
Abstract
Migraine is a common neurological disorder that affects approximately 12–20% of the general adult population. Migraine pathogenesis is complex and not wholly understood. Molecular genetic investigations, imaging and biochemical studies, have unveiled a number of interconnected neurological pathways which seem to have a cause and effect component integral to its cause. Much weight of migraine attack initiation can be placed on the initial trigger and the pathways involved in its neuronal counter reaction. Ion channels play a large role in the generation, portrayal and mitigation of the brains response to external triggers. Several genetic studies have identified and implicated a number of ion channelopathy genes which may contribute to this generalised process. This review will focus on the genetics of migraine with particular emphasis placed on the potentially important role genes HEPH (responsible for iron transport and homeostasis) and KCNK18 (important for the transport and homeostasis of potassium) play in migraine cause.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Migraine is a common neurological disorder affecting 12–20% of the general adult population (globally population dependent) and is approximately three times more prevalent in females than in males (Stovner and Andree 2010; Bolay et al. 2015). According to the World Health Organisation (WHO), migraine- and headache-related disorders are a public-health concern with a large amount of associated disability and financial cost to society (WHO 2011). Given that migraine is most prevalent in the productive years of life, financial cost to society from lost working hours and reduced productivity are significant (Semenov 2015). The WHO states that 25 million working or school days are lost every year in the United Kingdom due to migraine alone, a financial cost which can be compared to tension type headache (TTH) and chronic daily headache combined (WHO 2011). Similarly, the burden placed on primary healthcare due to migraine is high with physicians reporting that one-third of all neurological related consultations are due to migraine and related headache disorders (WHO 2011). Currently, migraine diagnosis is dependent on the presence of distinctive and concurrent episodic attacks. These attacks are classified based on the semblance of a severe pulsating (throbbing) headache with additional symptoms of nausea, phonophobia and/or photophobia. Symptoms of this calibre constitute the 70% of migraine sufferers without aura (MO). The remaining 30% of migraine patients have additional symptoms of transient, reversible, visual, sensory or motor neurological disturbances, better known as aura and termed migraine with aura (MA). Migraine can be attributed to a number of genetic, chemical and environmental interactions making its pathophysiology multifactorial in nature.
Most pathological states of head pain are predicted to be the result of disrupted and dysregulated systems of peripheral nerve excitation. Such excitation is tightly controlled by coordinated plasmalemmal ion channels whose main function is to generate a degree of neuronal action potential proportional to the strength of the external trigger (Du and Gamper 2013). This somatosensory nociceptive reflex has a cause and effect relationship with interconnected neurons projecting to different regions of the brain. The pathway begins with a generalised peripheral signal of external origin which first excites nociceptive fibres (V1/2/3). These fibres innervate the skin, periosteum and large cranial vessels including arteries, veins and sinuses as well as the meninges (Nicolson 2016). Information is then transmitted to a large ipsilateral nuclear group termed the ‘trigeminocervical complex’ (trigeminal ganglion). Neurons at this site cross over and project into the trigeminal nucleus caudalis (TNC) to become a division of second-order neurons. In the contralateral nucleus ventralis posteromedialas (VPM) of the thalamus, these second-order neurons terminate and third-order neurons originate to continue to transmit sensory information to the cortex. It is here where these peripheral nociceptive signals may be amplified centrally to reach pain-inducing intensity (Du and Gamper 2013). The generation, continuance and termination of the somatosensory reflex are brought about via balance in the concerted action of surrounding ion channels. This suggests that the pain felt during a migraine headache is potentially the result of uncontrolled nociceptive fibre innervations accompanied by changes in its localised environment mediated by ion channels rather than the brain itself.
This proposed pain pathway mirrors the trigeminovascular theory which currently defines the process of migraine pathophysiology. Previous migraine studies (Akerman et al. 2011; Noseda and Burstein 2013) have integrated the anatomy of nociceptive structures of the brain with concepts of central nervous system modulation. This correlation combines regions of the brain implicated during mammalian physiological studies with biochemical observations, i.e. neurotransmitter and inflammatory peptide release (Messlinger et al. 2011). These studies suggest that disturbances in brain areas of the subcortical aminergic sensory modulatory systems and other brainstem, hypothalamic and thalamic structures (Angelini et al. 2004) are key contributors to migraine cause (Sprenger and Goadsby 2010; Goadsby 2012). This model suggests that the ventrolateral periaquductal grey matter (PAG) and the posterior hypothalamic grey (PHG) modulatory regions are activated by nociceptive trigeminovascular input (Knight et al. 2002; Goadsby 2012).
Data supporting this include stimulated pain afferents in the superior sagittal sinus in cat studies found to consequently activate neuronal pathways in the PAG—resulting in subsequent inhibitory signals to the trigeminocervical complex (Knight et al. 2002). Concomitantly, other positron emission tomography (PET) investigations in migrainuers have shown recurrent PAG activation during continual migraine (Rocca et al. 2006; Sprenger and Goadsby 2010). In addition, the associated PHG area has been significantly implicated in migraine due to the appearance of dopaminergic involvement in clinical features of the premonitory phase of migraine and the accompanying neurological symptoms (Goadsby 2012). These include the identification of dopamine 2 (D2) receptors in significant quantities in rat trigeminocervical neurons (Goadsby 2012). Subsequent studies have demonstrated that dopamine-containing A11 neurons inhibit trigeminovascular nociceptive transmission through D2 receptor-mediated responses (Charbit et al. 2009) suggesting that the prevalence of lesions and dysfunction in this region facilitate pain transmission (Charbit et al. 2009).
The complex nature of migraine pathogenesis has to date limited the advancement of research, diagnosis and the identification of causative genes. Despite centuries of research, molecular diagnostic methods aiding a definitive diagnosis remain under-developed with the genetic and pathological mechanisms underpinning migraine pathogenesis yet to be fully elucidated. However, a number of potential factors contributing to the cause of migraine have been identified and, as a result, a number of theories have been developed regarding migraine pathophysiology.
Genetic epidemiology and twin studies
Migraine as a disorder can be considered ‘commonplace’ in the context of public awareness. Most people are quick to relate to the impact of the more subtle migraine-like symptoms or at the very least know someone who suffers with it. Migraine has a lifetime prevalence rate of 16–35% and a 1-year prevalence rate of 10–12% depending on racial culture (Russell et al. 1995; Zivadinov et al. 2001; Carson et al. 2004). As a reflection of racial liability, the disease has been found to have reduced prevalence in Hong Kong, Saudi Arabia and in African and Asian Americans (Leonardi et al. 2005).
