic syndrome in baboons. Cardiovasc Diabetol. 2009;8:22.
Guardado-Mendoza R, Davalli AM, Chavez AO, Hubbard GB, Dick EJ, Majluf-Cruz A, Tene-Perez CE, Goldschmidt L, Hart J, Perego C, Comuzzie AG, Tejero ME, Finzi G, Placidi C, La Rosa S, Capella C, Halff G, Gastaldelli A, DeFronzo RA, Folli F. Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc Natl Acad Sci U S A. 2009;106:13992–7.
Guardado-Mendoza R, Jimenez-Ceja L, Majluf-Cruz A, Kamath S, Fiorentino TV, Casiraghi F, Velazquez AO, DeFronzo RA, Dick E, Davalli A, Folli F. Impact of obesity severity and duration on pancreatic β- and α-cell dynamics in normoglycemic non-human primates. Int J Obes. 2013;37:1071–8.
Guardado Mendoza R, Perego C, Finzi G, La Rosa S, Capella C, Jimenez-Ceja LM, Velloso LA, Saad MJ, Sessa F, Bertuzzi F, Moretti S, Dick Jr EJ, Davalli AM, Folli F. Delta cell death in the islet of Langerhans and the progression from normal glucose tolerance to type 2 diabetes in non-human primates (baboon, Papio hamadryas). Diabetologia. 2015;58:1814–26.
Folli F, Saad MJ, Backer JM, Kahn CR. Regulation of phosphatidylinositol 3-kinase activity in liver and muscle of animal models of insulin-resistant and insulin-deficient diabetes mellitus. J Clin Invest. 1993;92:1787–94.
Saad MJ, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest. 1993;92:2065–72.
Saad MJ, Folli F, Araki E, Hashimoto N, Csermely P, Kahn CR. Regulation of insulin receptor, insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-F442A adipocytes. Effects of differentiation, insulin, and dexamethasone. Mol Endocrinol. 1994;8:545–57.
Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, Guarino PD, Lovejoy AM, Peduzzi PN, Conwit R, Brass LM, Schwartz GG, Adams Jr HP, Berger L, Carolei A, Clark W, Coull B, Ford GA, Kleindorfer D, JR O’L, Parsons MW, Ringleb P, Sen S, Spence JD, Tanne D, Wang D, winder TR for the IRIS trial investigators. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. 2016;374:1321–31.
Reaven G. Insulin resistance and coronary heart disease in nondiabetic individuals. Arterioscler Thromb Vasc Biol. 2012;32:1754–9.
Ferrannini E, Buzzigoli G, Bonadonna R, Giorico MA, Oleggini M, Graziadei L, Pedrinelli R, Brandi L, Bevilacqua S. Insulin resistance in essential hypertension. N Engl J Med. 1987;317:350–7.
Velloso LA, Folli F, Sun XJ, White MF, Saad MJ, Kahn CR. Cross-talk between the insulin and angiotensin signaling systems. Proc Natl Acad Sci U S A. 1996;93:12490–5.
Vicari AM, Monzani ML, Pellegatta F, Ronchi P, Galli L, Folli F. Platelet calcium homeostasis is abnormal in patients with severe arteriosclerosis. Arterioscler Thromb. 1994;14:1420–4.
Tripathy D, Daniele G, Fiorentino TV, Perez-Cadena Z, Chavez-Velasquez A, Kamath S, Fanti P, Jenkinson C, Andreozzi F, Federici M, Gastaldelli A, Defronzo RA, Folli F. Pioglitazone improves glucose metabolism and modulates skeletal muscle TIMP-3-TACE dyad in type 2 diabetes mellitus: a randomised, double-blind, placebo-controlled, mechanistic study. Diabetologia. 2013;56:2153–63.
Crook M. Type 2 diabetes mellitus: a disease of the innate immune system? An update Diabetic Medicine. 2004;21:203–7.
Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2001;280:E745–51.
Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of Nutrients, gut Microbiota, and metabolic inflammation. Endocr Rev. 2015;36:245–27.
Scherer PE. The multifaceted roles of adipose tissue- therapeutic targets for diabetes and beyond. Diabetes. 2016;65:1452–61.
