
Abstract
Glutamate is the predominant excitatory neurotransmitter in the mammalian central nervous system (CNS). A family of five Na+-dependent transporters maintain low levels of extracellular glutamate and shape excitatory signaling. Shortly after the research group of the person being honored in this special issue (Dr. Baruch Kanner) cloned one of these transporters, his group and several others showed that their activity can be acutely (within minutes to hours) regulated. Since this time, several different signals and post-translational modifications have been implicated in the regulation of these transporters. In this review, we will provide a brief introduction to the distribution and function of this family of glutamate transporters. This will be followed by a discussion of the signals that rapidly control the activity and/or localization of these transporters, including protein kinase C, ubiquitination, glutamate transporter substrates, nitrosylation, and palmitoylation. We also include the results of our attempts to define the role of palmitoylation in the regulation of GLT-1 in crude synaptosomes. In some cases, the mechanisms have been fairly well-defined, but in others, the mechanisms are not understood. In several cases, contradictory phenomena have been observed by more than one group; we describe these studies with the goal of identifying the opportunities for advancing the field. Abnormal glutamatergic signaling has been implicated in a wide variety of psychiatric and neurologic disorders. Although recent studies have begun to link regulation of glutamate transporters to the pathogenesis of these disorders, it will be difficult to determine how regulation influences signaling or pathophysiology of glutamate without a better understanding of the mechanisms involved.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glutamate as a Neurotransmitter
The amino acid, glutamate, is the predominant excitatory neurotransmitter in the mammalian central nervous system. It is also the most abundant neurotransmitter in the brain with levels that approximate 10 mmol/kg [for review, see 1]. Glutamatergic signaling is required for essentially all rapid excitatory synaptic transmission and contributes to learning, memory, and cognition (for reviews, see [2,3,4,5]). Glutamate is also likely used as a significant source of fuel through oxidative metabolism (for reviews, see [6,7,8,9]). In fact, the yield of ATP from oxidation of glutamate can be as high as two-thirds of that obtained from glycolysis/oxidation of glucose, and the levels of glutamate are approximately 20-fold higher than those of glucose [8, 10]. Glutamate is also one of the three amino acids required for synthesis of one of the major anti-oxidants, glutathione [11, 12]. Glutamatergic signaling is mediated by two families of receptors, ligand-gated ion channels also called ionotropic receptors (iGluRs) and G protein-coupled receptors also referred to as metabotropic receptors (mGluRs) (for reviews, see [13,14,15,16,17,18,19,20]). Some of these receptors mediate very rapid signaling with a time course of a few msec [21,22,23], while others have a slower time course that can last seconds [19, 20, 24,25,26,27]. These receptors are heterogeneously distributed around the synapse and on surrounding cells (e.g. astroglia). Over 30 years ago, several different groups demonstrated that excessive activation of glutamate receptors can cause neuronal loss and that blocking glutamate receptors in vivo can attenuate the damage observed in animal models of acute injury (stroke and trauma). Abnormal excitatory signaling has also been implicated in chronic neurodegenerative and psychiatric disorders (for reviews, see [3, 28,29,30,31,32,33,34]). Given the importance of excitatory signaling to information processing and several different neurologic/psychiatric disorders, it should not be surprising that the extracellular concentrations of glutamate are tightly controlled.
Using microdialysis, the extracellular levels of glutamate were originally estimated to be as high as 10 µM [35,36,37], but several of the glutamate receptors are activated by high nM or low micromolar concentrations of glutamate [19, 27]. At 10 µM, most glutamate receptors would be tonically activated and/or desensitized/inactivated, and there would be little or no glutamatergic signaling. More recent studies using glutamate receptor activation as a bioassay have placed the synaptic concentrations of glutamate closer to 25 nM [38]. While some have suggested that the concentrations of glutamate may vary in different extracellular compartments [39], there is no evidence for a gradient in hippocampal slices [40]. Nevertheless, it seems likely that local enrichment of transporters differentially shapes the kinetics of glutamate clearance. With total brain glutamate levels of 10 mmol/kg, this puts the transmembrane gradient at something like 250,000-fold, if glutamate were uniformly distributed inside cells. While this assumption is certainly wrong because glutamate is estimated to be as high as 100 mM in synaptic vesicles (for discussion, see [24]), neurons and glia expend a tremendous amount of energy to keep extracellular levels of glutamate very low. Therefore, glutamate can spill out of subcellular compartments and cells under conditions that deplete brain energy.
Importance of Glutamate Transport
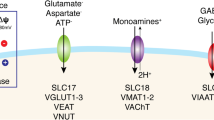
Extracellular concentrations of glutamate are controlled by a family of five Na+-dependent transporters and by a chloride-dependent transport system, called system \({\text{X}}_{\text{c}}^{-}\). The Na+-dependent transporters include: GLAST, GLT-1, and EAAC1 (also known as EAAT1-3, respectively), EAAT4, and EAAT5. The rodent genes for these transporters are referred to as Slc1a3, Slc1a2, Slc1a1, Slc1a6, and Slc1a7, respectively. These transporters are heterogeneously distributed in the nervous system [41,42,43]. GLT-1 and GLAST are enriched in astrocytes with higher levels of GLT-1 found in forebrain astrocytes and higher levels of GLAST found in cerebellar astrocytes (Bergmann glia) or retinal astrocytes (Müller glia) [41, 44,45,46,47]. The levels of GLT-1 increase dramatically during the period of synaptogenesis in both rodents and humans, and GLAST levels also increase during development [48, 49]. Although some investigators have observed EAAC1 immunoreactivity in astrocytes and oligodendroglia [50, 51], several commercial antibodies bind to tissue from EAAC1 knock-out animals [52]; at least some of these cross-react with tubulin [53]. When one uses antibodies that do not display this cross-reactivity, EAAC1 is restricted to cell bodies and dendrites of neurons [41, 54]. EAAT4 expression is highest in cerebellar Purkinje cells, but it is also found elsewhere in the nervous system [55,56,57,58]. EAAT5 was originally thought to be exclusively expressed in photoreceptors and bipolar cells of the retina, but several mRNA splicing variants of EAAT5 are found throughout the brain [59,60,61,62]. These transporters are not uniformly distributed on the plasma membrane. For example, expression of GLT-1 and GLAST is enriched on fine astrocyte processes that oppose excitatory synapses [44, 45]. EAAT4 is enriched in cell bodies and dendrites, and EAAT5 is enriched in presynaptic nerve terminals [56, 63].