Migraine preponderance generally follows a 1:3 male-to-female ratio (Stovner and Andree 2010). Epidemiologically this can be further refined to discriminate between the subtypes MA and MO. MO has a noted lifetime prevalence of 14.7% with 1:2.2 male-to-female ratio, while MA has a lower lifetime prevalence of 7.9% and a male-to-female ratio of 1:1.5 (Russell et al. 1996; Uygunoglu and Siva 2015). These differences in gender prevalence ratios highlight the increased risk in migraine susceptibility in females. Adding age as an antecedent epidemiological variable identifies significant female preponderance at all ages for MA (Russell et al. 1996; Uygunoglu and Siva 2015). In contrast, no distinguishable differences in gender predisposition have been identified for MO until the age of 13 (Russell et al. 1996; Ferrari et al. 2015), suggesting that both males and females have equal MO susceptibility risk during early childhood. As a result, female hormones have been postulated to play a role in migraine initiation and cause, particularly in MO (Russell et al. 1996). Moreover, migraine has been found to peak in women between the ages of 35–45 (Ferrari et al. 2015).
Family history plays a substantial role in migraine inheritance, with familial clustering evident in 37–91% of first degree relatives of migraine probands (Stewart et al. 2006; Lemos et al. 2009). In addition, correlation between early onset and the higher number of affected relatives with strong family history has been linked with migraine onset with those aged 20 and younger (Stewart et al. 2006). Migraine tends to prominently follow the maternal line, however, with paternal orientated inheritance also observed; migraine is suggested to arise via autosomal dominant/recessive rather than X-linked inheritance (Wang et al. 2008).
Examinations of disease risk conducted using various familial based studies suggest an overall increased risk in migraine susceptibility in relatives of migraine probands (Stewart et al. 2006). First degree relatives of MO probands have a threefold increase in MO susceptibility, while first degree relatives of MA probands have a twofold disease risk increase for both MA and MO subtypes (Russell et al. 1995; Montagna 2008). More refined analysis into associated disease risk in first degree relatives of probands identified a 1.4-fold increase in MO risk amongst spouses of MO probands (Russell et al. 1995; Montagna 2008). No associative disease risk increase was found for MA in spouses of MA probands (Montagna 2008). In general, the observed increased risk of migraine amongst first degree relatives strongly supports the role genetic factors play in migraine susceptibility, while the higher risk associated with proband spouses shows compatibility with assortive mating and/or shared environmental factors (Montagna 2008).
Twin studies are renowned for their ability to prove and define the genetic cause and environmental factor contribution to disease. In the case of migraine, twin studies have demonstrated higher concordance rates for migraine amongst monozygotic (MZ) twins when compared with dizygotic (DZ) twins (Gervil et al. 1999). A study conducted by Ziegler et al. found consistently high tetrachoric correlations in female twin pairs raised both together and apart, with a defined heritability estimate of 52% (Ziegler et al. 1998). A Finnish study by Kallela et al. further supports this, noting 40–50% of migrainous cause attributable to genetic factors (Kallela et al. 1999). No distinguishable inheritance differences were found between genders with the exception of the effects from presumed dominance (26% for men and 14% for women) (Kallela et al. 1999). In addition, a Danish twin study found a significantly high correlation between genetic liability and proband concordance rates in MZ twins with the MO migrainous subtype (Ulrich et al. 1999). In this study, 61% of MO cause was attributed to additive genetic effects and the remaining 39% due to individual-specific environmental effects (Ulrich et al. 1999). In MA classified twin sets, the correlation in genetic liability was identified to be 68% in MZ twins and 22% in DZ twins. A recurrence risk rate was also determined with MA found to have a 50% chance of reoccurring in MZ twins and only 21% in DZ twins (comparative with non-twin siblings at 27%) (Ulrich et al. 1999; Montagna 2008) highlighting that specific environmental factors encountered by twins do not directly influence MA.
These and other studies have provided plausible theories on both the cause and inheritance of migraine to identify that migraine has a multifactorial genetic pattern with varying clinical manifestations. The clinical characterisation of migraine may vary dependant on the effects of combinational additive genes and unshared environmental factors. Hence, further genetic investigations through candidate gene and genome-wide association studies are a priority.
Genome-wide association (GWA) studies
A recent meta-analysis conducted by Anttila et al. (2013) combined and analysed GWAS data from 29 clinic- and population-based studies, comparing 23,285 migraine case and 99,425 control samples (Anttila et al. 2013). The primary meta-analysis revealed 142 significant SNPs at 12 loci associated with migraine. The single most significant SNP rs11172113 in the LRP1 gene encodes for a low-density lipoprotein receptor-related protein-1 (LRP1) (Anttila et al. 2013). The LRP1 gene has been shown to exert regulatory effects on a number of correlated cellular events including amyloid precursor protein (APP) metabolism, kinase-dependent intracellular signalling, neuronal calcium signalling and neurotransmission (von Einem et al. 2010; Spuch et al. 2012; Nakajima et al. 2013). While not directly indicative of ion channel causation, the identification of LRP1 suggests a role for ion channel proteins involved in neuronal calcium signalling and potential glutamate neurotransmission through its direct interaction with N-methyl-d-aspartate (NMDA) receptors with migraine susceptibility (Spuch et al. 2012).
Additional genes previously reported from GWAS data (Anttila et al. 2010; Chasman et al. 2011) supporting the association of ion channels with migraine aetiology (either directly or indirectly) include: PRDM16 with high neuronal expression in GABAergic Amacrine Cells and linked involvement in oxidative stress (Chasman et al. 2011; Martin 2015; Chi and Cohen 2016); TRPM8 which encodes for a receptor-activated non-selective cation channel activated by cold environmental temperatures and modulated intracellular pH, with variants linked with both MA and MO subtypes (Anttila et al. 2010; Chasman et al. 2011; Anttila et al. 2013); and TGFBR2 which plays a role in homocysteine metabolism, glutamatergic neurotransmission, TGF-β signalling and synaptic/endothelial function (Freilinger et al. 2012; Martin 2015). Other loci highlighted with genome-wide significance include various genes involved in neuron and synapse development, brain vasculature, extracellular matrix processes and pain sensation (Eising et al. 2016).
