
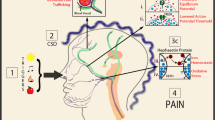
Abstract
Glutamate levels and lifetime in the brain extracellular space are dinamically regulated by a family of Na+- and K+-dependent glutamate transporters, which thereby control numerous brain functions and play a role in numerous neurological and psychiatric diseases. Migraine is a neurological disorder characterized by recurrent attacks of typically throbbing and unilateral headache and by a global dysfunction in multisensory processing. Familial hemiplegic migraine type 2 (FHM2) is a rare monogenic form of migraine with aura caused by loss-of-function mutations in the α2 Na/K ATPase (α2NKA). In the adult brain, this pump is expressed almost exclusively in astrocytes where it is colocalized with glutamate transporters. Knockin mouse models of FHM2 (FHM2 mice) show a reduced density of glutamate transporters in perisynaptic astrocytic processes (mirroring the reduced expression of α2NKA) and a reduced rate of glutamate clearance at cortical synapses during neuronal activity and sensory stimulation. Here we review the migraine-relevant alterations produced by the astrocytic glutamate transport dysfunction in FHM2 mice and their underlying mechanisms, in particular regarding the enhanced brain susceptibility to cortical spreading depression (the phenomenon that underlies migraine aura and can also initiate the headache mechanisms) and the enhanced algesic response to a migraine trigger.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glutamate levels and lifetime in extracellular space are finely regulated by a family of Na+- and K+-dependent glutamate transporters, which thereby control numerous brain functions, including termination of glutamate signaling at synapses, and play a role in numerous neurological and psychiatric diseases [1,2,3,4,5,6,7]. Here we will present a brief review of the role glutamate transporters play in migraine.
An Overview of Glutamate Transporters
Five glutamate transporters (GluTs) subtypes have been described: EAAC1 (EAAT3/Slc1a1/SLC1A1); GLT-1 (EAAT2/Slc1a2/SLC1A2); GLAST (EAAT1/Slc1a3/SLC1A3); EAAT4 (Slc1a6/SLC1A6/); and EAAT5 (Slc1a7/SLC1A7)Footnote 1. In the following, we will use the rodent nomenclature when reviewing data obtained in rodents and the human nomenclature when reviewing data obtained in humans. Reportedly, GluTs are trimers, with each monomer able to transport glutamate (and other substrates) and coupled ions. They comprise a “transport domain”, that binds and transports, and a “scaffold domain” that interacts with the membrane [8, 9]. In eukaryotic cell transporters, the stoichiometry of the process is the inward movement of one glutamate (which carries a negative charge), three Na+, and one H+, followed by the counter transport of one K+, resulting in the net influx of two positive charges per cycle [10, 11]. GluTs also mediate (in physiological conditions) a glutamate-dependent Cl− flux [12,13,14], whose function is poorly understood. The mechanisms of transport are similar among different GluTs, and for all of them the efficiency is about 50% [15]. The remaining 50% of glutamate molecules are released back in the extracellular space, and bind to glutamate receptors or transporters localized in the vicinity. Since the rates of glutamate binding to receptors and transporters are relatively similar, and the density of transporters inserted in membranes is four times higher than that of extrasynaptic receptors [16], in all likelihood glutamate molecules unbound from transporters will bound to other transporters [17]. Although the basic mechanisms of transport appear similar among GluTs, functional differences in their properties have been reported for, among the others, affinity for glutamate, steady state affinity, ratio of substrate transport versus anion permeation, and voltage-dependency [6].
Different subtypes of glutamate transporters are differentially expressed in neurons and glial cells and exhibit striking regional variations [3, 18]. For example, GLT-1 and GLAST are mostly expressed in astrocytes, EAAC1, EAAT4-5 in neurons, and EAAT4 and EAAT5 are by far more robustly expressed in cerebellum and retina than in all other brain regions [3, 19, 20]. EAAC1, GLT-1 and GLAST are widely expressed in the central nervous system, but their levels vary significantly across regions [18].
GLT-1 (EAAT2)
GLT-1 [21] has the highest density of expression in the vast majority of adult brain regions, and it is responsible for the largest proportion of glutamate transport [18]. Three splice variants of GLT-1 differing in the C-terminal domain are known: GLT-1a, the predominant variant, represents ca. 90% of total GLT-1 protein,GLT-1b, which represents ca. 6%; and GLT-1c, which accounts for ca. 1% of GLT-1 protein [22,23,24]. They have similar regional distribution. [5, 23, 25,26,27,28]. A large literature exists on GLT-1, which has been the subject of several excellent reviews [1, 3,4,5,6,7, 29]. For this reason, here we will briefly review only those aspects that are important for the understanding of the next sections.
Early localization studies showed that GLT-1 is expressed only by astrocytes (e.g., [18, 30,31,32,33,34]). These observations were somehow in conflict with the biochemical observation that high-affinity uptake of glutamate could be measured in a homogenate or in synaptosomal preparation, and in slices by autoradiography, strongly suggesting that the uptake was highly specific for glutamatergic nerve terminals [35]. Although a neuronal expression was reported (e.g., [34, 36,37,38,39,40,41]), the discrepancy was generally attributed to post-transcriptional mechanisms operating in neurons (e.g., [42]) or to the newly discovered isoforms [27, 43, 44]. An important breakthrough was the demonstration that optimal fixation conditions allow the detection of GLT1a protein in a fraction of hippocampal axons [26], a finding that has been replicated in neocortex [45]. Here, it was shown that 25% of GLT-1a positive profiles are axon terminals and that ca. 50% of GLT-1a positive profiles are in the vicinity of asymmetric synapses. Using pre-embedding electron microscopy, it was observed that 70% of GLT-1a located in the vicinity of asymmetric synapses is astrocytic and ca. 30% was neuronal. Post-embedding immunogold studies showed that the density of gold particles coding for GLT-1a is much higher in astrocytic processes than in axon terminals, a finding that was confirmed in GLT-1 eGFP BAC reporter transgenic adult mice [20]. In addition, it was shown that in both astrocytic processes and axon terminals most gold particles are localized in a membrane region extending for about 250 nm from active zone margin, with a peak at 140 nm for astrocytic processes and at 80 nm for axon terminals. These studies allowed to conclude, among others, that, although GLT-1a is expressed by both astrocytes and axon terminals, astrocytic GLT-1a predominates at asymmetric synapses, and that the perisynaptic localization of GLT-1a in cortex is well-suited to modulate glutamate concentrations at the cleft and also to restrict glutamate spillover.
Not only the astrocytic GLT-1 transporters mediate the majority of glutamate clearance during neuronal activity in the adult murine cortex [3, 46, 47], but the GLT-1-mediated Na+ influx is one of the major determinants for astrocytic Na+ signaling, which in turn is central to coordinate neuronal activity with the astrocytic homeostatic response [48, 49]. The large GLT-1-mediated Na+ influx that occurs in astrocytes during neuronal activity requires a rapid restoration of Na+ and K+ gradients, which is undertaken by Na+/K+-ATPase (NKA) pumps [50,51,52,53]. Three Na+/K+–ATPase α subunits (and 3 auxiliary β) are expressed in adult brain: α1 is considered the “housekeeping” pump expressed in all brain cells, while α3 is expressed only in neurons, and α2 almost exclusively in astrocytes [54,55,56]. Copurification by mass spectrometry and/or coimmunoprecipitation in forebrain synaptosome preparations showed that GLT-1 forms macromolecular complexes with α1, α2, and α3 NKAs [57,58,59]. Using several microscopy techniques, it has been recently shown that at excitatory synapses α1and α3 NKAs are exclusively neuronal (mainly in dendrites and in some axon terminals), while α2 NKA is almost exclusively astrocytic [60]. GLT-1 displays a differential colocalization with α1–3 NKAs. GLT-1 colocalizes with α2 NKA at perisynaptic astrocytic processes, and with α1 and α3 NKAs at axon terminals. GLT-1 and α2 NKA gold particles are ∼1.5–2 times closer than GLT-1/α1 and GLT-1/α3 particles. GLT-1/α2 complexes (edge to edge interdistance of gold particles ≤ 50 nm) concentrate at the perisynaptic region of distal astrocytic processes membranes, whereas neuronal GLT-1/α1 and GLT-1/α3 complexes are fewer and more uniformly distributed in axon terminals [60]. These data unveil different composition of GLT-1 and NKA α subunits complexes in the glial and neuronal domains of excitatory synapses. The spatial organization of GLT-1/α1–3 complexes suggests that GLT-1/NKA interaction is more efficient in astrocytes than in neurons, further supporting the dominant role of astrocytic GLT-1 in glutamate homeostasis.