Daniele G, Guardado Mendoza R, Winnier D, Fiorentino TV, Pengou Z, Cornell J, Andreozzi F, Jenkinson C, Cersosimo E, Federici M, Tripathy D, Folli F. The inflammatory status score including IL-6, TNF-a, osteopontin, fractalkine, MCP-1 and adiponectin underlies whole-body insulin resistance and hyperglycemia in type 2 diabetes mellitus. Acta Diabetol. 2014;51:123–31.
Federici M, Hribal ML, Menghini R, Kanno H, Marchetti V, Porzio O, Sunnarborg SW, Rizza S, Serino M, Cunsolo V, Lauro D, Mauriello A, Smookler DS, Sbraccia P, Sesti G, Lee DC, Khokha R, Accili D, Lauro R. Timp3 deficiency in insulin receptor-haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF-alpha. J Clin Invest. 2005;115:3494–505.
Monroy A, Kamath S, Chavez AO, Centonze VE, Veerasamy M, Barrentine A, Wewer JJ, Coletta DK, Jenkinson C, Jhingan RM, Smokler D, Reyna S, Musi N, Khokka R, Federici M, Tripathy D, DeFronzo RA, Folli F. Impaired regulation of the TNF-α converting enzyme/tissue inhibitor of metalloproteinase 3 proteolytic system in skeletal muscle of obese type 2 diabetic patients: a new mechanism of insulin resistance in human. Diabetologia. 2009;52:2169–81.
Pontiroli AE, Pizzocri P, Paroni R, Folli F. Sympathetic Overactivity, endothelial dysfunction, inflammation, and metabolic abnormalities cluster in grade III (World Health Organization) obesity. Diabetes Care. 2006;29:12.
Vicari AM, Taglietti MV, Pellegatta F, Spotti D, Melandri M, Galli L, Ronchi P, Folli F. Deranged platelet calcium homeostasis in diabetic patients with end-stage renal failure. A possible link to increased cardiovascular mortality? Diabetes Care. 1996;19:1062–6.
Pellegatta F, Folli F, Ronchi P, Caspani L, Galli L, Vicari AM. Deranged platelet calcium homeostasis in poorly controlled IDDM patients. Diabetes Care. 1993;16:178–83.
Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des. 2013;19:5695–703.
Hotamisligil GS, Shargill NS. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance., Spiegelman BM. Science. 1993;259:87–91.
Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–8.
Arruda AP, Hotamisligil GS. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 2015;22:381–97.
Li B, Baylink DJ, Deb C, Zannetti C, Rajaallah F, Xing W, Walter MH, Lau K-HW, Qin X. 1, 25-Dihydroxyvitamin D3 suppresses TLR8 expression and TLR8-mediated inflammatory responses in monocytes in vitro and experimental autoimmune encephalomyelitis in vivo. PLoS One. 2013;8:e58808.
Gao D, Trayhurn P, Bing C. 1, 25-Dihydroxyvitamin D3 inhibits the cytokine-induced secretion of MCP-1 and reduces monocyte recruitment by human preadipocytes. Int J Obes. 2013;37:357–65.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7.
Vlasova M, Purhonen A, Jarvelin M, Rodilla E, Pascual J, Herzig K. Role of adipokines in obesity-associated hypertension. Acta Physiol. 2010;200:107–27.
Calton EK, Keane KN, Newsholme P, Soares MJ. Impact of vitamin D levels on inflammatory status: a systematic review of immune cell studies. PLoS One. 2015;10:e0141770.
Gonzalez-Molero I, Rojo-Martinez G, Morcillo S, Gutierrez C, Rubio E, Perez-Valero V, et al. Hypovitaminosis D and incidence of obesity: a prospective study. Eur J Clin Nutr. 2013;
Landrier JF, Karkeni E, Marcotorchino J, Bonnet L, Tourniaire F. Vitamin D modulates adipose tissue biology: possible consequences for obesity? Proc Nutr Soc. 2016;75:38–46.
Cannell JJ, Grant WB, Holick MF. Vitamin D and inflammation. Dermato-Endocrinology. 2014;6:1.
Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins a and D take centre stage. Nat Rev Immunol. 2008;8:685–98.