Similar findings in corresponding biological pathways were identified in the most recent migraine meta-analysis. The study is the largest genetic study of migraine to date, with 59,674 cases compared with 316,078 controls from a combined total of 22 GWA studies (Gormley et al. 2016). Results of the study by Gormley et al. noted 44 independent SNPs associated with migraine risk, mapped to 38 distinct loci. This included the first loci situated on the X chromosome and an additional 28 loci not previously reported as associated with migraine risk (Gormley et al. 2016). Moreover, the study highlighted a significantly associated ion channel gene, KCNK5 (a family member of the KCNK18 gene) and also reported three additional genes SLC24A3, ITPK1 and GJA1 linked to cellular ion homeostasis (Gormley et al. 2016).
GWAS data by the International Headache Genetics Consortium (IHGC) (Anttila et al. 2013) have also been utilised in a recent gene-based analysis conducted by Eising et al. (2016). The study aimed to identify brain regions, cell types and pathways involved in migraine cause using IHGC GWAS data spatially mapped with expression data from 3702 samples across 6 normal human adult brains (from the Allen Human Brain Atlas group: http://human.brain-map.org/) (Eising et al. 2016). The study identified a number of strong migraine-associated genes. Genes were grouped according to enrichment and expression, creating five migraine-associated modules across regions within the brain. Two of the five modules (A and C) highlighted 3151 genes largely involved in neurotransmission and highly expressed within the cortex, of which 112 contribute to voltage-gated cation channel activity (Eising et al. 2016). Module A highlighted genes (including three ion channels genes) involved in glutamatergic system function (GLS, GRIK3, GRIN2A and GRM7) (Eising et al. 2016), a pathway previously linked to migraine through gene localisation (previous GWAS studies—MTDH, LRP1 and MEF2D) and functional observations (Ligthart et al. 2011; Goadsby 2012; Anttila et al. 2013). Genes grouped in module B include recognised gene expression regulators that show high expression in the cerebellum and notable expression throughout the cortex (Eising et al. 2016). Genes grouped in module D include those involved in myelin formation, specifically expressed in oligodendrocytes and highly expressed in numerous subcortical brain regions and white matter (Eising et al. 2016). Recent studies in cellular neurology have noted that action potentials propagating through axons can be rapidly regulated by oligodendrocytes, with all categories of glial cells, including oligodendrocytes, containing many of the same ion channels and neurotransmitter receptors as neurons (Fields 2008). Interestingly, the genes defined in module D are highly expressed in regions implicated in the trigeminovascular pathway (Eising et al. 2016): a pathway which is tightly regulated by surrounding ion channels, recently reported to have physiological pathological evidence for its role in migraine. This is further supported in work by Guyuron et al. presenting disrupted myelin sheets in the trigeminal nerve of migraine patients (Guyuron et al. 2014).
These combined analyses of gene ontologies and co-expression data have to date revealed susceptibility areas within functional pathways involved with cortical neurotransmission, gene transcription regulation in the cortex and cerebellum and in myelination and energy supply in subcortical areas with migraine aetiology. These findings support a key role for the contribution of ion channel dysfunction to the complex multi-system cause of migraine pathophysiology.
Familial hemiplegic migraine (FHM)
Familial hemiplegic migraine is considered a rare subtype of migraine with aura, characterised by the presence of temporary unilateral hemiparesis (numbness and/or muscle weakness). FHM has a clear autosomal dominant inheritance pattern and typically co-occurs within families (Pietrobon 2007). The prevalence of hemiplegic migraine is estimated to be around 0.01% in European populations, with familial forms attributed to 50% of the total incidence rate (Thomsen and Olesen 2004). Phenotypically, FHM has been found to show variable expressivity and genetic heterogeneity with an approximate 70–90% penetrance (Rothner 2003). It is believed that FHM may be associated with episodic, or in some cases, permanent cerebellar nystagmus and ataxia (at least 20% of pedigrees) (Montagna 2008). Symptoms of bilateral sensorimotor disturbances occur in approximately 25% of patients (Ducros et al. 2000); 40% of patients suffer with prolonged aura attacks which can result in consciousness impairment (variations between confusion and coma), agitation, fever and meningismus (Ducros et al. 2000); 15% of people present with migraine with non-hemiplegic aura alternating with hemiplegic attacks and a few patients have reported FHM-associated seizures during a severe episode (Ducros et al. 2000). FHM is often triggered by exogenous factors (stress, light, food and sound), head trauma, epileptic seizures, recurrent confusion, coma and psychosis (Montagna 2008).
FHM was first labelled as a monogenic disease in 1993, as it was first mapped to chromosome 19 by Joutel et al. (1993). In 1996, Ophoff et al. defined the first gene (CACNA1A) associated with its pathophysiology at region 19p13.1 (Ophoff et al. 1996). The CACNA1A gene encodes for the α1A subunit of a P/Q type voltage-dependent calcium channel expressed in neuronal presynaptic terminals (Ducros et al. 2000). Missense mutations within this gene account for approximately 50% of FHM (type 1) cases and have also been correlated with two other autosomal dominant neurological disorders, episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6)—which is particularly interesting given their overlapping clinical features (Ducros et al. 2000; Blumenfeld et al. 2016).
In 2003, the second gene found associated with FHM pathophysiology was the ATP1A2 gene (1q23.2) by De Fusco et al. (2003) The ATP1A2 gene belongs to the family of P-type cation transport ATPases and the Na+/K+ ATPases responsible for regulating electrochemical gradients within the CNS (De Fusco et al. 2003; Blumenfeld et al. 2016). The two families in which FHM2 was genetically defined experienced seizures as a result of recurrent FHM-like attacks (De Fusco et al. 2003). Subsequent reports of FHM2 diagnosis also noted the occurrence of seizures as an associated clinical feature (Jen 2015). Moreover, mutations of the ATP1A2 gene have been associated with alternating hemiplegia observed during childhood (Bassi et al. 2004; Blumenfeld et al. 2016).
The final gene known to be associated with FHM (type 3) was identified in 2005 as the SCN1A gene (2q24.3) (Dichgans et al. 2005). The SCN1A gene encodes for a voltage-gated sodium channel protein type 1α-subunit, with mutations in previous genetic studies found to be associated with paroxysmal epilepsy (Dichgans et al. 2005). The contributing pathogenesis of mutations found in these three implicated FHM causative genes are as yet not completely understood. Several mutations within these genes are believed to affect channel gating, alter neurotransmitter modulation and cause abnormal neuronal excitability—all of which are hypothesised mechanisms common to the pathophysiological cause of FHM.
Why look further into ion channel genes?