GLT-1 expression and function are modulated by several drugs (e.g., [61]). Although the mechanisms are at present still poorly understood (but see [62]), these observations indicate that GLT-1 may be a therapeutic target to treat various neurological and psychiatric disorders and/or a useful tool in experimental research. Compounds that up-regulate GLT-1 expression include ceftriaxone, tamoxifen, 17B-estradiol, EGF, 5-a-MCPA and L-AP3 (group III mGluR antagonists), and minocycline [61]. Of them, ceftriaxone has attracted considerable attention following the demonstration that the β-lactam antibiotic ceftriaxone induces GLT-1 up-regulation (but not of the other GluTs) by increasing transcription of the scl1a2 gene through the nuclear factor-κB signaling pathway [63,64,65]. GLT-1 up-regulation by ceftriaxone reduces glutamate levels at the cleft, as shown by the severe impairment of mGluR-dependent long-term depression at hippocampal mossy fiber-CA3 synapses [66]. Compounds that down-regulate GLT-1 expression and function include clozapine [67], cronic morphine [68], and endothelins [69].
Migraine
Migraine is a common episodic neurological disorder with complex pathophysiology that is characterized by recurrent attacks of typically throbbing and unilateral, often severe, headache with certain associated features and by a global dysfunction in multisensory information processing [70,71,72]. It is generally believed that the development of migraine headache depends on the activation and sensitization of trigeminal sensory afferents that innervate cranial tissues (in particular the meninges and their large blood vessels) and subsequent activation and sensitization of second order neurons in the trigeminocervical complex (comprising the trigeminal nucleus caudalis and the dorsal horn of the first cervical segments, indicated here for simplicity as TNC) and of higher order neurons in areas of the brainstem and forebrain to which the TNC projects directly or indirectly. These areas comprise structures involved in the sensory/discriminatory, salience/alerting, and affective/motivational aspects of pain, as well as structures involved in the response to pain and in the descending facilitatory/inhibitory modulation of pain [70,71,72].
But migraine is much more than a pain disorder. It is a complex brain disorder characterized by a global dysfunction in multisensory information processing and integration. Indeed, the majority of migraine attacks feature sensory amplifications: photophobia, phonophobia, osmophobia, and cutaneous allodynia (i.e., the perception of light, sound, smell, and normal touch as amplified or painful). Hypersensitivity to sensory stimuli may persist in the interictal period, during which the brain of migraineurs show altered processing of noxious and non-noxious sensory information. The magnitude of some of these alterations increases in the interictal period in temporal relation to the next attack and becomes maximal before the attack in coincidence with prodromal premonitory symptoms (such as difficulty with speech, reading, concentration, increased emotionality, irritability, sensory hypersensitivity) that in many migraineurs are highly predictive of the attack. In one-third of patients the headache is preceded by transient neurological symptoms that are most frequently visual but may involve other senses and speech, the so called migraine aura [72,73,74].
The neurophysiological correlate of migraine aura is considered to be cortical spreading depression (CSD), a self-sustaining, slowly propagating (2–5 mm/min) wave of nearly complete depolarization of a population of brain cells that lasts about one minute and silences brain electrical activity for several minutes [75, 76]. CSD can be induced in healthy brain tissue by intense depolarizing stimuli that increase the extracellular concentration of K+ ions, [K]e, above a critical threshold and release glutamate and other neurotransmitters [76]. There is evidence from animal studies that CSD can activate and sensitize the trigeminovascular pain pathway and hence initiate the headache mechanisms [70]. In fact, a single experimental CSD can lead to delayed sustained increases in dural blood flow and in ongoing activity of rat dural nociceptors and TNC trigeminovascular neurons as well as delayed sensitization of these neurons [77,78,79,80,81]. Moreover a single CSD can produce periorbital mechanical allodynia, which is aborted by sumatriptan, a drug that is effective in aborting migraine pain [82]. Repeated CSDs produce a more robust pain phenotype lasting at least several days, accompanied by facial grimace as a sign of spontaneous discomfort.
Delayed trigeminal activation after a single CSD may result from CSD-induced release of proinflammatory molecules in the meninges, e.g. as a consequence of a parenchimal inflammation initiated by CSD-induced opening of pannexin1 channels and inflammasome activation [83, 84] and/or as a consequence of CSD-induced pial and dural macrophage activation [85]. Recent clinical imaging studies using a sensitive PET inflammation marker have provided supporting evidence for the presence of parenchimal and meningeal inflammation in migraine with aura patients [86, 87]. The labeling in the meninges was most prominent over the occipital cortex generating the visual aura and lasted several days after a migraine attack [87].
While in most cases the primary cause of migraine lies in the brain, the neurobiological mechanisms of the primary brain dysfunction(s) underlying the initiation of a migraine attack and susceptibility to CSD remain largely unknown. Also unknown are the neurobiological mechanisms underlying the global dysfunction in multisensory information processing (and its periodicity) and how they relate to those underlying the primary migraine causative brain dysfunctions.
Familial Hemiplegic Migraine
Migraine is a complex polygenic genetic disorder, with heritability estimates as high as 50% [88, 89]. Genome-wide association studies (GWAS) have identified 38 susceptibility loci for migraine [90]. However, the study of the functional consequences of GWAS hits is very difficult, if not impossible, given also the fact that they generally lie in intronic or intergenic regions and therefore they likely influence gene regulation rather than directly protein function. In contrast, familial hemiplegic migraine (FHM), a rare monogenic form of migraine with aura, is caused by mutations that directly affect protein function, and the functional consequences of the disease-causing mutations can be studied in genetic mouse models of the disease [88, 91,92,93]. Apart from the motor weakness or hemiplegia during aura and the possible longer duration of the aura, typical FHM attacks (as those occurring in pure FHM) resemble the attacks of common migraine with aura and both types of attacks may alternate in patients and co-occur within families. FHM and migraine with aura are considered to be part of a clinical and genetic spectrum [88, 93, 94]. Some FHM patients can have “atypical” severe attacks and show additional ictal and/or permanent neurological features such as epilepsy, loss of consciousness, ataxia and cognitive impairment [88, 91, 93].
Three FHM causative genes have been identified: CACNA1A, encoding the pore-forming subunit of the voltage-gated calcium channel CaV2.1 (FHM1) [95], ATP1A2, encoding the α2 Na/K ATPase (FHM2) [96] and SCNA1A, encoding the pore-forming subunit of the voltage-gated sodium channel NaV1.1 (FHM3) [97]. The CaV2.1 channel is widely expressed in the brain, it is localized at the active zones of most brain synaptic terminals and plays a dominant role in controlling neurotransmitter release at most excitatory and inhibitory brain synapses ([98] and references therein). The α2 Na/K ATPase (α2 NKA) is expressed primarily in neurons during embryonic development and at the time of birth and almost exclusively in astrocytes in the adult brain [55, 60, 99, 100]. At cortical excitatory synapses, the α2 NKA is colocalized with the glutamate transporters GLT-1 and GLAST at perisynaptic astrocytic processes (PAPs) [55, 60, 101], where a large fraction of GLT-1/α2 NKA couples exhibit a separation distance indicative of physical coupling [60]. The α2 NKA, which is the only NKA pump significantly expressed in cortical astrocytes [60, 102], plays a key role in K+ and glutamate clearance during neuronal activity [101, 103, 104] (see next chapter). Moreover, being essential for several astrocytic transporters relying on Na+ influx, its function is also important for astrocytic Na+ homeostasis, Ca2+ homeostasis and pH regulation [48, 49, 51, 53]. The NaV1.1 channel is highly expressed in inhibitory interneurons in several brain areas where it is mainly localized at the axon initial segment and plays a key role in interneurons excitability, particularly in sustaining high-frequency firing [105,106,107].
FHM1 and FHM3 mutations produce gain of function of recombinant CaV2.1 and NaV1.1 channels, respectively [98, 108,109,110]. Accordingly, in genetic mouse models of FHM1 (knockin mice carrying a different FHM1 mutation) the CaV2.1 calcium current is increased in different types of excitatory neurons [111,112,113,114,115] leading to increased action potential-evoked glutamate release at different types of excitatory synapses, including different intracortical and thalamocortical synapses [111, 113, 116, 117]. In a genetic mouse model of FHM3 the NaV1.1 sodium current is increased in cortical fast-spiking interneurons [118]. The migraine-relevant alterations in the brain of the FHM1 knockin mice, including the enhanced susceptibility to experimentally-induced CSD and the causative link between increased glutamatergic transmission and enhanced CSD susceptibility as well as the differential effect of FHM1 mutations on cortical excitatory and inhibitory synaptic transmission and the alterations in the trigeminovascular pain pathway were recently reviewed [92] and will not be discussed here.