Giulietti A, van Etten E, Overbergh L, Stoffels K, Bouillon R, Mathieu C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile.1,25-DihydroxyvitaminD (3) works as anti-inflammatory. Diabetes Res Clin Pract. 2007;77:47–57.
Neve A, Corrado A. CantatoreF. Immunomodulatory effects of vitamin D in peripheral blood monocyte derived macrophages from patients with rheumatoid arthritis. Clin Exp Med. 2013:1–9.
Sloka S, Silva C, Wang J, Yong VW. Predominance of Th2 polarization by VitaminD through a STAT6 dependent mechanism. J Neuroinflammation. 2011;8
Gregori S, Casorati M, Amuchastegui S, Smiroldo S, Davalli AM, Adorini L. Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. J Immunol. 2001;167:1945–53.
Tezza S, Nasr MB, Vergani A, Valderrama Vasquez A, Maestroni A, Abdi R, Secchi A, Fiorina P. Novel immunological strategies for islet transplantation. Pharmacol Res. 2015;98:69–75.
Nasr MB, D'Addio F, Usuelli V, Tezza S, Abdi R, Fiorina P. The rise, fall, and resurgence of immunotherapy in type 1 diabetes. Pharmacol Res. 2015;98:31–8.
Bassi R, Fiorina P. Impact of islet transplantation on diabetes complications and quality of life. Current diabetes reports. 2011;11:355.
Calton EK, Keane KN, Newsholme P, Soares MJ. The impact of vitamin D levels on inflammatory status: a systematic review of immune cell studies. PLoS One. 2015; doi:10.1371/journal.pone.0141770.
Plum LA, DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov. 2010:941–55.
Harinarayan CV, Arvind S, Joshi S, Thennarasu K, Vedavyas V, Baindur A. Improvement in pancreatic β cell function with vitamin D and calcium supplementation in vitamin D deficient non-diabetic subjects. Endocr Pract. 2013;6:1–33.
Pittas AG, Lau J, Hu FB, Dawson-Hughes B. The role of vitamin D and calcium in type 2 diabetes. A systematic review and meta-analysis. J Clin Endocrinol Metab. 2007;92:2017–29.
Clemente-Postigo M, Muñoz-Garach A, Serrano M, Garrido-Sánchez L, Bernal-López MR, Fernández-García D, Moreno-Santos I, Garriga N, Castellano-Castillo D, Camargo A, Fernández-Real JM, Cardona F, Tinahones FJ, Macías-González M. Serum 25-hydroxyvitamin D and adipose tissue vitamin D receptor gene expression: relationship with obesity and type 2 diabetes. J Clin Endocrinol Metab. 2015;100:591–5.
Vin hquốcLu'o'ng K, Nguyễn LT. The beneficial role of vitamin D in obesity: possible genetic and cell signaling mechanisms. Nutr J. 2013:12–89.
Vitseva OI, Tanriverdi K, Tchkonia TT, Kirkland JL, McDonnell ME, Apovian CM, Freedman J, Gokce N. Inducible toll-like receptor and NF-jB regulatory pathway expression in human adipose tissue. Obesity (Silver Spring). 2008;16:932–7.
Khoo AL, Chai L, Koenen H, Sweep F, Joosten I, Netea M, van der Ven A. Regulation of cytokine responses by seasonality of vitamin D status in healthy individuals. Clin Exp Immunol. 2011;164:72–9.
Bolland MJ, Grey AB, Ames RW, Mason BH, Horne AM, Gamble GD, Reid IR. The effects of seasonal variation of 25-hydroxyvitamin D and fat mass on a diagnosis of vitamin D sufficiency. Am J Clin Nutr. 2007;86:959–64.
Simpson RU, Thomas GA, Arnold AJ. Identification of 1,25-dihydroxyvitamin D3 receptors and activities in muscle. J Biol Chem. 1985;260:8882–91.
Maestro B, Campión J, Dávila N, Calle C. Stimulation by 1,25-dihydroxyvitamin D3 of insulin receptor expression and insulin responsiveness for glucose transport in U-937 human promonocytic cells. Endocr J. 2000;47:383–39.