Generations of genetic-based studies have collectively noted a significant association between ion channelopathies, FHM and other migraine subtypes (Liu et al. 2013; Ferrari et al. 2015). As previously mentioned, several voltage-gated ion channel mutations (Ca2+/Na+ channel and Na+/K+ pump mediated) have been found to functionally alter neuronal ionic homeostasis, excitability and in some cases neurotransmission in relation to FHM pathogenesis. We will now focus on the potential correlation between common migraine and two particular ion channel genes, HEPH and KCNK18 based on the provisional link, defined by Maher et al. (2012) and Lafrenière et al. (2010), respectively.
Hephaestin and the HEPH gene
The hephaestin protein is a member of the ferroxidase protein family and is located at the Xq12 region on the X chromosome. It is a homologue of the free copper-carrying ceruloplasmin protein; however, unlike the soluble ceruloplasmin (due to the C-terminus transmembrane domain) hephaestin is transmembrane bound (Syed et al. 2002). The hephaestin protein is composed of 1135 amino acids with a total molecular weight of 130.4 kDa (Syed et al. 2002). Hephaestin contains 6 copper domains. Three copper atoms form a trinuclear metallic unit at domain 1 and 6; and 3 more copper atoms form mononuclear type 1 centres in domains 2, 4 and 6 amongst predicted atypical sites (Lindley et al. 1997). These predicted atypical sites have been noted to demonstrate ferroxidase activity (Chen et al. 2004).
The functional role of hephaestin is to transcribe an iron transport protein involved in cellular iron export, by oxidising ferrous to ferric iron for uptake by transferrin or other iron carriers (Fig. 1) (Nadadur et al. 2008). The HEPH gene has been determined to be a ceruloplasmin (CP) homologue expressed neuronally and has previously been determined essential for iron homeostasis. HEPH gene expression is also present throughout the human GI tract, pancreatic islets and enteric nerves (Ranganathan et al. 2012). Studies using rat models have identified that both the CP gene and its HEPH homologue play a role in iron efflux from cells in peripheral tissues as well as in the CNS (Qian et al. 2007). Given that the hephaestin protein plays a key role in the transport of iron from the intracellular to extracellular environment via the export pathway, it is considered the physiological checkpoint for whole body iron homeostasis.

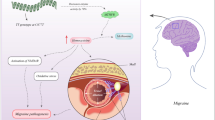
A schematic representation outlining some of the predicted mechanisms contributing to migraine pathogenesis. 1 A number of various endogenous and exogenous triggers, i.e. light, food, sound and smell may simultaneously disturb the subcortical aminergic sensory modulatory systems within the brain. Disturbance within these systems ultimately leads to the nociceptive activation of the first division of the trigeminal nervous system (V1/2/3). 2 Stimulation of these fibres ignites the transmission of nociceptive information and initiates a CSD wave—causing Cortical Hyperexcitability. 3a As a response, Cortical Hyperexcitability enhances somatotopic activity—which results in an increase in the release of endogenous bio-regulators, i.e. glutamate, neuropeptides and prostaglandins. These factors induce a regional sterile neurogenic inflammation, leading to an increase in pain trafficking. 3b Mutations in the KCNK18 gene and consequent TRESK dysfunction further exacerbate Cortical Hyperexcitability and pain transmission. (i) under normal circumstances, the fully functioning TRESK protein effectively controls potassium conductance and stabilises the negative resting membrane potential (excitability adjustment and depolarisation counteraction) through the transport of potassium, which subsequently regulates both Na+ and Ca2+ ions (potassium equilibrium potential). Dysfunction in the TRESK protein (ii) disrupts this Potassium Equilibrium Potential through prohibiting potassium efflux, ultimately lowering the action potential threshold of the neuron, enhancing the transmission of nociceptive information and the release of various neurotransmitters (Lafrenière et al. 2010; Andres-Enguix et al. 2012). 3c The HEPH gene encodes the hephaestin protein, which is functionally responsible for cellular iron homeostasis through the generation of an iron transport-appropriate protein, by oxidising Fe2+ to Fe3+ for the uptake by transferrin in neighbouring cells. Maintaining iron homeostasis ensures normal cellular oxidative metabolism (iii). Dysfunction of the hephaestin protein during the period of Cortical Hyperexcitability (iv) may cause excessive iron accumulation which could cause iron-catalysed free radical cell damage in the repeatedly activated nociceptive networks—essentially enhancing the perception of pain during a migraine attack and altering cell-to-cell anti-nociceptive communication (Welch et al. 2001; Nadadur et al. 2008). 4 The combination and contribution of factors 1–3b combined with those not yet understood lead to the formation of the characteristic head pain experienced during a migraine attack. The ‘proposed pain pathway’ was adapted and summarised from Nicolson (2016) and the cause-and-effect pathophysiology associated with KCNK18 and HEPH dysfunction was interpreted from Lafrenière et al. (2010), Andres-Enguix et al. (2012), Welch et al. (2001) and Nadadur et al. (2008) respectively
HEPH protein regulation
Iron homeostasis is essential for normal metabolic and neurological function (Jiang et al. 2015). Neuronal iron accumulation has been reported to contribute to multiple neurodegenerative diseases, namely Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and others (Ponka 2004; Zecca et al. 2004; Hametner et al. 2013). The elucidation of the functional and regulatory mechanisms of hephaestin may identify the genetic susceptibility of individuals to migraine and other iron-related co-morbidities.
A study conducted by Chen et al. (2004) identified that dietary iron concentrations play a key role in the regulation of HEPH gene transcription. Decreased levels of dietary iron resulted in increased hephaestin protein and protein activity per enteric cell (Chen et al. 2004). Moreover, the iron-dependent regulation of the hephaestin protein may be the result of a novel post-transcriptional mechanism, as current research has failed to identify an iron response element (IRE) on the hephaestin transcript (Chen et al. 2004). Currently, it is not known if the increased protein levels represent an increase in translation, an increase in protein stability or a combination of the two (Chen et al. 2004).
An additional study by Hinoi et al. (2005) identified a significant link between the homeobox transcription factor CDX2 and hephaestin gene expression associated with intracellular iron levels. The study revealed that the activation of CDX2 rapidly induced hephaestin expression, and RNA interference-mediated inhibition of CDX2 resulted in lower HEPH expression (Hinoi et al. 2005). The study focused on the hephaestin reporter gene construct using a chromatin immunoprecipitation (CHiP) approach—suggesting that CDX2 directly regulates HEPH transcription in the intestinal epithelium (Hinoi et al. 2005). CDX2 expression has previously been found in the epithelium of the choroid plexus and localised brain tumours; however, its expression elsewhere in the central nervous system is yet to be determined (Beschorner et al. 2009). In addition, the relationship between CDX2 and hephaestin-mediated iron levels in the brain are yet to be fully elucidated. Of note, CDX2 activation is mediated by the activator protein Fos, with Fos expression observed in the trigeminocervical complex after meningeal irritation with blood, drawing a link between iron regulation and migraine (Goadsby 2012).