FHM2 mutations cause the complete or partial loss-of-function of recombinant α2 NKAs [91, 119]. Accordingly, the brain expression of the α2 NKA is about 50% reduced in two different heterozygous knockin mice carrying FHM2 mutations, which produce complete loss-of-function and impaired membrane targeting of recombinant α2 NKAs [120, 121]. In the next chapter, we will review the consequences of FHM2 mutations on the expression and function of astrocytic glutamate transporters and the consequent migraine-relevant alterations produced in the brain of FHM2 knockin mice, in particular regarding the susceptibility to CSD and the algesic response to a migraine trigger.
Astrocytic Glutamate Transport Dysfunction in FHM2 and Consequent Migraine-Relevant Brain Alterations
In the somatosensory (S1) cortex of heterozygous knock-in mice carrying the α2 NKA W887R mutation, which causes pure FHM2 (FHM2 mice), the membrane density of the glutamate transporter GLT-1 in PAPs surrounding excitatory synapses is reduced by 50% [101]. The reduction of GLT-1 expression in PAPs mirrors the aforementioned 50% reduction in the expression of the α2 NKA protein in FHM2 mice [121]. Interestingly, the membrane density of GLT-1 in axon terminals, which do not express the α2 NKA, is unaltered in FHM2 mice [60, 101]. Conversely, administration of the GLT-1 up-regulator ceftriaxone [65] to the FHM2 mice did not result in an increase in GLT-1 at PAP membranes, but in an increase in GLT-1 at axon terminals [101]. The reduced density of GLT-1 in PAPs likely underlies the important finding that the rate of glutamate clearance during neuronal activity, elicited either by extracellular stimulation in cortical slices or by whisker stimulation in vivo in awake animals, is reduced in the S1 cortex of FHM2 mice [101, 122, 123]. The relative impairment of glutamate clearance by astrocytes in FHM2 mice is activity dependent, being larger after a train of pulses than after a single pulse stimulation and increasing with increasing stimulation frequency in cortical slices [101]. A reduced rate of glutamate clearance was also found in the cingulate cortex of FHM2 mice [124]. The expression of glutamate transporter GLAST, which appears implicated in astrocyte-mediated glutamate uptake in this cortical region [125], is reduced in the cingulate cortex of the FHM2 mutants [124]. In both somatosensory and cingulate cortices, also the rate of clearance of K+ ions released during neuronal activity is reduced in FHM2 mice [101] [124], showing the active role of the α2 NKA in K+ ions clearance.
Imaging extracellular glutamate using two-photon microscopy in awake mice revealed the presence of spontaneous focal, large glutamate transients (glutamatergic plumes, spreading from a central origin) in FHM2 mice, which were uncorrelated with the sensory stimulation [123]. Three findings support a key role of impaired glutamate clearance by astrocytes for plume generation: (1) spontaneous plumes occur in FHM2 mice but are almost completely absent in wild-type (WT) mice; (2) the spontaneous plumes in FHM2 mice are predominantly located in the superficial layer 1 (putative L1a), where, in comparison with L2/3, the coverage of glutamatergic synapses by PAPs expressing GLT-1 is reduced (as shown by the reduced density of GLT-1 + astrocyte processes and the increased percentage of excitatory synapses lacking GLT-1 + PAPs); and (3) plumes occur in WT mice after pharmacological inhibition of glutamate transporters [123]. The glutamatergic plumes are driven by both astrocytic glutamate clearance impairment and neuronal action-potential-independent release [123]. The conclusion that plumes glutamate is released from neuronal synapses in a calcium-dependent but action potential-independent manner is based on (1) pharmacological evidence showing that, although unaffected by TTX, plumes occurrence is strongly reduced by compounds that inhibit synaptic release and enhanced by compounds that increase synaptic release, and (2) the observation of spontaneous neural calcium transients, which mimicked the morphology and spatial spread of plumes, in L1a in FHM2 mice expressing a genetically encoded calcium sensor targeted to neurons [123]. Consistent with neuronal release of plumes glutamate is also the finding that manipulations causing intracellular Ca2+ increases in astrocytes failed to induce plumes in FHM2 mice [123]
A key migraine-relevant phenotype shown by FHM2 mice is their increased susceptibility to experimentally induced CSD, as revealed by a lower stimulation threshold for initiation of CSD and a higher rate of CSD propagation in vivo and in vitro [101, 121, 123]. The reduced rate of glutamate clearance can account for most of the facilitation of CSD initiation and for a large part of the facilitation of CSD propagation in FHM2 mice [101]. The remaining facilitation is likely due to the reduced rate of K+ clearance uncovered in these mutants [101]. Imaging of glutamate at the site of CSD induction in awake mice revealed that an increase in plumes frequency and in basal glutamate precedes and may predict CSD initiation [123]. Although an increase in plumes frequency and glutamate rise occurred with a smaller CSD-inducing stimulus in FHM2 compared to wild-type (WT) mice, the plumes frequency and the basal glutamate rise just preceding CSD ignition were similar in WT and FHM2 mice [123]. This finding supports the idea of a threshold level of glutamate and glutamatergic plumes necessary for CSD ignition regardless of genotype. Due to the reduced rate of glutamate clearance this threshold level is reached with a stimulus of lower intensity in FHM2, thus accounting for the facilitation of CSD initiation.
Further insights into the molecular and cellular mechanisms underlying enhanced susceptibility to CSD in FHM2 mice were provided by the finding that the relatively small slowing of glutamate clearance [101] results in increased amplitude and slowing of both rise time and decay of the NMDA receptor (NMDAR) EPSC elicited in L2/3 pyramidal cells of barrel cortex by (even low frequency) stimulation of neuronal afferents in L1 [122]. In agreement with the activity-dependence of the slowing of glutamate clearance, the increased activation of NMDARs in FHM2 mice is neuronal activity-dependent [101, 122]. The increased and prolonged activation of NMDARs in FHM2 mice is due to specific activation of extrasynaptic diheteromeric GluN1-N2B NMDARs, which do not contribute to the NMDAR EPSC elicited in L2/3 pyramidal cells in WT mice [122]. This is in line with the proposed role for extrasynaptic GluN1-N2B NMDARs as primary detectors of glutamate spillover [126, 127], which is enhanced at FHM2 synapses as a consequence of the impaired glutamate uptake by astrocytes. It is also consistent with the higher affinity for glutamate (and lower susceptibility to Mg block) of GluN1-N2B compared to other NMDA receptors, including the triheteromeric GluN1-N2A-N2B receptors [128, 129], which appear to be the main synaptic NMDARs at the L2/3 pyramidal cell synapses [122]. Interestingly, inhibition of diheteromeric GluN1-N2B NMDARs rescues most of the facilitation of CSD in FHM2 mice, as it increases the CSD threshold and reduces the velocity of CSD propagation to values close to those measured in WT mice, suggesting that the enhanced susceptibility to CSD in FHM2 mice is mainly due to specific activation of extrasynaptic GluN1-N2B NMDA receptors [122].
Confirming the key role of impaired glutamate clearance by GLT-1 transporters in facilitation of CSD initiation and propagation uncovered in FHM2 mice, an increased susceptibility to experimentally induced CSD and an increased rate of CSD propagation were recently reported in conditional GLT-1 knockout mice with about 70% reduction of GLT-1 expression in the brain [130]. In contrast, CSD induction and propagation were not altered in GLAST and EAAC1 knockout mice [130]. On the other hand, an increased rate of CSD propagation but unaltered stimulation threshold for CSD induction were recently reported in knockin mice carrying a different FHM2 mutation (E700K rather than W887R) [131]. In contrast with the complete loss-of-function and impaired membrane targeting of recombinant α2 NKAs produced by the W887R mutation, whereby the α2 NKA protein expression is reduced to half the WT level in the cortex of heterozygous FHM2 mice [121], the E700K mutation produces a decreased catalytic turnover rate of the α2 NKA, not a complete loss of function [132], and it is unclear whether the expression of the pump is reduced in the brain of heterozygous E700K knockin mice. If it is not reduced, then one expects a smaller impairment of the rate of glutamate clearance and a smaller facilitation of CSD initiation in the E700K compared to the W887R FHM2 knockin mice. This, together with the large variability, might explain the fact that the CSD threshold was reduced only as a trend in female E700K knockin mice [131]. However, E700K FHM2 knockin mice showed a larger c-Fos upregulation in the amygdala produced by CSD spread to this region compared to WT mice [131]. This seems interesting also in light of the fact that in a SHIRPA primary screening assessing sensory, motor and neuropsychiatric functions, the FHM2 mice showed an increased level of fear/anxiety as the only behavioural anomaly [121].