These transporters are thought to share the same stoichiometry with one H+ and three Na+ ions accompanying the inward movement of a single molecule of glutamate combined with the outward movement of one K+ ion [64,65,66]. These transporters also have a substrate-activated Cl− conductance that is uncoupled from transport [59, 67,68,69,70]. Compared to the rapid ms signaling that can occur at glutamatergic synapses, these transporters are relatively slow with cycle times that range from 10 to ~ 300 ms [71,72,73]. The ‘slower’ transporters, EAAT4 and EAAT5, tend to have a larger Cl− conductance, consistent with the notion that these transporters function more as glutamate-activated inhibitory ‘receptors’ [63, 71, 74]. The faster transporters, GLT-1, GLAST and EAAC1, have cycle times of 10 to 50 ms [71,72,73, 75, 76], but this is still too slow to influence the kinetics of the faster forms of excitatory signaling [72]. Thus, unlike cholinergic signaling that is terminated by acetylcholinesterase, a diffusion rate limited enzyme that can cleave a molecule of acetylcholine in about 50 µs [77, 78], the time-course of rapid glutamatergic signaling is not likely controlled by transporter cycling. The levels of GLT-1 are as high as 1% of brain protein, and the concentrations are high enough to bind all the glutamate in a single vesicle [79]. In fact, Huang and colleagues demonstrated that transporters control excitatory signaling by binding glutamate, thus functioning as ‘buffers’ to limit the amplitude of excitatory responses rather than controlling the time-course ([80] for recent, discussion see [81]). Several more recent electrophysiologic studies have shown that the effects of transport on excitatory signaling are more complicated; the relative locations of transporters and subtypes of receptors combined with diffusion shape excitatory signaling [72, 82,83,84].
Under conditions that impair the supply of energy needed to fuel the transmembrane ion gradients, these transporters fail to clear extracellular glutamate. In addition, these transporters can operate in reversed-direction releasing cytosolic glutamate into the extracellular space. This transport-dependent increase in extracellular glutamate has been observed after stroke-like insults [85,86,87]. Thus, although glutamate transport is generally considered beneficial, there is evidence that these transporters can contribute to damage observed with these insults (for review, see [88]).
Glutamate transporter function, localization, transcriptional regulation, and relationships to disease have been the topic of several reviews [42, 43, 68, 70, 89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109] and there is one recent review of the post-translational regulation of GLT-1 [110]. In this review, we will focus on the rapid regulation of each member of this transporter family.
Methodological Considerations
Before we begin, we need to provide some background information regarding complications with the systems used to study this regulation and with the interpretation of measures of glutamate uptake. One would ideally like to study regulation in a native preparation in case the effects are dependent upon cellular context. As indicated above, GLT-1 and GLAST are enriched in astrocytes in vivo, but astrocytes in culture do not express much or any GLT-1 [111,112,113,114]. They do express GLT-1 after treatment with compounds like dibutyryl cAMP, epidermal growth factor, etc. [for review, see 106] or by co-culturing them with neurons or endothelia [112,113,114,115,116], but GLT-1-mediated activity, defined using compounds like dihydrokainate that selectively inhibit GLT-1 [117], represents less than 10 % of the total activity even under these conditions. This does, however, make it relatively easy to study regulation of GLAST using cultured astrocytes. One limitation of these analyses is that unlike astrocytes in vivo that are polarized with processes that sheath blood vessels or extend to synapses, these cultured astrocytes are not polarized [118], and they are molecularly quite different from astrocytes found in vivo [119]. Synaptosomes have also been used. These subcellular fractions are relatively enriched in nerve terminals, but they also contain resealed glial membranes [120]. The pharmacology of glutamate transport in synaptosomes from forebrain closely parallels that observed for GLT-1 and is different from that observed with the other members of this family (for review, see [90]). In fact, complete deletion of GLT-1 from mice reduces glutamate uptake to 5% of control in synaptosomes prepared from cortex [121]. Originally, we and others thought this meant that glial GLT-1 mediated essentially all of the uptake in synaptosomes because most GLT-1 immunoreactivity is observed in astrocytes in vivo (for a review, see [92]). This conclusion was not consistent with older findings of direct accumulation of glutamate transport substrates into nerve terminals [122,123,124,125,126]. In fact, low levels of GLT-1 mRNA and protein had also been detected in neurons in vitro and in vivo [127,128,129,130,131]. More recently, GLT-1 has been selectively deleted from either neurons or astrocytes in vivo using cell-selective, Cre-recombinase-induced gene excision [132, 133]. In these studies, deletion of GLT-1 from neurons has a minimal effect on synaptosomal GLT-1 protein levels (90–95% of control), but it reduces glutamate uptake in synaptosomes to ~ 50% of control. Deletion of GLT-1 from astrocytes reduces GLT-1 immunoreactivity in synaptosomes to 20% of control and only has a small effect on uptake (to 85% of control). While