HEPH and migraine
As previously outlined, both twin and FHM studies support the notion that the inheritance of migrainous genes contributes to the high prevalence of migraine worldwide. A recent X chromosome association scan of the Norfolk Island (NI) genetic isolate population provided convincing evidence for a novel migraine susceptibility locus mapping to chromosome Xq12 (Maher et al. 2012). More importantly, analysis of the X chromosomal data identified a cluster of associated SNPs at the Xq12 locus associated with migraine in the NI cohort (Maher et al. 2012). The NI population provides a valuable resource for discovering variants involved in complex disease susceptibility given the reduced genetic and environmental heterogeneity. Subsequent studies by Maher et al. (2012) showed that most of the identified SNPs were also significantly associated with migraine in an outbred Australian Caucasian cohort of female migraineurs, thus identifying the HEPH locus as an important migraine susceptibility region for common migraine subtypes. The Xq12 locus identified in the study spans a 377 kb region and contains two genes—the hephaestin gene involved in iron transport and the V-set gene involved in immune response. The most significant SNP identified in the study was rs1028348, located in the 5’UTR of HEPH suggesting a particularly important role for HEPH in migraine pathogenesis (Maher et al. 2012).
The concept of the iron–migraine relationship is not new. Previous studies have reported an observed increase in iron levels in the periaqueductal grey matter (PAG) (Welch et al. 2001), the putamen, globus pallidus and red nucleus (Welch 2009) in migraine patients. These areas are of particular importance in the descending anti-nociceptive neural network, a pathway currently implicated in migraine pathophysiology. A high-resolution magnetic resonance imaging (MRI) study conducted by Welch et al. (2001) observed the highest concentration of transferrin receptors (required for iron transport) in the PAG than in any other brain regions (Welch et al. 2001). The study findings suggest that high transferrin receptor density may be a marker of the cellular requirements for iron during oxidative metabolism (Welch et al. 2001). During a migraine attack, an increase in oxidative metabolism may serve as the cause for neuronal vulnerability due to oxidative stress (Fig. 1). Increased oxidative stress levels have also been implicated in other brain stem structures activated during a migraine attack (Welch et al. 2001). A recent GWAS study also supports the genetic association of the migraine–oxidative stress relationship, with 6 new genes linked with oxidative stress reported (Gormley et al. 2016). Overexpression of transferrin receptors or malfunctions in iron cell-to-cell transport (a pathway in which hephaestin is involved) may subject these cranial tissues to excessive iron accumulation, iron toxicity and iron-catalysed free radical cell damage, potentially accentuated by repeated episodes of hyperoxia during MO or MA and with chronic daily headache (Welch et al. 2001; Hametner et al. 2013).
At this point, the role for iron accumulation in migraine pathogenesis has not been defined in detail; however, the study by Welch (2009) identified an increase in iron that may reflect free radical damage in repeatedly activated networks involved in nociception. Alternatively, impaired modulator function at an alternate locus in the nociceptive system may result in abnormal excitability of connected structures enhancing the perception of pain (Welch 2009) combined with additive effects of impaired iron homeostasis and associated neuronal dysfunction or damage (Welch et al. 2001).
Overall, there is considerable evidence suggesting an association between iron accumulation and migraine-related symptoms, providing the fundamental basis to prioritise the investigation into HEPH, in an attempt to successfully identify the functional variant(s) that may influence the genetic basis of migraine.
TRESK channel and the KCNK18 gene
TRESK channel structure
The TWIK-related spinal cord potassium (TRESK) channel is encoded by the KCNK18 gene and is the most recent discovery of the potassium two pore (K2P) family (Enyedi and Czirják 2015). The general properties and structure of TRESK are similar to those of other K2P channels; however, the protein shares only a 19% homology in amino acid sequence amongst its K2P relatives, indicative of sub-family status (Enyedi et al. 2012; Enyedi and Czirják 2015).
The TRESK protein is composed of four transmembrane segments (TMS) and two extracellular re-entrant pore loops located between the 1st and 2nd, and 3rd and 4th TMS, respectively (Enyedi et al. 2012). The complete protein is thought to function as a dimer of subunits with the long intracellular loop between TMS 2 and 3 containing two docking sites for protein interaction amongst additional regulatory regions (Enyedi et al. 2012).
The two pore potassium channel TRESK is unique in both its function and regulation. It is the only member in its family whose mechanism of activation is dependent on calcium activation (calcineurin modulated) and/or M1 muscarinic receptor stimulation (Enyedi et al. 2012; Enyedi and Czirják 2015). TRESK is abundantly expressed in the dorsal root ganglion and other sensory ganglia such as the trigeminal ganglion with its role to control potassium conductance and stabilisation of the negative resting membrane potential in relation to excitability adjustment, depolarisation counteraction and potassium transport across the plasma membrane (Fig. 1) (Enyedi et al. 2012).
The main requirement essential to TRESK activation is the direct, non-catalytic protein–protein interaction that occurs between the TRESK intracellular docking domain and calcineurin phosphatase. Upon a sudden influx of calcium, neuronal calmodulin is activated; this sequence of events in turn causes the activation of calcineurin. Calcineurin binds to the TRESK docking domain, resulting in dephosphorylation of one of the TRESK main regulatory regions and subsequent activation of the TRESK channel (Enyedi et al. 2012; Enyedi and Czirják 2015). Protein Kinase A is responsible for the opposing action of calcineurin and, as a consequence, inhibits channel function and facilitates the return of neuronal membrane potential to its quiescent state preceding stimulation (Enyedi et al. 2012; Enyedi and Czirják 2015).
A possible alternate route for TRESK activation is proposed following stimulation via M1 muscarinic receptors. A study conducted by Czirjak et al. (2004) examined electrochemical currents in oocytes co-expressing both TRESK channels and M1 muscarinic receptors. Initial current measurements were made without M1 muscarinic receptor stimulation followed by measurements after M1 muscarinic receptor stimulation with carbachol. The results demonstrated TRESK channel activation with a subsequent decrease and equalisation in electrochemical gradient, suggestive of M1 muscarinic receptor interplay and TRESK activation (Czirjak et al. 2004).