An increased susceptibility to experimentally induced CSD, as revealed by an increased frequency of CSDs elicited by prolonged epidural high KCl application in vivo, was reported in heterozygous knockin mice carrying the G301R FHM2 mutation [133]. In humans, this mutation causes a severe clinical syndrome with atypical attacks that may include, in addition to hemiplegic migraine, prolonged coma/torpor or confusional state, epileptic seizures, elevated temperature, cerebral edema and transient cerebellar signs [134, 135]. In agreement with and possibly contributing to the severe phenotype, full tonic-clonic seizures were frequently observed after a certain number of CSDs elicited by continuous KCl application in heterozygous G301R knockin mice [133]. The G301R mutation produces complete loss of function and lack of membrane targeting of recombinant α2 NKAs [134] and the α2 NKA protein expression is reduced (36–78%) in the brain of heterozygous G301R knockin mice [120]. Heterozygous G301R knockin mice revealed several behavioural alterations, which included increased startle response to aversive acoustic stimuli, stress-induced depression-like phenotypes, decreased sociability and increased compulsive behavior [120]. The increase in compulsive behavior was female-specific and reverted by progestin-only contraceptive treatment and also by memantine, a low-affinity open channel blocker of NMDARs [120]. At low therapeutic doses, memantine has been reported to preferentially block extrasynaptic NMDARs while relatively sparing synaptic NMDARs [121, 136, 137]. Memantine also rescued the increased compulsive repetitive behavior shown by mice with a 60–80% reduction of GLT-1 expression (after conditional knockout in adolescents) [138]. Thus, most likely, although not directly measured, in heterozygous G301R knockin mice the density of GLT-1 receptors in PAPs surrounding excitatory synapses is reduced resulting in impaired clearance of glutamate and increased activation of NMDARs as a consequence of glutamate spillover (possibly more, at certain synapses, than in pure FHM2 mice, which did not show compulsive behavior).
Besides increased susceptibility to CSD, another key migraine relevant phenotype shown by FHM2 mice is increased nociceptive response to facial mechanical stimulation after systemic injection of the nitric oxide donor nitroglycerin (NTG) [124]. In migraineurs, NTG administration induces a delayed migraine-like headache with associated features such as premonitory symptoms and allodynia [139, 140]. The NTG-induced hyperalgesia to facial mechanical stimulation in animals provides a behavioral model of the NTG-induced allodynia observed in migraineurs during the attack [141]. FHM2 mice developed facial mechanical hypersensitivity upon NTG doses that were ineffective in the WT littermates and, at higher NTG doses, showed a larger orofacial pain score than WT mice [124]. As the NTG-induced mechanical hyperalgesia was responsive to the migraine abortive drug sumatriptan in WT mice [141], the enhanced nociceptive response in FHM2 mice can be taken as evidence for an enhanced algesic response to a migraine trigger.
Romanos et al. [124] obtained interesting insights into the mechanisms underlying the enhanced sensitivity of FHM2 mice to a head pain trigger by investigating the consequences on neuronal function of the slowing of glutamate and K+ clearance in the cingulate cortex. The cingulate cortex plays a key role in pain processing, particularly in nociceptive hypersensitivity and sensitization [142, 143], and it is activated during both spontaneous migraine attacks and during the premonitory phase of the migraine-like headache induced by NTG [139, 144]. The slowdown of glutamate clearance by astrocytes in the cingulate cortex of FHM2 mice was shown to have two effects on neural function [124]: (1) it facilitated the generation of NMDA spikes in the tuft dendrites of layer 5 pyramidal cells and (2) it increased the output somatic burst firing promoted by these long-lasting dendritic regenerative depolarizations, which involve activation of extrasynaptic NMDARs by glutamate spillover [145, 146]. Local rescue of the astrocytic defect by expressing the WT Atp1a2 gene in the cingulate cortex of FHM2 mice, reversed the defective glutamate and K+ clearance by astrocytes as well as the facilitation of dendritic NMDA spike generation and the increased output firing induced by these spikes, thus showing a causal relationship between the local astrocyte malfunction and the observed NMDAR-mediated neuronal dysfunctions in the cingulate cortex [124]. Quite interestingly, astrocyte compensation of the defective ATPase in the cingulate cortex of FHM2 mice strongly reduced their increased nociceptive response upon NTG treatment [124].
The evidence that cingulate cortex astrocyte dysfunction in FHM2 mice leads to hypersensitivity to a migraine-relevant trigger suggests that this cortical area may be involved in pain generation and/or sensitization of the pain processing system in familial migraine. In fact, some of the downstream regions to which the cingulate cortex is connected (eg the rostral ventromedial medulla) are involved in descending modulatory pathways that play an important role in central sensitization of trigeminovascular neurons in the TNC [72, 73, 143]. Clinical observations and animal studies support the idea that facial allodynia in migraine reflects sensitization of trigeminovascular neurons in the TNC receiving convergent input from the meningeal nociceptors and facial skin [72]. Interestingly, facial mechanical allodynia measured in rats after repeated applications of an inflammatory soup to the dura (to model the recurrent trigeminovascular activation assumed to occur in chronic migraine) involves a strong decrease of GLT-1 expression, increased glutamate and increased tyrosine phosphorylation of GluN2B in the TNC [147, 148]. Increasing GLT-1 expression alleviated the facial mechanical allodynia, and decreased glutamate and phosphorylated GluN2B in the TNC [148].
While the facilitation of NMDA spikes in the apical dendrites of pyramidal cells and the consequent increase in output firing of these neurons in the cingulate cortex of FHM2 mice has important implications in the context of migraine pain [124], a dysfunctional regulation of NMDA spikes generation in apical dendrites of pyramidal cells in sensory cortices might contribute to the altered processing of sensory information revealed in migraineurs during the interictal period [72,73,74]. In fact, dendritic NMDA spikes evoked in vivo during sensory input strongly influence context-dependent sensory gain and perception [149,150,151,152]. Dendritic NMDA spikes are also involved in associative LTP and experience-dependent synaptic plasticity in vivo [153,154,155]. Indeed, long-term-potentiation induced by high frequency stimulation at the hippocampal perforant path synapses in the dentate gyrus was found to be enhanced in FHM2 mice; in contrast, long-term potentiation was unaltered at stratum radiatum-CA1 synapses, possibly in correlation with the differential expression of GLT-1 in the dentate gyrus and CA1 area [156].
In light of the findings that FHM2-associated astrocytic glutamate transporters dysfunction in particular brain regions may engender different migraine-relevant functional consequences (in particular enhanced susceptibility to CSD and hypersensitivity to a migraine pain trigger), it is interesting that a genome-wide association study identified a significant association between migraine and a single nucleotide polymorphism in astrocyte elevated gene-1 (AEG-1), which inhibits the promoter activity of EAAT2 encoding glutamate transporter GLT-1 in astrocytes [157].
Data Availability
Enquiries about data availability should be directed to the authors.
Notes
* EAAC1, GLT1, GLAST, EAAT4-5 refer to proteins in rodents; EAAT1-5 to proteins in humans; Slc1a1, Slc1a2, Slc1a3, Slc1a6, Slc1a7 to genes in rodents; and SLC1A1, SLC1A2, SLC1A3, SLC1A6, SLC1A7 to genes in humans.