the fact that the effects on uptake do not add up to 100% suggest that deletion of neuronal or glial GLT-1 may result in a compensatory up-regulation of GLT-1 in the other cell, these studies show that synaptosomal uptake of glutamate represents a mixture of both glial and neuronal pools of GLT-1 (for reviews, see [134, 135]). While heterologous expression systems are potentially useful for detailed analyses of mechanisms, there are complications. First, Na+-dependent glutamate transporters are widely distributed and expressed by most cells used for expression analyses [136,137,138]. This complicates analyses of transport activity that is mediated by both the endogenous and the exogenous transporter. While over-expression could enrich the contribution of an exogenous transporter, there is evidence that over-expression can blunt protein kinase C-dependent internalization of the dopamine transporter [139], making it hard to relate results to a native system. Finally, heterologous expression systems may lack a protein or express a protein that changes the way a transporter is regulated (for an example, see "Regulation of EAAC1 by PKC" below). Glutamate transport activity is generally measured using radioactive substrates, l-[3 H]-glutamate or d-[3 H]-aspartate. There are several ways to indirectly change Na+-dependent glutamate uptake. First, the uptake of these substrates is dependent on the availability of ATP that is required to support the Na+ gradients that drive transport activity [140]. Glutamate uptake is also activated by ATP-activated inward rectifying K+ channels (Kir4.1) [141, 142]. This means that changes in uptake can be caused by indirect effects on electrochemical gradients/K+ channels. Second, these transporters can function as exchangers. Under these conditions, radioactive substrate is exchanged for non-radioactive substrate at 1:1 stoichiometry across the plasma membrane. There is evidence that exchange is faster than net inward flux (for discussion, see [71]), but this was not observed with reconstituted GLT-1 [143]. Nevertheless, changing intracellular pools of glutamate or aspartate can change the driving force and the rate at which radioactive substrate accumulates inside the cell (for further discussion, see [134]). Therefore, changes in uptake may or may not be indicative of a selective effect on a glutamate transporter.
In the following sections, we will describe what is known about the signaling pathways that rapidly regulate the activity, subcellular localization, and/or levels of the various glutamate transporters. For the purposes of this review, we define rapid as effects that occur within minutes to a few hours and are presumably independent of transcription/translation. In at least a few cases, the results are complicated and/or contradictory. In most of these cases, we include citations to more than one study with the goal of demonstrating that many of these results are reproducible and likely informative. The differences may be a reflection of cellular milieu, and it may be possible to identify proteins that contribute to this differential regulation.
Regulation by Protein Kinase C
Protein kinase C (PKC) belongs to the AGC super family of Ser/Thr kinases. There are three different subfamilies of PKC and each has subtypes within these subfamilies, including conventional PKC (PKCα, β, and γ), novel (PKC δ, ε, η, and θ) and atypical (PKC ζ and ι) (for review, see [144]). The different subfamilies are activated by different stimuli. Conventional PKCs are activated by Ca2+ and diacylglycerol, novel PKCs are activated by diacylglycerol only, and atypical PKCs use a scaffolding protein for activation. Phorbol esters activate the first two families of PKC, but phorbol esters can have effects that are independent of PKC [145]. Dozens of laboratories, including our own, have published analyses of the linkages of PKC to glutamate transport activity. Many of the initial studies sought to define the mechanisms by which PKC regulates glutamate transport activity, including testing for correlations with changes in the numbers of transporters in the plasma membrane. More recently, there has been an emphasis on linking PKC-dependent regulation of these transporters to disease. In the sections below, we will address various themes that have emerged, including the fact that PKC regulates glutamate transport in ways that are difficult to reconcile with simple models.
Regulation of GLAST by PKC
The effects of phorbol ester-dependent activation of PKC on GLAST-mediated uptake have been examined using astrocytes prepared from cortex, cerebellum (Bergmann glia), and retina (Müller cells). In Bergmann glia [146, 147] and Müller cells [148, 149], activation of PKC with phorbol esters decreases the Vmax for glutamate transport. The time course for this effect is on the order of 15 min to an hour, and the effects are blocked by PKC antagonists. Activation of PKC had no effect on Na+-dependent glycine or GABA uptake, indicating that the decrease in glutamate uptake is not due to a decrease in the Na+-electrochemical gradient. In these cells, where GLAST is the only subtype of Na+-dependent glutamate transporter expressed, the effects of phorbol esters on glutamate uptake are correlated with a redistribution of GLAST from the cell surface to an intracellular compartment [146, 148, 149]. With longer incubations (6 h), activation of PKC causes a decrease in total GLAST protein levels [146, 148]. These results are consistent with the notion that prolonged activation of PKC can trigger internalization and degradation of GLAST (see Fig. 1 for schematic).