Dunlop TW, Väisänen S, Frank C, Molnár F, Sinkkonen L, Carlberg C. The human peroxisome proliferator-activated receptor delta gene is a primary target of 1alpha,25-dihydroxyvitamin D3 and its nuclear receptor. J Mol Biol. 2005;349:248–60.
Luquet S, Gaudel C, Holst D, Lopez-Soriano J, Jehl-Pietri C, Fredenrich A, Grimaldi PA. Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochim Biophys Acta. 1740;2005:313–7.
Wei Y, Sowers JR, Clark SE, Li W, Ferrario CM, Stump CS. Angiotensin IIinduced skeletal muscle insulin resistance mediated by NF-kappaB activation via NADPH oxidase. Am J Physiol Endocrinol Metab. 2008;294:E345–51.
Wright DC, Hucker KA, Holloszy JO, Han DH. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes. 2004;53:330–5.
Muscogiuri G, Chavez AO, Gastaldelli A, Perego L, Tripathy D, Saad MJ, Velloso L, Folli F. The crosstalk between insulin and renin-angiotensin-aldosterone signaling systems and its effect on glucose metabolism and diabetes prevention. Curr Vasc Pharmacol. 2008;6:301–12.
Altieri B, Grant WB, Casa SD, Orio F, Pontecorvi A, Colao A, Sarno G, Muscogiuri G. Vitamin D and pancreas: the role of sunshine vitamin in the pathogenesis of diabetes mellitus and pancreatic cancer. Crit Rev Food Sci Nutr. 2016; doi:10.1080/10408398.2015.1136922.
Galbiati F, Polastri L, Thorens B, Dupraz P, Fiorina P, Cavallaro U, Christofori G, Davalli AM. Molecular pathways involved in the antineoplastic effects of calcitriol on insulinoma cells. Endocrinology. 2003;144:1832–41.
Galbiati F, Polastri L, Gregori S, Freschi M, Casorati M, Cavallaro U, Fiorina P, Bertuzzi F, Zerbi A, Pozza G, Adorini L, Folli F, Christofori G, Davalli AM. Antitumorigenic and antiinsulinogenic effects of calcitriol on insulinoma cells and solid beta-cell tumors. Endocrinology. 2002;143:4018–30.
Khoo A-L, Chai LY, Koenen HJ, Kullberg B-J, Joosten I, van der Ven AJ, Netea MG. 1, 25-Dihydroxyvitamin D3 modulates cytokine production induced by Candida albicans: impact of seasonal variation of immune responses. J Infect Dis. 2011;203:122–30.
Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr. 2006;83:754–9.
Petchey WG, Johnson DW, Isbel NM. Shining D’light on chronic kidney disease: mechanisms that may underpin the cardiovascular benefit of vitamin D. Nephrology (Carlton). 2011;16:351–67.
Chiu KC, Chu A, Go VL, Saad MF. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr. 2004;79:820–5.
Dutta D, Maisnam I, Shrivastava A, Sinha A, Ghosh S, Mukhopadhyay P, et al. Serum vitamin-D predicts insulin resistance in individuals with prediabetes. Indian J Med Res. 2013;138:853–60.
Manickam B, Neagu V, Kukreja SC, Barengolts E. Relationship between glycated hemoglobin and circulating 25-hydroxyvitamin D concentration in African American and Caucasian American men. Endocr Pract. 2013;19:73–80.
Scragg R, Sowers M, Bell C. Serum 25-hydroxyvitamin D, diabetes, and ethnicity in the third National Health and Nutrition examination survey. Diabetes Care. 2004;27:2813–8.
Chandler PD, Scott JB, Drake BF, Ng K, Manson JE, Rifai N, et al. Impact of vitamin D supplementation of inflammatory markers in African Americans: results of a four-arm, randomized, placebo-controlled trial. Cancer Prev Res. 2014;7:218–25.
Laird E, McNulty H, Ward M, Hoey L, McSorley E, Wallace JM, et al. Vitamin D deficiency is associated with inflammation in older Irish adults. J Clin Endocrinol Metab. 2014;99:1807–15.
Ganji V, Zhang X, Shaikh N, Tangpricha V. Serum 25-hydroxyvitamin D concentrations are associated with prevalence of metabolic syndrome and various cardiometabolic risk factors in US children and adolescents based on assay-adjustedserum25-hydroxyvitaminD. Am J Clin Nutr. 2011;94:225–33.