References
Beart PM, O’Shea RD (2007) Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150:5–17. https://doi.org/10.1038/sj.bjp.0706949
Conti F, Weinberg RJ (1999) Shaping excitation at glutamatergic synapses. Trends Neurosci 22:451–458. https://doi.org/10.1016/s0166-2236(99)01445-9
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105. https://doi.org/10.1016/s0301-0082(00)00067-8
Lauriat TL, McInnes LA (2007) EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders. Mol Psychiatry 12:1065–1078. https://doi.org/10.1038/sj.mp.4002065
Maragakis NJ, Dykes-Hoberg M, Rothstein J (2004) Altered expression of the glutamate transporter EAAT2b in neurological disease. Ann Neurol 55:469–477. https://doi.org/10.1002/ana.20003
Ryan RM, Ingram SL, Scimemi A (2021) Regulation of glutamate, GABA and dopamine transporter uptake, surface mobility and expression. Front Cell Neurosci 15:670346. https://doi.org/10.3389/fncel.2021.670346
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51:333–355. https://doi.org/10.1016/j.neuint.2007.03.012
Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445:387–393. https://doi.org/10.1038/nature05455
Reyes N, Ginter C, Boudker O (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462:880–885. https://doi.org/10.1038/nature08616
Levy LM, Warr O, Attwell D (1998) Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J Neurosci 18:9620–9628. https://doi.org/10.1523/jneurosci.18-23-09620
Zerangue N, Kavanaugh MP (1996) Flux coupling in a neuronal glutamate transporter. Nature 383:634–637. https://doi.org/10.1038/383634a0
Picaud S, Larsson HP, Wellis DP, Lecar H, Werblin F (1995) Cone photoreceptors respond to their own glutamate release in the tiger salamander. Proc Natl Acad Sci USA 92:9417–9421. https://doi.org/10.1073/pnas.92.20.9417
Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP (1995) Kinetics of a human glutamate transporter. Neuron 14:1019–1027. https://doi.org/10.1016/0896-6273(95)90340-2
Eliasof S, Jahr CE (1996) Retinal glial cell glutamate transporter is coupled to an anionic conductance. Proc Natl Acad Sci USA 93:4153–4158. https://doi.org/10.1073/pnas.93.9.4153
Tzingounis AV, Wadiche JI (2007) Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 8:935–947. https://doi.org/10.1038/nrn2274
Lehre KP, Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757. https://doi.org/10.1523/jneurosci.18-21-08751.1998
Leary GP, Holley DC, Stone EF, Lyda BR, Kalachev LV, Kavanaugh MP (2011) The central cavity in trimeric glutamate transporters restricts ligand diffusion. Proc Natl Acad Sci USA 108:14980–14985. https://doi.org/10.1073/pnas.1108785108
Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13:713–725. https://doi.org/10.1016/0896-6273(94)90038-8
Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18:3606–3619. https://doi.org/10.1523/jneurosci.18-10-03606.1998
de Vivo L, Melone M, Rothstein JD, Conti F (2010) GLT-1 promoter activity in astrocytes and neurons of mouse hippocampus and somatic sensory cortex. Front Neuroanat 3:31. https://doi.org/10.3389/neuro.05.031.2009
Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI (1992) Cloning and expression of a rat brain L-glutamate transporter. Nature 360:464–467. https://doi.org/10.1038/360464a0
Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA (2002) Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 22:2142–2152. https://doi.org/10.1523/JNEUROSCI.22-06-02142.2002
Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG et al (2009) The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience 162:1055–1071. https://doi.org/10.1016/j.neuroscience.2009.03.048
Rauen T, Wiessner M, Sullivan R, Lee A, Pow DV (2004) A new GLT1 splice variant: cloning and immunolocalization of GLT1c in the mammalian retina and brain. Neurochem Int 45:1095–1106. https://doi.org/10.1016/j.neuint.2004.04.006
Bassan M, Liu H, Madsen KL, Armsen W, Zhou J, Desilva T et al (2008) Interaction between the glutamate transporter GLT1b and the synaptic PDZ domain protein PICK1. Eur J Neurosci 27:66–82. https://doi.org/10.1111/j.1460-9568.2007.05986.x
Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA (2004) The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24:1136–1148. https://doi.org/10.1523/JNEUROSCI.1586-03.2004
Schmitt A, Asan E, Lesch KP, Kugler P (2002) A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience 109:45–61. https://doi.org/10.1016/s0306-4522(01)00451-1
Gonzalez-Gonzalez IM, Garcia-Tardon N, Cubelos B, Gimenez C, Zafra F (2008) The glutamate transporter GLT1b interacts with the scaffold protein PSD-95. J Neurochem 105:1834–1848. https://doi.org/10.1111/j.1471-4159.2008.05281.x
Gegelashvili G, Schousboe A (1997) High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol 52:6–15. https://doi.org/10.1124/mol.52.1.6
Danbolt NC, Storm-Mathisen J, Kanner BI (1992) An [Na+ + K+] coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 51:295–310. https://doi.org/10.1016/0306-4522(92)90316-t
Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC (1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15:1835–1853. https://doi.org/10.1523/JNEUROSCI.15-03-01835.1995
Levy LM, Lehre KP, Rolstad B, Danbolt NC (1993) A monoclonal antibody raised against an [Na+ + K+] coupled L-glutamate transporter purified from rat brain confirms glial cell localization. FEBS Lett 317:79–84. https://doi.org/10.1016/0014-5793(93)81495-l
Minelli A, Barbaresi P, Reimer RJ, Edwards RH, Conti F (2001) The glial glutamate transporter GLT-1 is localized both in the vicinity of and at distance from axon terminals in the rat cerebral cortex. Neuroscience 108:51–59. https://doi.org/10.1016/s0306-4522(01)00375-x
Torp R, Danbolt NC, Babaie E, Bjoras M, Seeberg E, Storm-Mathisen J, Ottersen OP (1994) Differential expression of two glial glutamate transporters in the rat brain: an in situ hybridization study. Eur J Neurosci 6:936–942. https://doi.org/10.1111/j.1460-9568.1994.tb00587.x
Fonnum F (1984) Glutamate: a neurotransmitter in mammalian brain. J Neurochem 42:1–11. https://doi.org/10.1111/j.1471-4159.1984.tb09689.x
Brooks-Kayal AR, Munir M, Jin H, Robinson MB (1998) The glutamate transporter, GLT-1, is expressed in cultured hippocampal neurons. Neurochem Int 33:95–100. https://doi.org/10.1016/s0197-0186(98)00018-7
Mennerick S, Dhond RP, Benz A, Xu W, Rothstein JD, Danbolt NC, Isenberg KE, Zorumski CF (1998) Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J Neurosci 18:4490–4499. https://doi.org/10.1523/JNEUROSCI.18-12-04490.1998
Plachez C, Danbolt NC, Recasens M (2000) Transient expression of the glial glutamate transporters GLAST and GLT in hippocampal neurons in primary culture. J Neurosci Res 59:587–593. https://doi.org/10.1002/(SICI)1097-4547(20000301)59:5%3c587::AID-JNR1%3e3.0.CO;2-L
Rauen T, Kanner BI (1994) Localization of the glutamate transporter GLT-1 in rat and macaque monkey retinae. Neurosci Lett 169:137–140. https://doi.org/10.1016/0304-3940(94)90375-1
Schmitt A, Asan E, Püschel B, Jöns T, Kugler P (1996) Expression of the glutamate transporter GLT1 in neural cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience 71:989–1004. https://doi.org/10.1016/0306-4522(95)00477-7
Torp R, Hoover F, Danbolt NC, Storm-Mathisen J, Ottersen OP (1997) Differential distribution of the glutamate transporters GLT1 and rEAAC1 in rat cerebral cortex and thalamus: an in situ hybridization analysis. Anat Embryol 195:317–326. https://doi.org/10.1007/s004290050051
Danbolt NC, Chaudhry FA, Dehnes Y, Lehre KP, Levy LM, Ullensvang K, Storm-Mathisen J (1998) Properties and localization of glutamate transporters. Progress in Brain Research, vol 116. Elsevier, Amsterdam, pp 23–43
Reye P, Sullivan R, Fletcher EL, Pow DV (2002) Distribution of two splice variants of the glutamate transporter GLT1 in the retinas of humans, monkeys, rabbits, rats, cats, and chickens. J Comp Neurol 445:1–12. https://doi.org/10.1002/cne.10095
Sullivan R, Rauen T, Fischer F, Wiessner M, Grewer C, Bicho A, Pow DV (2004) Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: implications for CNS glutamate homeostasis. Glia 45:155–169. https://doi.org/10.1002/glia.10317