Schematic model of PKC-dependent regulation of GLAST. Activation of PKC has two distinct effects on GLAST-mediated activity. It can increase GLAST activity by a mechanism that has not yet been defined (dashed arrow). Activation can also decrease GLAST mediated activity by a mechanism that depends on Nedd4-2 and presumably ubiquitination/internalization of GLAST. It is not known if the effects of PKC are dependent upon direct phosphorylation of Nedd4-2. It is not known if or how the fate of GLAST is regulated after internalization, but it can be recycled back to the plasma membrane, targeted for degradation, or incorporated into vesicles for release into the extracellular space. Interleukin 1B or endothelin-1 also couple to decreases in GLAST via activation of PKC. Dashed arrows depict mechanisms that have not been defined
In cortical astrocytes, the story is more complicated. Some laboratories find that activation of PKC causes an increase in glutamate uptake [147, 150, 151]. This increase occurs relatively rapidly (within 15 to 30 min), it is blocked by inhibitors of PKC, and it is associated with an increase in Vmax. Na+-dependent glycine uptake is not altered by PKC [151], indicating that this effect is not caused by an increase in the Na+-electrochemical gradient. In other studies using seemingly the same cells, phorbol esters decrease [152, 153] or have no significant effect on glutamate transport [154]. Endothelin-1, a receptor agonist that can activate PKC, also causes a decrease in glutamate uptake [155]. These decreases in uptake occur on a similar timescale (15–30 min), are associated with a decrease in Vmax, and are blocked by inhibitors of PKC. Under the conditions used for both sets of studies, these cortical cultures exclusively express GLAST, and this was confirmed in several of these studies. In some of these studies, activation of PKC is associated with a loss of GLAST at the cell surface and a decrease in total GLAST immunoreactivity [151, 156], and it has also been associated with the appearance of GLAST in extracellular vesicles [153]. The different effects on GLAST activity have even been observed within the same study. For example, Bernabé and colleagues found that activati n of PKC decreases glutamate transport in cerebellar astrocytes, and it has the opposite effect in cortical astrocytes [147]. Similarly, Susarla and colleagues observed a PKC-dependent increase in transport activity in the same cultures that they documented a loss of GLAST protein [151]. Together these studies show that PKC can both increase and decrease GLAST-mediated activity, but activation of PKC causes a decrease in the levels of GLAST in the plasma membrane.
In Xenopus oocytes or human embryonic kidney cells that exogenously express GLAST, activation of PKC decreases glutamate uptake [157]. This effect is associated with incorporation of radioactive phosphate into GLAST immunoprecipitates. In this same study, this group found that mutation of consensus sites for PKC phosphorylation do not abolish PKC-dependent regulation of GLAST uptake, nor do they abolish incorporation of phosphate. Finally, they observed no evidence of intracellular accumulation of GLAST; this could be consistent with the rapid loss of GLAST immunoreactivity observed by Susarla in astrocytes [151], the secretion of GLAST into vesicles observed by Gosselin [153], or an effect of over-expression, which as mentioned above can blunt PKC-dependent internalization of the dopamine transporter [139].
More recently, some investigators have extended these analyses to animal models of disease that are associated with decreased GLAST protein. For example, in a rat model of neuropathic pain in which the sciatic nerve is partially ligated, there is a decrease in glutamate uptake in spinal cord synaptosomes measured using radioactive glutamate and in astrocytes where uptake was measured by recording glutamate transporter evoked currents [158]. This group also showed that interleukin-1 beta or phorbol ester cause a decrease in glutamate uptake in this system, and that a PKC antagonist blocks both of these effects. They used an inhibitor of dynamin and actin disrupting agents to implicate endocytosis in these effects and observed lower levels of GLAST (and GLT-1) in subcellular fractions enriched in plasma membranes and higher levels in cytosolic fractions. The decreases in glutamate uptake were rapidly reversed in many of these experiments, suggesting that these transporters are not necessarily targeted for degradation; they may be available for recycling back to the plasma membrane. In animals treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a toxin used to model Parkinson’s Disease, or in astrocytes treated with the active toxin, 1-methyl-4-phenylpyridinium (MPP+), there is a loss of GLAST protein and a decrease in glutamate uptake. Both of these effects are blocked by knocking down expression of the ubiquitin ligase, Nedd4-2 [159]. This would seemingly implicate ubiquitination in the loss of GLAST, but they were unable to detect GLAST in ubiquitin immunoprecipitates (even though they detected GLT-1 in these immunoprecipitates). It is possible that the GLAST antibody was not sufficiently sensitive or that ubiquitinated GLAST is rapidly degraded. Finally, decreased GLAST expression is observed in a bone cancer-induced model of pain; this effect is associated with increased PKC expression [160].
A schematic summary of PKC-dependent regulation of GLAST is provided in Fig. 1. While PKC can clearly cause internalization of GLAST and a decrease in transport activity, there are several issues that remain unresolved. First, the target of PKC has not been identified. No site on GLAST has been identified that abolishes this effect. Although Nedd4-2 is also phosphorylated by PKC [161], no specific sites have been identified that upon appropriate mutation either mimic or block the effects of PKC on GLAST. Second, although the ubiquitin ligase, Nedd4-2, has been implicated in the regulation of GLAST in oocytes [162] and in models of Parkinson’s Disease [159], incorporation of ubiquitin into GLAST has not been documented. Third, it is not clear why some observe a PKC-dependent increase in GLAST-mediated activity while others observe a PKC-dependent decrease in activity. There are no obvious differences in the methods used to prepare these cultures, and the cultures are consistently enriched in cells that express glial fibrillary acidic protein, a marker of astrocytes. Nevertheless, it seems likely that the differential effects of PKC are dependent on an un-identified protein(s) that are differentially expressed in cultures prepared in different laboratories. As a related issue, it is not clear how PKC increases GLAST-mediated uptake. It seems likely that the effects of PKC are dependent on some other protein. One possibility is mitochondrial ATP-activated K+ channels. Openers of these channels increase glutamate uptake in cortical astrocytes, and these channels are activated by PKC [163]. Finally, it is not clear how, or if, the fate of internalized GLAST is determined, but it can be targeted for degradation, for exocytosis in vesicles, or recycling back to the plasma membrane.