Tepper S, Shahar D, Geva D etal. Identifying the threshold for vitamin D insufficiency in relation to cardio-metabolic markers. Nutr Metab Cardiovasc Dis 2014; 24: 489–494.
Knekt P, Laaksonen M, Mattila C, et al. Serum vitaminD and subsequent occurrence of type 2 diabetes. Epidemiology. 2008;19:666–71.
Pittas AG, Dawson-Hughes B, Li T, et al. Vitamin D and calcium intake in relation to type 2 diabetes in women. Diabetes Care. 2006;29:650–6.
Liu S, Song Y, Ford ES, Manson JE, Buring JE, Ridker PM. Dietary calcium, vitaminD, and the prevalence of metabolic syndrome in middle-aged and older U.S. women. Diabetes Care. 2005;28:2926–32.
Kayaniyil S, Vieth R, Retnakaran R, et al. Association of vitamin D with insulin resistance and beta-cell dysfunction in subjects at risk for type 2 diabetes. DiabetesCare. 2010;33:1379–81.
Broder AR, Tobin JN, Putterman C. Disease-specific definitions of vitamin D deficiency need to be established in autoimmune and non-autoimmune chronic diseases: a retrospective comparison of three chronic diseases. Arthritis Res Ther. 2010;12:R191.
Hypponen E, Boucher BJ, Berry DJ, et al. 25-hydroxyvitamin D, IGF-1, and metabolic syndrome at 45 years of age: a cross-sectional study in the 1958 British birth cohort. Diabetes. 2008;57:298–305.
Lu L, Yu Z, Pan A, et al. Plasma 25-hydroxyvitamin D concentration and metabolic syndrome among middle-aged and elderly Chinese individuals. Diabetes Care. 2009;32:1278–83.
Heaney RP, French CB, Nguyen S, Ferreira M, Baggerly LL, Brunel L, Veugelers PA. Novel Approach Localizes the Association of Vitamin D Status With Insulin Resistance to One Region of the 25-Hydroxyvitamin D Continuum. Adv Nutr. 2013;4:303–10.
Garcia OP, Long KZ, Rosado JL. Impact of micronutrient deficiencies on obesity. Nutr Rev. 2009;67:559–72.
Cheng S, Massaro JM, Fox CS, et al. Adiposity, cardiometabolic risk, and vitamin D status: the Framingham heart study. Diabetes. 2010;59:242–8.
Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D supplementation on serum 25OHD in thin and obese women. J Steroid Biochem Mol Biol. 2013;136:195–200.
Pereira-Santos M, Costa PR, Assis AM, et al. Obesity and vitamin D deficiency: a systematic review and meta-analysis. Obes Rev. 2015;16:341–9.
Dhaliwal R, Mikhail M, Feuerman M, Aloia JF. The vitamin D dose response in obesity. Endocr Pract. 2014;20:1258–64.
Wright CS, Weinheimer-Haus EM, Fleet JC, Peacock M, Campbell WW. The apparent relation between plasma25-hydroxyvitaminD and insulin resistance is largely attributable to central adiposity in overweight and obese adults. JNutr. 2015;145:2683–9.
Wortsman J, Matsuoka LY, Chen TC, et al. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr. 2000;72:690–3.
Drincic AT, Armas LA, Van Diest EE, et al. Volumetric dilution, rather than sequestration best explains the low vitamin d status of obesity. Obesity (Silver Spring). 2012;20:1444–8.
Oh J, Weng S, Feltonetal SK. 1,25(OH) 2 vitamin D inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation. 2009;120:687–98.
Joergensen C, Gall MA, Schmedes A, Tarnow L, Parving HH, Rossing P. Vitamin D levels and mortality in type 2 diabetes. Diabetes Care. 2010;33:2238–43.
Astorri E, Fiorina P, Gavaruzzi G, Astorri A, Magnati G. Left ventricular function in insulin-dependent and in non-insulin-dependent diabetic patients: radionuclide assessment. Cardiology. 1997;88:152–5.