Melone M, Bellesi M, Conti F (2009) Synaptic localization of GLT- 1a in the rat somatic sensory cortex. Glia 57:108–117. https://doi.org/10.1002/glia.20744
Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA (2015) Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 35:5187–5201. https://doi.org/10.1523/JNEUROSCI.4255-14.2015
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichichara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702. https://doi.org/10.1126/science.276.5319.1699
Rose CR, Verkhratsky A (2016) Glial ionic excitability: the role for sodium. Glia 64:1609–1610. https://doi.org/10.1002/glia.23012
Rose CR, Verkhratsky A (2016) Principles of sodium homeostasis and sodium signalling in astroglia. Glia 64:1611–1627. https://doi.org/10.1002/glia.22964
Kirischuk S, Kettenmann H, Verkhratsky A (2007) Membrane currents and cytoplasmic sodium transients generated by glutamate transport in Bergmann glial cells. Pflügers Arch 454:245–252. https://doi.org/10.1007/s00424-007-0207-5
Kirischuk S, Parpura V, Verkhratsky A (2012) Sodium dynamics: another key to astroglial excitability? Trends Neurosci 35:497–506. https://doi.org/10.1016/j.tins.2012.04.003
Rose CR, Karus C (2013) Two sides of the same coin: sodium homeostasis and signaling in astrocytes under physiological and pathophysiological conditions. Glia 61:1191–1205. https://doi.org/10.1002/glia.22492
Verkhratsky A, Nedergaard M (2018) Physiology of astroglia. Physiol Rev 98:239–389. https://doi.org/10.1152/physrev.00042.2016
Cameron R, Klein L, Shyjan AW, Rakic P, Levenson R (1994) Neurons and astroglia express distinct subsets of Na, K-ATPase α and β subunits. Mol Brain Res 21:333–343. https://doi.org/10.1016/0169-328x(94)90264-x
Cholet N, Pellerin L, Magistretti PJ, Hamel E (2002) Similar perisynaptic glial localization for the Na+, K+-ATPase α2 subunit and the glutamate transporters GLAST and GLT-1 in the rat somatosensory cortex. Cereb Cortex 12:515–525. https://doi.org/10.1093/cercor/12.5.515
McGrail KM, Phillips JM, Sweadner KJ (1991) Immunofluorescent localization of three Na, K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na, K-ATPase. J Neurosci 11:381–391. https://doi.org/10.1523/JNEUROSCI.11-02-00381.1991
Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, Spruce LA, Xiao R, Guo W, Putt M et al (2011) Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 31:18275–18288. https://doi.org/10.1523/JNEUROSCI.3305-11.2011
Illarionava NB, Brismar H, Aperia A, Gunnarson E (2014) Role of Na, K-ATPase α1 and α2 isoforms in the support of astrocyte glutamate uptake. PLoS One. 9:e98469. https://doi.org/10.1371/journal.pone.0098469
Rose EM, Koo JC, Antflick JE, Ahmed SM, Angers S, Hampson DR (2009) Glutamate transporter coupling to Na K-ATPase. J Neurosci 29:81438155. https://doi.org/10.1523/JNEUROSCI.1081-09.2009
Melone M, Ciriachi C, Pietrobon D, Conti F (2019) Heterogeneity of astrocytic and neuronal GLT-1 at cortical excitatory synapses, as revealed by its colocalization with Na+/K+-ATPase α isoforms. Cereb Cortex 29:3331–3350. https://doi.org/10.1093/cercor/bhy203
Soni N, Reddy BV, Kumar P (2014) GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol Biochem Behav 127:70–81. https://doi.org/10.1016/j.pbb.2014.10.001
Guillem AM, Krizman EN, Robinson MB (2022) Rapid regulation of glutamate transport: where do we go from here? Neurochem Res 47:61–84. https://doi.org/10.1007/s11064-021-03329-7
Bellesi M, Melone M, Gubbini A, Battistacci S, Conti F (2009) GLT-1 upregulation impairs prepulse inhibition of the startle reflex in adult rats. Glia 57:703–713. https://doi.org/10.1002/glia.20798
Lee S-G, Zhao-Zhong S, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB (2008) Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem 283:13116–23. https://doi.org/10.1074/jbc.M707697200
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB (2005) β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433:73–77. https://doi.org/10.1038/nature03180. (PMID: 15635412)
Omrani A, Melone M, Bellesi M, Safiulina V, Aida T, Tanaka K, Cherubini E, Conti F (2009) Up-regulation of GLT-1 severely impairs LTD at mossy fibre–CA3 synapses. J Physiol 587:4575–4588. https://doi.org/10.1113/jphysiol.2009.177881
Melone M, Vitellaro-Zuccarello L, Vallejo-Illarramendi A, Pérez-Samartin A, Matute C, Cozzi A, Pellegrini-Giampietro DE, Rothstein JD, Conti F (2001) The expression of glutamate transporter GLT-1 in the rat cerebral cortex is down-regulated by the antipsychotic drug clozapine. Mol Psychiatry 6:380–386. https://doi.org/10.1038/sj.mp.4000880
Lim G, Wang S, Mao J (2005) cAMP and protein kinase A contribute to the downregulation of spinal glutamate transporters after chronic morphine. Neurosci Lett 376:9–13. https://doi.org/10.1016/j.neulet.2004.11.016
Rozyczka J, Figiel M, Engele J (2004) Endothelins negatively regulate glial glutamate transporter expression. Brain Pathol 14:406–414. https://doi.org/10.1111/j.1750-3639.2004.tb00084.x
Burstein R, Noseda R, Borsook D (2015) Migraine: multiple processes, complex pathophysiology. J Neurosci 35:6619–6629. https://doi.org/10.1523/JNEUROSCI.0373-15.2015
Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S (2017) Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev 97:553–622. https://doi.org/10.1152/physrev.00034.2015
Pietrobon D, Moskowitz MA (2013) Pathophysiology of migraine. Annu Rev Physiol 75:365–391. https://doi.org/10.1146/annurev-physiol-030212-183717
Brennan KC, Pietrobon D (2018) A systems neuroscience approach to migraine. Neuron 97:1004–1021. https://doi.org/10.1016/j.neuron.2018.01.029
de Tommaso M, Ambrosini A, Brighina F, Coppola G, Perrotta A, Pierelli F, Sandrini G, Valeriani M, Marinazzo D, Stramaglia S, Schoenen J (2014) Altered processing of sensory stimuli in patients with migraine. Nat Rev Neurol 10:144–155. https://doi.org/10.1038/nrneurol.2014.14
Ayata C, Lauritzen M (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95:953–993. https://doi.org/10.1152/physrev.00027.2014
Pietrobon D, Moskowitz MA (2014) Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 15:379–393. https://doi.org/10.1038/nrn3770
Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA (2002) Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8:136–142. https://doi.org/10.1038/nm0202-136
Melo-Carrillo A, Noseda R, Nir R-R, Schain AJ, Stratton J, Strassman AM, Burstein R (2017) Selective inhibition of trigeminovascular neurons by Ffemanezumab: a humanized monoclonal anti-CGRP antibody. J Neurosci 37:7149–7163. https://doi.org/10.1523/JNEUROSCI.0576-17.2017
Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R (2010) Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30:8807–8814. https://doi.org/10.1523/JNEUROSCI.0511-10.2010
Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R (2011) Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 69:855–865. https://doi.org/10.1002/ana.22329
Zhao J, Levy D (2016) Cortical spreading depression promotes persistent mechanical sensitization of intracranial meningeal afferents: implications for the intracranial mechanosensitivity of migraine. eNeuro 3(6):ENEURO.0287-16.2016. https://doi.org/10.1523/ENEURO.0287-16.2016
Harriott AM, Chung DY, Uner A, Bozdayi RO, Morais A, Takizawa T, Qin T, Ayata C (2021) Optogenetic spreading depression elicits trigeminal pain and anxiety behavior. Ann Neurol 89:99–110. https://doi.org/10.1002/ana.25926
Erdener ŞE, Kaya Z, Dalkara T (2021) Parenchymal neuroinflammatory signaling and dural neurogenic inflammation in migraine. J Headache Pain 22:138. https://doi.org/10.1186/s10194-021-01353-0
Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD, Dalkara T (2013) Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339:1092–1095. https://doi.org/10.1126/science.1231897
Schain AJ, Melo-Carrillo A, Borsook D, Grutzendler J, Strassman AM, Burstein R (2018) Activation of pial and dural macrophages and dendritic cells by cortical spreading depression. Ann Neurol 83:508–521. https://doi.org/10.1002/ana.25169