Regulation of GLT-1 by PKC
The results of analyses of acute regulation of GLT-1 by PKC are similarly contradictory, but more progress has been made with certain mechanistic aspects. In astrocytes grown under conditions in which GLT-1-mediated activity can be measured using a GLT-1 selective inhibitor (dihydrokainate), activation of the mGluR5 subtype of glutamate receptor causes a PKC-dependent increase in GLT-1-mediated activity [164,165,166]. This increase in glutamate uptake requires PKCε [166] (see Fig. 2 for schematic). In synaptosomes prepared from different forebrain regions, where GLT-1 mediates up to 95% of the total uptake [121], activation of PKC has no effect on glutamate uptake in striatal synaptosomes [167], decreases uptake in hippocampal synaptosomes [168], and has no effect on uptake in synaptosomes prepared from whole forebrain [169]. As mentioned above, synaptosomal glutamate uptake in forebrain is mediated by both neuronal and glial GLT-1 [132, 133]. In this regard, Daniels and colleagues found that in contrast to activation of PKC having no effect on uptake measured in synaptosomes, it increased the Vmax of glutamate uptake in gliosomes prepared from forebrain [169]. Perhaps the variable effects observed in these synaptosomal studies are related to the relative contributions of glial and neuronal pools of GLT-1 to total uptake, but this has not been investigated. To our knowledge, it has not been determined if these effects are related to changes in the cell surface expression of GLT-1. In platelets, activation of PKC also causes an increase in GLT-1 mediated uptake [170]. In a very early study, Casado and colleagues found that activation of PKC increases the incorporation of radioactive phosphate into GLT-1 and increases glutamate uptake measured in Hela cells transfected with wild-type GLT-1 [150]. They also demonstrated that mutation of serine-113 to asparagine resulted in a transporter that was not activated by PKC. Recently, Chen and colleagues demonstrated that the neurosteroid, dehydroepiandrosterone, causes a PKC-dependent increase in GLT-1-mediated currents recorded from astrocytes in hippocampal slices [171]. They also showed that this effect was associated with an increase in the amount of GLT-1 in the plasma membrane, as measured using a membrane impermeant biotinylation reagent. Together these studies show that activation of PKC can increase GLT-1 mediated uptake.

Schematic model of PKC-dependent regulation of GLT-1. mGluR5 activation causes an increase in GLT-1 mediated activity; this effect requires PKCε. Direct activation of PKC causes an increase in GLT-1 mediated activity; this effect requires serine-113. Activation of PKC can also cause a decrease in GLT-1-mediated activity. This effect is dependent upon PKCα, and GLT-1 forms immunoprecipitable complexes with PKCα. Internalization requires Nedd4-2 and ubiquitination of any one of several different lysine residues on GLT-1. It is not known if Nedd4-2 is directly modified by PKC. Flotillin has also been implicated as a possible downstream target of PKC. PPARα triggers internalization and degradation of GLT-1. After internalization, GLT-1 is either recycled back to the plasma membrane, targeted for degradation, or incorporated into vesicles for release. Sonic hedgehog (Shh) causes internalization and degradation by a mechanism that is dependent upon PKCδ. The de-ubiquitinating enzyme (DUB), ubiquitin C-terminal hydrolase‐L1, contributes to GLT-1 recycling. Dashed arrows depict mechanisms that have not been defined
In other systems, activation of PKC has no effect on GLT-1 mediated uptake [137], causes a decrease in Km that might look like an increase in transport if transport activity is only measured at lower concentrations of glutamate [172], or causes a decrease in GLT-1 mediated uptake [173,174,175,176]. The PKC-induced decreases in GLT-1-mediated transporter activity are associated with clustering of GLT-1 in the plasma membrane and redistribution of GLT-1 from the plasma membrane to internal compartment [156, 173, 177,178,179,180]. Using inhibitors that selectively block subtypes of PKC, PKCα was implicated in this regulation, and PKCα forms immunoprecipitable complexes with GLT-1 [178]. Although these effects are associated with an increase in phosphorylated GLT-1, they have not been linked to the mutation of any particular serine residue on GLT-1 [161, 173]. In fact, mutation of serine-520 abolishes the incorporation of phosphate into GLT-1 but does not block PKC-dependent redistribution of GLT-1 [161]. This suggests that PKC-dependent internalization of GLT-1 is not dependent on direct phosphorylation of GLT-1. Activation of PKC causes an increase in incorporation of ubiquitin into GLT-1 and mutation of all 11 lysine residues in the amino and carboxy termini of GLT-1 abolishes PKC-dependent internalization. Re-introduction of any one of several different lysine residues in carboxyl terminal is sufficient to restore PKC-dependent internalization [179, 180]. Co-expression of the ubiquitin ligase, Nedd4-2, with GLT-1 in Xenopus oocytes reduces GLT-1 mediated uptake while expression of a mutant variant that lacks ligase activity has no effect [161, 181]. In COS cells, PKC-dependent decreases in GLT-1-mediated activity and cell surface expression are blocked by siRNA-mediated knock-down of Nedd4-2 [161]. In this same study, activation of PKC increases the amount of GLT-1 that is found in Nedd4-2 immunocomplexes and vice versa. They also showed that activation of PKC increases the amount of radioactive phosphate incorporated into Nedd4-2 immunoprecipitates. Together these studies raise the possibility that PKC phosphorylates and activates Nedd4-2, but this has not been directly demonstrated. After internalization, GLT-1 is recycled through endosomes, and inhibition of the de-ubiquitinating enzyme (DUB), ubiquitin C-terminal hydrolase‐L1, blocks the recycling of GLT-1 back to the plasma membrane [182].