Perseghin G, Fiorina P, De Cobelli F, Scifo P, Esposito A, Canu T, Danna M, Gremizzi C. Cross-sectional assessment of the effect of kidney and kidney-pancreas transplantation on resting left ventricular energy metabolism in type 1 diabetic-uremic patients: a phosphorous-31 magnetic resonance spectroscopy study. Journal of the American College of Cardiology. 2005;46:1085–92.
Lee TI, Kao YH, Chen YC, Tsai WC, Chung CC, Chen YJ. Cardiac metabolism, inflammation, and peroxisome proliferator-activated receptors modulated by 1,25-dihydroxyvitamin D3 in diabetic rats. Int J Cardiol. 2014;176:151–7.
Sugden JA, Davies JI, Witham MD, Morris AD, Struthers AD. Vitamin D improves endothelial function in patients withType2diabetes mellitus and low vitamin D levels. Diabet Med. 2008;25:20–325.
Forouhi N, Menon R, Sharp S, et al. Effects of vitamin D2 or D3 supplementation on glycaemic control and cardiometabolic risk among people at risk of type 2 diabetes: results of a randomized double-blind placebo-controlled trial. Diabetes Obes Metab. 2015;18:392–400.
Witham MD, Dove FJ, Dryburgh M, et al. The effect of different doses of vitamin D (3) on markers of vascular health in patients with type 2 diabetes: a randomised controlled trial. Diabetologia. 2010;53:2112–9.
Asemi Z, Samimi M, Tabassi Z, Shakeri H, Esmaillzadeh A. Vitamin D supplementation affects serum high-sensitivity C-reactive protein, insulin resistance, and biomarkers of oxidative stress in pregnant women. J Nutr. 2013;143:1432–8.
Lind L, Pollare T, Hvarfner A, Lithell H, Sorensen OH, Ljunghall S. Long-term treatment with active vitamin D (alphacalcidol) in middle-age men with impaired glucose tolerance; effects on insulin secretion and sensitivity, glucose tolerance and blood pressure. Diabetes Res. 1989;11:141–7.
Afzal S, Bojesen SE, Nordestgaard BG. Low 25-hydroxyvitamin D and risk of type 2 diabetes: a prospective cohort study and metaanalysis. Clin Chem. 2013;59:381–91.
Song Y, Wang L, Pittas AG, et al. Blood 25-Hydroxy vitamin D levels and incident type 2 diabetes: a meta-analysis of prospective studies. Diabetes Care. 2013;36:1422–8.
Pittas AG, Harris SS, Stark PC, et al. The effects of calcium and vitamin D supplementation on blood glucose and markers of inflammation in nondiabetic adults. Diabetes Care. 2007;30:980–6.
Mitri J, Dawson-Hughes B, Hu FB, et al. Effects of vitamin D and calcium supplementation on pancreatic beta cell function, insulin sensitivity, and glycemia in adults at high risk of diabetes: the calcium and vitamin D for diabetes mellitus (CaDDM) randomized controlled trial. Am J Clin Nutr. 2011;94:486–94.
Zanetti M, Harris SS, Dawson-Hughes B. Ability of vitamin D to reduce inflammation in adults without acute illness. Nutr Rev. 2014;72:95–8.
Tzotzas T, Papadopoulou FG. Tziomalos K etal. Rising serum 25-hydroxy-vitamin D levels after weight loss in obese women correlate with improvement in insulin resistance. J Clin Endocrinol Metab. 2010;95:4251–7.
Shab-Bidar S, Neyestani TR, Djazayery A, et al. Regular consumption of vitamin D-fortified yogurt drink (Doogh) improved endothelial biomarkers in subjects with type 2 diabetes: a randomized double-blind clinical trial. BMC Med. 2011;9:125.
Jorde R, Figenschau Y. Supplementation with cholecalciferol does not improve glycaemic control in diabetic subjects with normal serum 25-hydroxyvitamin D levels. Eur J Nutr. 2009;48:349–54.