TRESK and migraine
Given the functional role of TRESK in neuronal electrochemical excitability and its local expression in both the DRG and TG, it is conceivable that the TRESK channel interacts with the nociceptive pathways during migraine pathogenesis. Some evidence exists for this in rat models, which have demonstrated that down-regulation of TRESK expression increases sensitivity to painful stimuli, with TRESK overexpression in DRG neurons shown to attenuate injury-induced pain sensitisation (Tulleuda et al. 2011; Zhou et al. 2013). Notable TRESK mutations which have been identified in migraine patients that may contribute to these effects are outlined below. Reference has been made to the Exome Aggregation Consortium (ExAC: http://exac.broadinstitute.org/). The ExAC database congregates exome data from large-scale sequencing projects, making variant allele frequencies available across disease-specific and population genetic studies (60,706 unrelated individuals). This database can serve as reference of mutation novelty or prevalence.
Frameshift mutation F139Wfsx24
A study conducted by Lafrenière et al. (2010) identified a rare dominant-negative frame shift mutation (F139Wfsx24, ExAC: 0.0006) in the KCNK18 gene, utilising a large, multigenerational pedigree genome-wide linkage approach (Lafrenière et al. 2010). The 2-bp deletion in KCNK18 was found to segregate wholly amongst migrainuers with typical MA, and presented with a significant genome-wide linkage LOD score of 3.0 (Lafrenière et al. 2010). In addition, results from a functional analysis of the frame shift mutation demonstrated absolute loss of TRESK function, while a truncated version resulted in significant impairment of TRESK activity in wild-type individuals (Andres-Enguix et al. 2012). This observation supports a role for KCNK18 in migraine, and defined a possible relationship between TRESK dysfunction and its contribution to familial MA.
Investigation of migraine pathophysiology has highlighted the high expression levels of KCNK18 in trigeminal sensory neurons implicating the dominant-negative TRESK mutation in both migraine and its associated symptoms of aura (MA subtype). Integrated theories suggest that cortical spreading depression is the direct cause of aura, and ultimately initiates the subsequent phases of migraine—generally resulting in dural vasodilation, oedema and headache (Enyedi et al. 2012). Functional investigations aimed at defining the link between migraine and TRESK mutations found it highly unlikely that TRESK mutations alone lead to the disturbance of trigeminal sensory neurons. Rather the functional role of TRESK activity appears to prevent the pathological activation of neurons during the early stages of CSD (Enyedi et al. 2012). These data suggest the neuro-protective mechanism regulated by TRESK is lost in patients who have dominant-negative mutations (Enyedi et al. 2012).
Additional KCNK18 mutations
Andres-Enguix et al. (2012) conducted additional screening for KCNK18 mutations in unrelated migraine case (n = 479) and control (n = 496) cohorts. Their study identified a number of additional missense variants including R10G (ExAC: 0.087), A34V (ExAC: 0.000008), C110R (ExAC: 0.007), S231P (ExAC: 0.060 and A233V (ExAC: 0.009). Interestingly, the A233V mutation was only found in a few control samples, while the variant—A34V was found in a single Australian migraine proband and not in any control samples (Andres-Enguix et al. 2012). According to Andres-Enguix et al. (2012), R10G can be found within the TRESK N-terminus, A34V within Transmembrane domain 1 (TM1) and C110R can be found relatively close to the selectivity filter in the first pore domain with both S231P and A233V assumed to be located within the regulatory loop between transmembrane domains 2 and 4. A relationship between the two variants A34V and C110R and the TRESK selectivity filter has been suggested, identifying a possible functional effect on the regulation of potassium channel gating (Andres-Enguix et al. 2012).
As with the F139Wfsx24 frame shift mutation, a functional analysis was conducted on these additional mutations with the variants R10G, S231P and A233V shown to have no apparent effect on TRESK function. This appears consistent with the notion that they do not contribute to migraine pathophysiology as they were found in both cases and healthy controls (Andres-Enguix et al. 2012; Rainero et al. 2014). Functional studies examining the effects of the two variants A34V and C110R have revealed conflicting results, hence their contribution to migraine remains unknown (Andres-Enguix et al. 2012; Guo et al. 2014). Currently, the role of TRESK in migraine is not entirely understood. The dominant-negative mutation may serve a complex role in migraine pathogenesis, while other non-functional variants illustrate no obvious affect. It is these gaps in our understanding, which highlight the importance of further investigations into the genetic enigma of migraine pathogenesis.
TRESK dysfunction and related co-morbidities
Several genetic studies investigating links between K2P channelopathies and disease have identified a relationship between K2P dysfunction and Episodic Ataxia Type-1. A non-functional mutation in gene TM6 (V408A) demonstrated that dominant-negative co-assembly of mutants with wild type samples significantly reduced potassium channel activity (Adelman et al. 1995). Similar to TRESK studies, these results conferred a significant decrease in channel activity, suppressing K2P function by up to 90% (Andres-Enguix et al. 2012).
Additional studies focusing on other disorders of defective neuronal excitability such as epilepsy suggest that ion channel mutations which cause the complete dysfunction of channel activity contribute significantly to an increased associated disease risk (Schorge and Kullmann 2010). As a result, disease severity is suggested to be dependent on other channel mutations and their combined impact (Schorge and Kullmann 2010).
This review provides support for the continued research into the role of the HEPH and KNCK18 genes, further supporting continued efforts to identify new variants, which may in combination contribute to varying degrees of HEPH/KCNK18 dysfunction in relation to migraine. It is important that future research determines the dominant or recessive nature of these variants and their interaction given that individuals may experience the effects of varying penetrance and symptomology, which ultimately complicate migraine classification and treatment. It is hoped that the classification of genetic factors and the identification of additional candidate genes will assist in the development of improved prophylactic and abortive treatment regimens to further alleviate both the personal and economic burden of migraine.