In a series of recent studies, Perez-Jimenez and colleagues have linked Lafora disease to decreased cell surface expression of GLT-1 [183]. Lafora disease is caused by loss-of-function mutations in genes that code for a glucan phosphatase and malin, an E3-ubiquiting ligase. It is associated with late onset progressive myoclonus epilepsy. They show that malin also causes ubiquitination of GLT-1 at a location other than the carboxy terminus, and this is associated with retention of GLT-1 at the plasma membrane. They also show malin causes ubiquitination of arrestin. These studies suggest that somehow Nedd4-2 and malin differentially control cell surface expression of GLT-1; it is possible that ubiquitination by malin targets arrestin for degradation, providing a brake for Nedd4-2-dependent ubiquitination and internalization of GLT-1.
Pharmacologic and genetic approaches have been used to identify the endocytic routes that contribute to PKC-dependent redistribution of GLT-1. Hypertonic sucrose or a dominant-negative variant of dynamin block PKC-dependent redistribution of GLT-1 [177, 184]. Although these reagents affect both clathrin-dependent and clathrin-independent endocytosis [185], the results are consistent with the notion that PKC increases endocytosis of GLT-1. Co-expression of a dominant-negative inhibitor of the clathrin heavy chain attenuates PKC-dependent internalization of GLT-1 [184], but this may be different depending on the cellular context. In a study focused on PKC-dependent internalization of the dopamine transporter, Cremona and colleagues showed PKC-dependent internalization of GLT-1 is blocked by knocking down expression of flotillin, a component of a clathrin-independent endocytic pathway [186]. They also showed that PKC-dependent internalization of the dopamine transporter is accompanied by phosphorylation of flotillin and that mutation of serine 315 of flotillin reduces PKC-dependent phosphorylation of flotillin and internalization of the dopamine transporter. Although these later studies were not extended to analyses of GLT-1, they are consistent with the possibility that PKC causes internalization of GLT-1 by phosphorylating flotillin and triggering flotillin-dependent endocytosis. Shortly after this study was published, a different group showed that endocytosis of the dopamine transporter is independent of flotillin, but that depletion of flotillin increases lateral diffusion of the dopamine transporter [187]. This group suggested that flotillin is required for recruitment of the dopamine transporter to lipid rafts. The fact that GLT-1 is known to be associated with lipid rafts in vivo [188] suggests that PKC may cause clustering of GLT-1 with flotillin in lipid rafts, but this has not been examined.
As is observed with GLAST, longer incubations with phorbol ester cause a loss of GLT-1, and this loss is blocked by inhibitors of lysosomal degradation [152, 184]. More recently, sonic hedgehog has been shown to decrease GLT-1-mediated activity [176]. This effect is blocked by inhibitors of PKC and is associated with degradation of GLT-1. This loss of GLT-1 was blocked by an inhibitor of proteosomal degradation and dependent upon PKCδ. Finally, peroxisome proliferator-activated receptor-alpha (PPARα) agonists also trigger internalization and degradation of GLT-1 [189]. These effects are blocked by inhibitors of PKC, implicating another receptor in PKC-dependent regulation of GLT-1. It is not known if these effects are dependent on Nedd4-2.
Together these studies demonstrate that GLT-1 activity and cell surface expression are acutely regulated. A schematic summary of PKC-dependent regulation of GLT-1 is provided in Fig. 2. As is observed with GLAST, there is clear evidence that PKC can increase GLT-1-mediated activity. It seems likely that this is a direct effect of phosphorylation of serine 113 [190], but this needs to be tested in different systems. There is also clear evidence that activation of PKC can cause internalization of GLT-1. It seems that this later effect is independent of direct transporter phosphorylation and instead depends on ubiquitination of GLT-1. The fact that GLT-1 is the most abundant of these transporters [54, 79] and that genetic knock-out of GLT-1 causes a severe phenotype [121] provides a compelling rationale for detailed mechanistic studies that result in the generation of specific tools that can be used to both determine the conditions under which this regulation occurs in vivo and the physiologic/pathologic consequences of this regulation.
Regulation of EAAC1 by PKC
Activation of PKC causes an increase in EAAC1-mediated activity in C6 glioma, a cell line that expresses none of the other Na+-dependent glutamate transporters [191, 192]. In fact, activation of PKC doubles L-[3 H]-glutamate uptake within 2 min. This effect is due to an increase in Vmax and not associated with a change in Na+-dependent glycine uptake [193]. A PKC-dependent increase in EAAC1-mediated activity has been observed in several studies and is consistently associated with a redistribution of EAAC1 from intracellular stores to the plasma membrane using C6 glioma [156, 192, 194,195,196]. A PKC-induced increase in EAAC1 surface expression has been observed in other systems that endogenously express EAAC1, including human embryonic kidney cells [197], human SH-SY5Y neuroblastoma [198], and primary neuronal cultures [156, 199]. This is consistent with the existence of high levels of intracellular EAAC1 immunoreactivity that has been observed in neurons in vivo [54]. Interestingly, the volatile anesthetic, isoflurane, also increases EAAC1-mediated activity and cell surface expression in C6 glioma or synaptosomes [200]. This effect of isoflurane is blocked by PKC antagonists or knockdown of PKCα. The PKC-induced increase in EAAC1 activity and cell surface expression is mimicked by platelet-derived growth factor (PDGF), but this increase is dependent upon activation of phosphatidylinositol kinase (PI3K) not PKC and is smaller than that observed with PKC [201]. Neurotensin and endothelin-1 also increase EAAC1 surface expression, but these effects are not blocked by inhibitors of PKC or PI3K [202, 203]. Together these studies show that activation of PKC (and other signaling pathways) can cause an increase in EAAC1-mediated activity that is associated with a redistribution of EAAC1 from subcellular compartments to the plasma membrane.