Albrecht DS, Mainero C, Ichijo E, Ward N, Granziera C, Zürcher NR, Akeju O, Bonnier G, Price J, Hooker JM, Napadow V, Loggia ML, Hadjikhani N (2019) Imaging of neuroinflammation in migraine with aura: A [(11)C]PBR28 PET/MRI study. Neurology 92:e2038–e2050. https://doi.org/10.1212/WNL.0000000000007371
Hadjikhani N, Albrecht DS (2020) Extra-axial inflammatory signal in parameninges in migraine with visual aura. Ann Neurol 87:939–949. https://doi.org/10.1002/ana.25731
Ferrari MD, Klever RR, Terwindt GM, Ayata C, van den Maagdenberg AMJM (2015) Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol 14:65–80. https://doi.org/10.1016/S1474-4422(14)70220-0
Sutherland HG, Griffiths LR (2017) Genetics of migraine: Insights into the molecular basis of migraine disorders. Headache 57:537–569. https://doi.org/10.1111/head.13053
Gormley P et al (2016) Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 48:856–866. https://doi.org/10.1038/ng.3598
Pietrobon D (2007) Familial hemiplegic migraine. Neurotherapeutics 4:274–284. https://doi.org/10.1016/j.nurt.2007.01.008
Pietrobon D, Brennan KC (2019) Genetic mouse models of migraine. J Headache Pain 20:79. https://doi.org/10.1186/s10194-019-1029-5
Russell MB, Ducros A (2011) Sporadic and familial hemiplegic migraine: pathophysiological mechanisms, clinical characteristics, diagnosis, and management. Lancet Neurol 10:457–470. https://doi.org/10.1016/S1474-4422(11)70048-5
Gormley P et al (2018) Common variant burden contributes to the familial aggregation of migraine in 1,589 families. Neuron 99:1098. https://doi.org/10.1016/j.neuron.2018.08.029
Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SMG, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Hommen G-JB, Hofker MH, Ferrari MD, Frants RR (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87:543–552. https://doi.org/10.1016/s0092-8674(00)81373-2
De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 33:192–196. https://doi.org/10.1038/ng1081
Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366:371–377. https://doi.org/10.1016/S0140-6736(05)66786-4
Pietrobon D (2013) Calcium channels and migraine. Biochim Biophys Acta 1828:1655–1665. https://doi.org/10.1016/j.bbamem.2012.11.012
Ikeda K, Onaka T, Yamakado M, Nakai J, Ishikawa TO, Taketo MM, Kawakami K (2003) Degeneration of the amygdala/piriform cortex and enhanced fear/anxiety behaviors in sodium pump alpha2 subunit (Atp1a2)-deficient mice. J Neurosci 23:4667–4676. https://doi.org/10.1523/JNEUROSCI.23-11-04667.2003
Moseley AE, Lieske SP, Wetzel RK, James PF, He S, Shelly DA, Paul RJ, Boivin GP, Witte DP, Ramirez JM, Sweadner KJ, Lingrel JB (2003) The Na, K-ATPase alpha 2 isoform is expressed in neurons, and its absence disrupts neuronal activity in newborn mice. J Biol Chem 278:5317–5324. https://doi.org/10.1074/jbc.M211315200
Capuani C, Melone M, Tottene A, Bragina L, Crivellaro G, Santello M, Casari G, Conti F, Pietrobon D (2016) Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol Med 8:967–986. https://doi.org/10.15252/emmm.201505944
Stoica A, Larsen BR, Assentoft M, Holm R, Holt LM, Vilhardt F, Vilsen B, Lykke-Hartmann K, Olsen ML, MacAulay N (2017) The alpha2beta2 isoform combination dominates the astrocytic Na+/K+-ATPase activity and is rendered nonfunctional by the alpha2.G301R familial hemiplegic migraine type 2-associated mutation. Glia 65:1777–1793. https://doi.org/10.1002/glia.23194
Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N (2014) Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62:608–622. https://doi.org/10.1002/glia.22629
Verkhratsky A, Semyanov A (2022) The great astroglial metabolic revolution: mitochondria fuel astrocyte homeostatic support and neuroprotection. Cell Calcium 104:102583. https://doi.org/10.1016/j.ceca.2022.102583
Hedrich UBS, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J, Escayg A, Dihné M, Beck H, Mantegazza M, Lerche H (2014) Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J Neurosci 34:14874–14889. https://doi.org/10.1523/JNEUROSCI.0721-14.2014
Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K (2007) Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27:5903–5914. https://doi.org/10.1523/JNEUROSCI.5270-06.2007
Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9:1142–1149. https://doi.org/10.1038/nn1754
Cestèle S, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M (2013) Nonfunctional NaV1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc Natl Acad Sci USA 110:17546–17551. https://doi.org/10.1073/pnas.1309827110
Pietrobon D (2018) Ion channels in migraine disorders. Curr Opin Physiol 2:98–108. https://doi.org/10.1016/j.cophys.2018.02.001
Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D (2002) Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci USA 99:13284–13289. https://doi.org/10.1073/pnas.192242399
Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, van den Maagdenberg AMJM, Ferrari MD, Pietrobon D (2009) Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 61:762–773. https://doi.org/10.1016/j.neuron.2009.01.027
van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, van de Ven RC, Tottene A, van der Kaa J, Plomp JJ, Frants RR, Ferrari MD (2004) A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41:701–710. https://doi.org/10.1016/s0896-6273(04)00085-6
Di Guilmi MN, Wang T, Inchauspe CG, Forsythe ID, Ferrari MD, van den Maagdenberg AM, Borst JG, Uchitel OD (2014) Synaptic gain-of-function effects of mutant Cav2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. J Neurosci 34:7047–7058. https://doi.org/10.1523/JNEUROSCI.2526-13.2014
Fioretti B, Catacuzzeno L, Sforna L, Gerke-Duncan MB, van den Maagdenberg AMJM, Franciolini F, Connor M, Pietrobon D (2011) Trigeminal ganglion neuron subtype-specific alterations of CaV2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J Physiol 589:5879–5895. https://doi.org/10.1113/jphysiol.2011.220533
van den Maagdenberg AM et al (2010) High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol 67:85–98. https://doi.org/10.1002/ana.21815
Marchionni I, Pilati N, Forli A, Sessolo M, Tottene A, Pietrobon D (2022) Enhanced feedback inhibition due to increased recruitment of somatostatin-expressing interneurons and enhanced cortical recurrent excitation in a genetic mouse model of migraine. J Neurosci 42:6654–6666. https://doi.org/10.1523/JNEUROSCI.0228-22.2022
Tottene A, Favero M, Pietrobon D (2019) Enhanced thalamocortical synaptic transmission and dysregulation of the excitatory-inhibitory balance at the thalamocortical feedforward inhibitory microcircuit in a genetic mouse model of migraine. J Neurosci 39:9841–9851. https://doi.org/10.1523/JNEUROSCI.1840-19.2019
Auffenberg E et al (2021) Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J Clin Invest 131(21):e142202. https://doi.org/10.1172/JCI142202
Bøttger P, Doğanlı C, Lykke-Hartmann K (2012) Migraine- and dystonia-related disease-mutations of Na+/K+-ATPases: relevance of behavioral studies in mice to disease symptoms and neurological manifestations in humans. Neurosci Biobehav Rev 36:855–871. https://doi.org/10.1016/j.neubiorev.2011.10.005
Bøttger P, Glerup S, Gesslein B, Illarionova NB, Isaksen TJ, Heuck A, Clausen BH, Füchtbauer E-M, Gramsbergen JB, Gunnarson E, Aperia A, Lauritzen M, Lambertsen KL, Nissen P, Lykke-Hartmann K (2016) Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci Rep 6:22047. https://doi.org/10.1038/srep22047
Leo L, Gherardini L, Barone V, De Fusco M, Pietrobon D, Pizzorusso T, Casari G (2011) Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet 7:e1002129. https://doi.org/10.1371/journal.pgen.1002129
Crivellaro G, Tottene A, Vitale M, Melone M, Casari G, Conti F, Santello M, Pietrobon D (2021) Specific activation of GluN1-N2B NMDA receptors underlies facilitation of cortical spreading depression in a genetic mouse model of migraine with reduced astrocytic glutamate clearance. Neurobiol Dis 156:105419. https://doi.org/10.1016/j.nbd.2021.105419
Parker PD, Suryavanshi P, Melone M, Sawant-Pokam PA, Reinhart KM, Kaufmann D, Theriot JJ, Pugliese A, Conti F, Shuttleworth CW, Pietrobon D, Brennan KC (2021) Non-canonical glutamate signaling in a genetic model of migraine with aura. Neuron 109:611-628.e618. https://doi.org/10.1016/j.neuron.2020.11.018