References
Adelman JP, Bond CT, Pessia M, Mayliet J (1995) Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 15:1449–1454
Akerman S, Holland PR, Goadsby PJ (2011) Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci 12:570–584
Andres-Enguix I, Shang L, Stansfeld PJ et al (2012) Functional analysis of missense variants in the TRESK (KCNK18) K+ channel. Sci Rep. doi:10.1038/srep00237
Angelini L, de Tommaso M, Guido K et al (2004) Steady-state visual evoked potentials and phase synchronization in migraine patients. Phys Rev Lett 93:038103. doi:10.1103/PhysRevLett.93.038103
Anttila V, Stefansson H, Kallela M et al (2010) Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 42:869–873
Anttila V, Winsvold BS, Gormley P et al (2013) Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet 45:912–917
Bassi MT, Bresolin N, Tonelli A et al (2004) A novel mutation in the ATP1A2 gene causes alternating hemiplegia of childhood. J Med Genet 41:621–628
Beschorner R, Mittelbronn M, Mugler M, Meyermann R, Schittenhelm J (2009) Immunohistochemical analysis of CDX2 expression in normal choroid plexus epithelium and choroid plexus tumors. Histol Histopathol 24:1507–1514
Blumenfeld AE, Victorio MC, Berenson FR (2016) Complicated Migraines. Semin Pediatr Neurol 23:18–22
Bolay H, Ozge A, Saginc P et al (2015) Gender influences headache characteristics with increasing age in migraine patients. Cephalalgia 35:792–800
Carson AP, Rose KM, Sanford CP et al (2004) Lifetime prevalence of migraine and other headaches lasting 4 or more hours: the Atherosclerosis Risk in Communities (ARIC) study. Headache 44:20–28
Charbit AR, Akerman S, Holland PR, Goadsby PJ (2009) Neurons of the dopaminergic/calcitonin gene-related peptide A11 cell group modulate neuronal firing in the trigeminocervical complex: an electrophysiological and immunohistochemical study. J Neurosci 29:12532–12541
Chasman DI, Schurks M, Anttila V et al (2011) Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet 43:695–698
Chen H, Attieh ZK, Su T et al (2004) Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood 103:3933–3939
Chi J, Cohen P (2016) The multifaceted roles of PRDM16: adipose biology and beyond. Trends Endocrinol Metab 27:11–23
Czirjak G, Toth ZE, Enyedi P (2004) The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J Biol Chem 279:18550–18558
De Fusco M, Marconi R, Silvestri L et al (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 33:192–196
Dichgans M, Freilinger T, Eckstein G et al (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366:371–377
Du X, Gamper N (2013) Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol 11:621–640
Ducros A, Denier C, Tournier-Lasserve E (2000) Genetics of familial hemiplegic migraine. J Headache Pain 1:S129–S134
Eising E, Huisman SM, Mahfouz A et al (2016) Gene co-expression analysis identifies brain regions and cell types involved in migraine pathophysiology: a GWAS-based study using the Allen Human Brain Atlas. Hum Genet 135:425–439
Enyedi P, Czirják G (2015) Properties, regulation, pharmacology, and functions of the K(2)p channel, TRESK. Pflugers Archiv 467:945–958
Enyedi P, Braun G, Czirják G (2012) TRESK: the lone ranger of two-pore domain potassium channels. Mol Cell Endocrinol 353:75–81
Ferrari MD, Klever RR, Terwindt GM, Ayata C, van den Maagdenberg AM (2015) Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol 14:65–80
Fields RD (2008) Oligodendrocytes changing the rules: action potentials in glia and oligodendrocytes controlling action potentials. Neuroscientist 14:540–543
Freilinger T, Anttila V, de Vries B et al (2012) Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet 44:777–782
Gervil M, Ulrich V, Kaprio J, Olesen J, Russell MB (1999) The relative role of genetic and environmental factors in migraine without aura. Neurology 53:995–999
Goadsby PJ (2012) Pathophysiology of migraine. Ann Indian Acad Neurol 15:15–22
Gormley P, Anttila V, Winsvold BS et al (2016) Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 48:856–866
Guo Z, Liu P, Ren F, Cao YQ (2014) Nonmigraine-associated TRESK K+ channel variant C110R does not increase the excitability of trigeminal ganglion neurons. J Neurophysiol 112:568–579
Guyuron B, Yohannes E, Miller R, Chim H, Reed D, Chance MR (2014) Electron microscopic and proteomic comparison of terminal branches of the trigeminal nerve in patients with and without migraine headaches. Plast Reconstr Surg 134:796e–805e
Hametner S, Wimmer I, Haider L, Pfeifenbring S, Brück W, Lassmann H (2013) Iron and neurodegeneration in the multiple sclerosis brain. Ann Neurol 74:848–861
Hinoi T, Gesina G, Akyol A et al (2005) CDX2-regulated expression of iron transport protein hephaestin in intestinal and colonic epithelium. Gastroenterology 128:946–961
Jen JC (2015) Familial hemiplegic migraine. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews. NCBI, University of Washington, Seattle
Jiang R, Hua C, Wan Y et al (2015) Hephaestin and ceruloplasmin play distinct but interrelated roles in iron homeostasis in mouse brain. J Nutr 145:1003–1009
Joutel A, Bousser MG, Biousse V et al (1993) A gene for familial hemiplegic migraine maps to chromosome 19. Nat Genet 5:40–45
Kallela M, Wessman M, Färkkilä M, Palotie A, Koskenvuo M, Honkasalo ML, Kaprio J (1999) Clinical characteristics of migraine in a population-based twin sample: similarities and differences between migraine with and without aura. Cephalalgia 19:151–158
Knight YE, Bartsch T, Kaube H, Goadsby PJ (2002) P/Q-type calcium-channel blockade in the periaqueductal gray facilitates trigeminal nociception: a functional genetic link for migraine? J Neurosci 22:1–6
Lafrenière RG, Cader MZ, Poulin JF et al (2010) A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16:1157–1160
Lemos C, Castro MJ, Barros J, Sequeiros J, Pereira-Monteiro J, Mendonҫa D, Sousa A (2009) Familial clustering of migraine: further evidence from a Portuguese study. Headache 49:404–411
Leonardi M, Steiner TJ, Scher AT, Lipton RB (2005) The global burden of migraine: measuring disability in headache disorders with WHO’s Classification of Functioning, Disability and Health (ICF). J Headache Pain 6:429–440
Ligthart L, de Vries B, Smith AV et al (2011) Meta-analysis of genome-wide association for migraine in six population-based European cohorts. Eur J Hum Genet 19:901–907
Lindley PF, Card G, Zaitseva I, Zaitsev V, Reinhammar B, Selin-Lindgren E, Yoshida K (1997) An X-ray structural study of human ceruloplasmin in relation to ferroxidase activity. J Biol Inorg Chem 2:454–463