Changes in cell surface expression can be caused by increasing in the rate of delivery of transporter to the plasma membrane or by decreasing the rate of endocytosis (Fig. 3). Both of these rates can be measured using tricks with biotinylation and are easier to measure when there is a substantial intracellular pool that rapidly cycles into and out of the plasma membrane. Delivery to the plasma membrane can be measured by determining the rate at which the amount of biotinylated transporter increases when cells are maintained at 37 °C (conditions that allow constitutive recycling). In both C6 glioma and primary neuronal cultures, the amount of biotinylated transporter doubles within 15 min, consistent with relatively rapid recycling of the transporter into and out of the plasma membrane with a half-life of between 5 and 7 min [199]. The rate of endocytosis can be measured using a biotinylating reagent that contains a disulfide bond that can be cleaved. With this reagent, cell surface proteins are labelled under conditions that prevent trafficking (4 °C), and then cells are warmed for various periods of time before re-cooling and stripping biotin from those proteins that remain on the cell surface. A comparable half-life of 5–7 min is observed using this strategy [199, 204]. These studies suggest that, as has been observed for subtypes of glutamate receptors [205, 206] and insulin-regulated GLUT4 glucose transporter [207,208,209], EAAC1 constitutively recycles into and out of the plasma membrane.

Schematic model of PKC-dependent regulation of EAAC1. There is evidence that activation of PKC increases EAAC1-mediated activity by three different mechanisms. First, there is an increase in EAAC1-mediated activity that is dependent upon PKCε and is associated with Arl6ip1 being recruited into a complex with GTRAP3-18 (Arl6ip5). It is not known if this dimer remains associated with EAAC1. PKC increases the rate of EAAC1 delivery into the plasma membrane. This effect is mimicked by PDGF/PI3K. PKC also slows the rate of endocytosis of EAAC1. Activation of PKC increases clustering and co-immunoprecipitation of EAAC1 with PKCα. In MDCK cells (lower part of figure), regulation of EAAC1 has some overlapping features (e.g. regulation by Arl6ip5), but there are several differences. The steady state levels of EAAC1 are higher in the plasma membrane compared to C6 glioma and neurons. In MDCK cells, NHERF3 interacts with EAAC1 and stabilizes it on the plasma membrane; NHERF3 is not expressed in brain. Activation of PKC causes a decrease in EAAC1-mediated activity and internalization of EAAC1. This effect is dependent upon recruitment of the clathrin adaptor, Numb, into a complex with EAAC1. In both C6 glioma and MDCK cells, the effects of PKC are dependent upon several different motifs within the carboxy terminal tail of EAAC1
Defining the mechanisms involved in the regulation of EAAC1 by PKC is complicated by the fact that there are three different reasons to believe that PKC has more than one effect on EAAC1 in C6 glioma. First, the increase in uptake is consistently larger than the increase in cell surface expression (uptake increases > 2-fold above baseline, cell surface expression increases ~ 50 %) [192]. Second, the PKC antagonist, Gö6976, completely blocks the PKC-induced increase in cell surface expression, but only partially blocks the PKC-induced increase in glutamate uptake at concentrations that selectively block the classical subtypes of PKCs [210]. Third, activation of PKC causes both an increase the rate of delivery of transporters to the plasma membrane and decreases the rate of endocytosis of EAAC1, suggesting that PKC affects two different aspects of EAAC1 trafficking [199]. This is not observed with all signals that increase EAAC1 surface expression because PDGF only increases the rate of delivery of EAAC1 to the plasma membrane and has no effect on the rate of endocytosis [199]. These analyses of the effects of PKC on the kinetics of transporter trafficking are complemented by analyses of mutant variants of EAAC1. In these studies, mutation of amino acids 502 to 504 (YVN) to alanine residues only partially blocks PKC-induced increases in EAAC1 cell surface expression, but completely block PDGF-induced increases [195]. Together these studies suggest that PKC could have three different effects on EAAC1-mediated activity: one that is independent of changes in cell surface expression, a second that is dependent on accelerated delivery of transporters from subcellular vesicles to the plasma membrane, and a third that is dependent upon decreases in the rate of constitutive endocytosis.