Avenell A, Cook JA, Mac Lennan GS, Mc Pherson GC. Vitamin D supplementation and type 2 diabetes: a substudy of a randomised placebo-controlled trial in older people. Age Ageing. 2009;38:606–9.
de Boer IH, Tinker LF, Connelly S, Curb JD, Howard BV, Kestenbaum B, Larson JC, Manson JE, Margolis KL, Siscovick DS. Weiss NS; Women's Health Initiative investigators. Calcium plus vitamin D supplementation and the risk of incident diabetes in the Women's Health Initiative. Diabetes Care. 2008;31:701–7.
Parekh D, Sarathi V, Shivane VK, Bandgar TR, Menon PS, Shah NS. Pilot study to evaluate the effect of short-term improvement in vitamin D status on glucose tolerance in patients with type 2 diabetes mellitus. Endocr Pract. 2010;16:600–8.
Heshmat R, Tabatabaei-Malazy O, Abbaszadeh-Ahranjani S, et al. Effect of vitamin D on insulin resistance and anthropometric parameters in Type 2 diabetes; a randomized double-blind clinical trial. Daru: journal of Faculty of Pharmacy, Tehran University of Medical Sciences. 2012; 20:10.
Kampmann U, Mosekilde L, Juhl C, Moller N, Christensen B, Rejnmark L, Wamberg L, Orskov L. Effects of 12 weeks high dose vitamin D3 treatment on insulin sensitivity, beta cell function, and metabolic markers in patients with type 2 diabetes and vitamin D insufficiency - a double-blind, randomized, placebo-controlled trial. Metabolism. 2014;63:1115–24.
Tepper S, Shahar DR, Geva D, Ish-Shalom S. Differences in homeostatic model assessment (HOMA) values and insulin levels after vitamin D supplementation in healthy men: a double-blind randomized controlled trial. Diabetes Obes Metab. 2016;18:633–7.
Von Hurst PR, Stonehouse W, Coad J. Vitamin D supplementation reduces insulin resistance in south Asian women living in New Zealand who are insulin resistant and vitamin D deficient-a randomised, placebo-controlled trial. Br J Nutr. 2010;103:549–55.
Nazarian S, St Peter JV, Boston RC, et al. Vitamin D3 supplementation improves insulin sensitivity in subjects with impaired fasting glucose. Transl Res. 2011;158:276–81.
Davidson MB, Duran P, Martin LL, Friedman TC. High-DoseVitaminDSupplementation inPeopleWithPrediabetesand Hypovitaminosis. Diabetes Care. 2013;36:260–6.
Ljunghall S, Lind L, Lithell H, et al. Treatment with one-alpha-hydroxycholecalciferol in middle-aged men with impaired glucose tolerance - a prospective randomized double-blind study. Acta Med Scand. 1987;222:361–7.
Tai K, Need AG, Horowitz CIM. Glucose tolerance and vitamin D: effects of treating vitamin D deficiency. Nutrition. 2008;24:950–6.
George PS, Pearson ER, Witham MD. Effect of vitamin D supplementation on glycaemic control and insulin resistance: a systematic reviewand meta-analysis. Diabet Med. 2012;29:e142–50.
Seida JC, Mitri J, Colmers IN, Majumdar SR, Davidson MB, Edwards AL, Hanley DA, Pittas AG, Tjosvold L, Johnson JA. Clinical review: effect of vitamin D3 supplementation on improving glucose homeostasis and preventing diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2014;99:3551–60.
Daniele G, Winnier D, Mari A, Bruder J, Fourcaudot M, Pengou Z, Tripathy D, Jenkinson C, Folli F. Sclerostin and insulin resistance in Prediabetes: evidence of a cross talk between bone and glucose metabolism. Diabetes Care. 2015;38:1509–17.
Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS, Karsenty G. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J Clin Invest. 2014;124:1–13.
Palsgaard J, Emanuelli B, Winnay JN, Sumara G, Karsenty G, Kahn CR. Cross-talk between insulin and Wnt signaling in preadipocytes. Role of Wnt co-receptor LDL receptor-related protein-5 (LRP5). J Biol Chem. 2016;291:16878.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Rights and permissions
About this article

Cite this article
Garbossa, S.G., Folli, F. Vitamin D, sub-inflammation and insulin resistance. A window on a potential role for the interaction between bone and glucose metabolism. Rev Endocr Metab Disord 18, 243–258 (2017). https://doi.org/10.1007/s11154-017-9423-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-017-9423-2