Romanos J, Benke D, Pietrobon D, Zeilhofer HU, Santello M (2020) Astrocyte dysfunction increases cortical dendritic excitability and promotes cranial pain in familial migraine. Sci Adv 6(23):eaaz1584. https://doi.org/10.1126/sciadv.aaz1584
Romanos J, Benke D, Saab AS, Zeilhofer HU, Santello M (2019) Differences in glutamate uptake between cortical regions impact neuronal NMDA receptor activation. Commun Biol 2:127. https://doi.org/10.1038/s42003-019-0367-9
Scimemi A, Fine A, Kullmann DM, Rusakov DA (2004) NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci 24:4767–4777. https://doi.org/10.1523/JNEUROSCI.0364-04.2004
Scimemi A, Tian H, Diamond JS (2009) Neuronal transporters regulate glutamate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. J Neurosci 29:14581–14595. https://doi.org/10.1523/JNEUROSCI.4845-09.2009
Stroebel D, Carvalho S, Grand T, Zhu S, Paoletti P (2014) Controlling NMDA receptor subunit composition using ectopic retention signals. J Neurosci 34:16630–16636. https://doi.org/10.1523/JNEUROSCI.2736-14.2014
Stroebel D, Casado M, Paoletti P (2018) Triheteromeric NMDA receptors: from structure to synaptic physiology. Curr Opin Physiol 2:1–12. https://doi.org/10.1016/j.cophys.2017.12.004
Aizawa H, Sun W, Sugiyama K, Itou Y, Aida T, Cui W, Toyoda S, Terai H, Yanagisawa M, Tanaka K (2020) Glial glutamate transporter GLT-1 determines susceptibility to spreading depression in the mouse cerebral cortex. Glia 68:2631–2642. https://doi.org/10.1002/glia.23874
Tang C, Unekawa M, Shibata M, Tomita Y, Izawa Y, Sugimoto H, Ikeda K, Kawakami K, Suzuki N, Nakahara J (2020) Characteristics of cortical spreading depression and c-Fos expression in transgenic mice having a mutation associated with familial hemiplegic migraine 2. Cephalalgia 40:1177–1190. https://doi.org/10.1177/0333102420929028
Schack VR, Holm R, Vilsen B (2012) Inhibition of phosphorylation of Na+, K+-ATPase by mutations causing familial hemiplegic migraine. J Biol Chem 287:2191–2202. https://doi.org/10.1074/jbc.M111.323022
Kros L, Lykke-Hartmann K, Khodakhah K (2018) Increased susceptibility to cortical spreading depression and epileptiform activity in a mouse model for FHM2. Sci Rep 8:16959. https://doi.org/10.1038/s41598-018-35285-8
Santoro L, Manganelli F, Fortunato MR, Soldovieri MV, Ambrosino P, Iodice R, Pisciotta C, Tessa A, Santorelli F, Taglialatela M (2011) A new Italian FHM2 family: clinical aspects and functional analysis of the disease-associated mutation. Cephalalgia 31:808–819. https://doi.org/10.1177/0333102411399351
Spadaro M, Ursu S, Lehmann-Horn F, Liana V, Giovanni A, Paola G, Frontali M, Jurkat-Rott K (2004) A G301R Na+/K+-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 5:177–185. https://doi.org/10.1007/s10048-004-0183-2
Gideons ES, Kavalali ET, Monteggia LM (2014) Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci USA 111:8649–8654. https://doi.org/10.1073/pnas.1323920111
Xia P, H-sV C, Zhang D, Lipton SA (2010) Memantine rreferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 30:11246–11250. https://doi.org/10.1523/JNEUROSCI.2488-10.2010
Aida T, Yoshida J, Nomura M, Tanimura A, Iino Y, Soma M, Bai N, Ito Y, Cui W, Aizawa H, Yanagisawa M, Nagai T, Takata N, Tanaka KF, Takayanagi R, Kano M, Gotz M, Hirase H, Tanaka K (2015) Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology 40:1569–1579. https://doi.org/10.1038/npp.2015.26
Maniyar FH, Sprenger T, Monteith T, Schankin C, Goadsby PJ (2013) Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain 137:232–241. https://doi.org/10.1093/brain/awt320
Thomsen LL, Kruuse C, Iversen HK, Olesen J (1994) A nitric oxide donor (nitroglycerin) triggers genuine migraine attacks. Eur J Neurol 1:73–80. https://doi.org/10.1111/j.1468-1331.1994.tb00053.x
Bates EA, Nikai T, Brennan KC, Fu YH, Charles AC, Basbaum AI, Ptacek LJ, Ahn AH (2010) Sumatriptan alleviates nitroglycerin-induced mechanical and thermal allodynia in mice. Cephalalgia 30:170–178. https://doi.org/10.1111/j.1468-2982.2009.01864.x
Bliss TV, Collingridge GL, Kaang BK, Zhuo M (2016) Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci 17:485–496. https://doi.org/10.1038/nrn.2016.68
Tan LL, Pelzer P, Heinl C, Tang W, Gangadharan V, Flor H, Sprengel R (2017) A pathway from midcingulate cortex to posterior insula gates nociceptive hypersensitivity. Nat Neurosci 20:1591–1601. https://doi.org/10.1038/nn.4645
Afridi SK, Giffin NJ, Kaube H, Friston KJ, Ward NS, Frackowiak RS, Goadsby PJ (2005) A positron emission tomographic study in spontaneous migraine. Arch Neurol 62:1270–1275. https://doi.org/10.1001/archneur.62.8.1270
Chalifoux JR, Carter AG (2011) Glutamate spillover promotes the generation of NMDA spikes. J Neurosci 31:16435–16446. https://doi.org/10.1523/JNEUROSCI.2777-11.2011
Major G, Larkum ME, Schiller J (2013) Active properties of neocortical pyramidal neuron dendrites. Annu Rev Neurosci 36:1–24. https://doi.org/10.1146/annurev-neuro-062111-150343
Wang X-Y, Zhou H-R, Wang S, Liu C-Y, Qin G-C, Fu Q-Q, Zhou J-Y, Chen L-X (2018) NR2B-Tyr phosphorylation regulates synaptic plasticity in central sensitization in a chronic migraine rat model. J Headache Pain 19:102. https://doi.org/10.1186/s10194-018-0935-2
Zhou X, Liang J, Wang J, Fei Z, Qin G, Zhang D, Zhou J, Chen L (2020) Up-regulation of astrocyte excitatory amino acid transporter 2 alleviates central sensitization in a rat model of chronic migraine. J Neurochem 155:370–389. https://doi.org/10.1111/jnc.14944
Manita S, Miyakawa H, Kitamura K, Murayama M (2017) Dendritic spikes in sensory perception. Front Cell Neurosci 11:29. https://doi.org/10.3389/fncel.2017.00029
Smith SL, Smith IT, Branco T, Häusser M (2013) Dendritic spikes enhance stimulus selectivity in cortical neurons in vivo. Nature 503:115–120. https://doi.org/10.1038/nature12600
Takahashi N, Oertner TG, Hegemann P, Larkum ME (2016) Active cortical dendrites modulate perception. Science 354:1587–1590. https://doi.org/10.1126/science.aah6066
Takahashi N, Ebner C, Sigl-Glöckner J, Moberg S, Nierwetberg S, Larkum ME (2020) Active dendritic currents gate descending cortical outputs in perception. Nat Neurosci 23:1277–1285. https://doi.org/10.1038/s41593-020-0677-8
Brandalise F, Carta S, Helmchen F, Lisman J, Gerber U (2016) Dendritic NMDA spikes are necessary for timing-dependent associative LTP in CA3 pyramidal cells. Nature Commun 7:13480. https://doi.org/10.1038/ncomms13480
Cichon J, Gan W-B (2015) Branch-specific dendritic Ca2+ spikes cause persistent synaptic plasticity. Nature 520:180–185. https://doi.org/10.1038/nature14251
Gambino F, Pagès S, Kehayas V, Baptista D, Tatti R, Carleton A, Holtmaat A (2014) Sensory-evoked LTP driven by dendritic plateau potentials in vivo. Nature 515:116–119. https://doi.org/10.1038/nature13664
Iure A, Mazzocchetti P, Bastioli G, Picconi B, Costa C, Marchionni I, Casari G, Tozzi A, Pietrobon D, Calabresi P (2019) Differential effect of FHM2 mutation on synaptic plasticity in distinct hippocampal regions. Cephalalgia 39:1333–1338. https://doi.org/10.1177/0333102419839967
Anttila V et al (2010) Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 42:869–873. https://doi.org/10.1038/ng.652
Acknowledgements
We are indebted to our present and past collaborators who contributed much to the studies on glutamate transporters and migraine reported here. We acknowledge the funding organizations cited in the original papers for their invaluable support.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript, and that they have no financial interests.
Author information
Authors and Affiliations
Contributions
Both authors contributed to the study conception and design. The first draft of the manuscript was written by FC and DP and both authors commented on previous versions of the manuscript, read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Conti, F., Pietrobon, D. Astrocytic Glutamate Transporters and Migraine. Neurochem Res 48, 1167–1179 (2023). https://doi.org/10.1007/s11064-022-03849-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-022-03849-w