Liu P, Xiao Z, Ren F, Guo Z, Chen Z, Zhao H, Cao YQ (2013) Functional analysis of a migraine-associated TRESK K+ channel mutation. J Neurosci 33:12810–12824
Maher BH, Lea RA, Benton M et al (2012) An X chromosome association scan of the Norfolk Island genetic isolate provides evidence for a novel migraine susceptibility locus at Xq12. PLoS One 7:e37903
Martin VT (2015) Newer research and its significance. In: Diamond S (ed) Headache and migraine biology and management, 1st edn. Academic, Cambridge, pp 283–286
Messlinger K, Fischer MJ, Lennerz JK (2011) Neuropeptide effects in the trigeminal system: pathophysiology and clinical relevance in migraine. Keio J Med 60:82–89
Montagna P (2008) Migraine genetics. Expert Rev Neurother 8:1321–1330
Nadadur SS, Srirama K, Mudipalli A (2008) Iron transport & homeostasis mechanisms: their role in health & disease. Indian J Med Res 128:533–544
Nakajima C, Kulik A, Frotscher M, Herz J, Schäfer M, Bock HH, May P (2013) Low density lipoprotein receptor-related protein 1 (LRP1) modulates N-methyl-d-aspartate (NMDA) receptor-dependent intracellular signaling and NMDA-induced regulation of postsynaptic protein complexes. J Biol Chem 288:21909–21923
Nicolson SE (2016) Differential diagnosis and treatment of headaches. In: Stern TA, Fava M, Rosenbaum JF, Wilens TE (eds) Massachusetts general hospital comprehensive clinical psychiatry, 2nd edn. Elsevier Health Sciences, Philadelphia, pp 839–851
Noseda R, Burstein R (2013) Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain 154(Suppl):1. doi:10.1016/j.pain.2013.07.021
Ophoff RA, Terwindt GM, Vergouwe MN et al (1996) A 3-Mb region for the familial hemiplegic migraine locus on 19p13.1-p13.2: exclusion of PRKCSH as a candidate gene. Dutch Migraine Genetic Research Group. Eur J Hum Genet 4:321–328
Pietrobon D (2007) Familial hemiplegic migraine. Neurotherapeutics 4:274–284
Ponka P (2004) Hereditary causes of disturbed iron homeostasis in the central nervous system. Ann N Y Acad Sci 1012:267–281
Qian ZM, Chang YZ, Leung G et al (2007) Expression of ferroportin 1, hephaestin and ceruloplasmin in rat heart. Biochim Biophys Acta 1772:527–532
Rainero I, Rubino E, Gallone S et al (2014) KCNK18 (TRESK) genetic variants in Italian patients with migraine. Headache 54:1515–1522
Ranganathan PN, Lu Y, Fuqua BK, Collins JF (2012) Immunoreactive hephaestin and ferroxidase activity are present in the cytosolic fraction of rat enterocytes. Biometals 25:687–695
Rocca MA, Ceccarelli A, Falini A et al (2006) Brain gray matter changes in migraine patients with T2-visible lesions: a 3-T MRI study. Stroke 37:1765–1770
Rothner AD (2003) Complicated migraine and migraine variants. Headache 43:427–428
Russell MB, Rasmussen BK, Thorvaldsen P, Olesen J (1995) Prevalence and sex-ratio of the subtypes of migraine. Int J Epidemiol 24:612–618
Russell MB, Rasmussen BK, Fenger K, Olesen J (1996) Migraine without aura and migraine with aura are distinct clinical entities: a study of four hundred and eighty-four male and female migraineurs from the general population. Cephalalgia 16:239–245
Schorge S, Kullmann DM (2010) Sodium channel mutations and epilepsy: association and causation. Exp Neurol 226:8–10
Semenov IA (2015) Migraine headaches. Dis Mon 61:218–222
Sprenger T, Goadsby PJ (2010) What has functional neuroimaging done for primary headache… and for the clinical neurologist? J Clin Neurosci 17:547–553
Spuch C, Ortolano S, Navarro C (2012) LRP-1 and LRP-2 receptors function in the membrane neuron. Trafficking mechanisms and proteolytic processing in Alzheimer’s disease. Front Physiol 3:269–282
Stewart WF, Bigal ME, Kolodner K, Dowson A, Liberman JN, Rb Lipton (2006) Familial risk of migraine: variation by proband age at onset and headache severity. Neurology 66:344–348
Stovner LJ, Andree C (2010) Prevalence of headache in Europe: a review for the Eurolight project. J Headache Pain 11:289–299
Syed BA, Beaumont NJ, Patel A et al (2002) Analysis of the human hephaestin gene and protein: comparative modelling of the N-terminus ecto-domain based upon ceruloplasmin. Protein Eng 15:205–214
Thomsen LL, Olesen J (2004) Sporadic hemiplegic migraine. Cephalalgia 24:1016–1023
Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X (2011) TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain 7:30–46
Ulrich V, Gervil M, Kyvik KO, Olesen J, Russell MB (1999) Evidence of a genetic factor in migraine with aura: a population-based Danish twin study. Ann Neurol 45:242–246
Uygunoglu US, Siva A (2015) Epidemiology of headache. In: Mitsikostas DD, Paemeleire K (eds) Pharmacological management of headaches, 1st edn. Spinger International Publishing, Cham, pp 7–18
von Einem B, Schwanzar D, Rehn F et al (2010) The role of low-density receptor-related protein 1 (LRP1) as a competitive substrate of the amyloid precursor protein (APP) for BACE1. Exp Neurol 225:85–93
Wang XP, Ding HL, Geng CM, Jiang YM (2008) Migraine as a sex-conditioned inherited disorder: evidences from China and the world. Neurosci Bull 24:110–116
Welch KM (2009) Iron in the migraine brain; a resilient hypothesis. Cephalalgia 29:283–285
Welch KMA, Nagesh V, Aurora SK, Gelman N (2001) Periaqueductal gray matter dysfunction in migraine and chronic daily headache may be due to free radical damage. J Headache Pain 2(S1):s33–s41. doi:10.1007/s101940170007
WHO (2011) Atlas of headache disorders and resources in the world 2011. World Health Organization. http://www.who.int/mental_health/management/atlas_headache_disorders/en/. Accessed 20 Feb 2016
Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5:863–873
Zhou J, Yang CX, Zhong JY, Wang HB (2013) Intrathecal TRESK gene recombinant adenovirus attenuates spared nerve injury-induced neuropathic pain in rats. NeuroReport 24:131–136
Ziegler DK, Hur YM, Bouchard TJ Jr, Hassanein RS, Barter R (1998) Migraine in twins raised together and apart. Headache 38:417–422
Zivadinov R, Willheim K, Jurjevic A, Sepic-Grahovac D, Bucuk M, Zorzon M (2001) Prevalence of migraine in Croatia: a population-based survey. Headache 41:805–812
Acknowledgements
Cassie Albury is the recipient of an Australian Postgraduate Award (APA).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Rights and permissions
About this article

Cite this article
Albury, C.L., Stuart, S., Haupt, L.M. et al. Ion channelopathies and migraine pathogenesis. Mol Genet Genomics 292, 729–739 (2017). https://doi.org/10.1007/s00438-017-1317-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-017-1317-1