In an early analysis of potential interacting proteins, Lin and colleagues used 87 amino acids of the carboxy terminal tail (amino acids 438–524) as bait for a yeast 2-hybrid screen to identify EAAC1 interacting proteins. They isolated a protein they called GTRAP3-18 that decreases EAAC1-mediated activity in co-transfection studies in HEK293 cells; this effect is associated with an increase in Km [211]. This GTRAP3-18-dependent inhibition of EAAC1 has been observed by others [212]. Up-regulation of GTRAP3-18 blocks PKC-dependent increases in EAAC1-mediated activity without blocking the PKC-dependent increases in EAAC1 cell surface expression in HEK 293 cells [197] and in cerebellar granule cells [213]. Activation of PKC increases co-localization of GTRAP3-18 with EAAC1 at the plasma membrane [213]. Thus GTRAP3-18 can function as a dominant-negative inhibitor of EAAC1 activity. GTRAP3-18 is also called JWA and was also cloned as an mRNA that is highly (three to fourfold) up-regulated in amygdala after chronic administration of morphine and was termed addicsin [214]. This protein is also called ADP-ribosylation factor-like GTPase 6 interacting protein 5 (Arl6ip5). In a search for GTRAP3-18 interacting proteins, Akiduki et al. identified Arl6ip1 [215]. Using C6 glioma engineered to express Arl6ip5 and Arl6ip1 under tight transcriptional control, they show that PKC-dependent increases in EAAC1-mediated activity are blocked by increased expression of Arl6ip5 and that expression of Arl6ip1 relieves this inhibition. Together these studies provide a mechanism by which PKC can regulate EAAC1 in a manner that is independent of changing transporters at the plasma membrane. Using pharmacological approaches and exploiting the differential sensitivity of PKC subtypes to down-regulation, PKCε has been implicated in the regulation of EAAC1 activity that happens in the absence of redistribution to the plasma membrane [210] (see Fig. 3).
The alpha subtype of PKC has been implicated in the PKC-dependent redistribution of EAAC1 using a variety of strategies. First, the PKC-dependent effects on cell surface expression are blocked by Gö6976 at concentrations that selectively block classical PKCs, and PKCα is the only classical PKC observed in C6 glioma [210]. Activation of PKC causes clustering of PKCα and EAAC1 at the plasma membrane and increases the amount of PKCα that co-immunoprecipitates with EAAC1 in both C6 glioma and synaptosomes [216]. Finally, the isoflurane-induced increase in EAAC1 is blocked by Gö6976 at concentrations that selectively block classical PKCs or by antisense mediated knockdown of PKCα [200]. Isoflurane also increases the amount of PKCα that co-immunoprecipitates with EAAC1 [200].
Using stably transfected COS7 cells, Huang and colleagues identified serine 465 as being required for isoflurane-induced increases in glutamate transport, redistribution to the plasma membrane, and phosphorylation [217]. This group found that this same residue was required for PKC-dependent activation of EAAC1 activity in Xenopus oocytes [218]. They also generated a peptide surrounding this region that blocks isoflurane-induced increases in biotinylated EAAC1, EAAC1-mediated uptake, and co-immunoprecipitation of PKCα with EAAC1 [219]. These studies strongly implicate serine 465 of EAAC1 as a target for PKC-dependent redistribution, but other motifs in this same region also contribute to the regulated redistribution of EAAC1.
When C6 glioma are transfected with either epitope-tagged EAAC1 or GLT-1, they respond differently to activation of PKC; EAAC1 increases at the plasma membrane and GLT-1 decreases at the plasma membrane [173, 195]. Using a family of GLT-1/EAAC1 chimeras, a ten amino acid carboxy terminal domain starting at amino acid 502 was identified that upon deletion completely abolishes phorbol ester- or PDGF-dependent increases in EAAC1 cell surface expression [195]. Mutation of 502YVN504 within this domain completely blocks the PDGF-induced increase in EAAC1 surface expression, but only partially blocks the PKC-induced increase in surface expression, suggesting that the YVN motif is required for the regulated delivery of EAAC1. The differential effects of deletion of ten amino acids compared to the effects of mutation of three amino acids implies that additional amino acids in this region also contribute to regulated trafficking of EAAC1, likely contributing to the control of endocytosis. It was not determined if introduction of this domain into GLT-1 is sufficient to confer PKC-dependent increase in surface expression. It is also not known how or if phosphorylation of serine-465 is mechanistically linked to the role of the YVN motif.
Several studies of the role of the carboxy terminus of EAAC1 in regulated trafficking have been conducted in Madin-Darby canine kidney (MDCK) cells. Before describing these analyses, it is important to mention that activation of PKC causes a decrease in EAAC1-mediated uptake and a redistribution of EAAC1 from the plasma membrane to an intracellular pool in MDCK cells [220, 221]. This is the exact opposite effect of that observed in C6 glioma and neurons. There is also a big difference in the rate at which EAAC1 constitutively recycles in the two systems. In C6 glioma and neurons, ~ 50% of the cell surface transporter is recycled in ~ 5 to 7 min [199, 204] while in MDCK cells, only 5% of EAAC1 is internalized after 15 min [222]. In neurons and C6 glioma, most EAAC1 is found in a non-biotinylated (intracellular fraction) [192, 199], and in MDCK cells, there is very little intracellular EAAC1 [222]. This is consistent with the observation that most EAAC1 is on the plasma membrane in kidney and most EAAC1 is intracellular in neurons in vivo [54].
Together these observations indicate that the effect of PKC on EAAC1 is dependent upon the cellular context, and the differences may provide mechanistic insights. Almost 20 years ago, the YVN motif was identified as part of a six amino acid domain, VNGGFA, within the carboxy terminus as being required for targeting of EAAC1 to the apical domain of MDCK cells or dendritic compartments of neurons [223]. More recently, the 502YVN504 motif of EAAC1 was identified as part of a tyrosine-based internalization signal that binds adapter proteins required for EAAC1 internalization [222]. This same group identified a consensus post-synaptic density-95/Discs large/Zonula occludens (PDZ) sequence of TSQF that is 16 amino acids downstream of this YVN motif. They showed that deletion of the PDZ domain increased the rate of constitutive endocytosis of EAAC1 and that mutation of the tyrosine residue in the YVN motif prevents clathrin-mediated endocytosis. They also showed EAAC1 interacts with Na+/H+-exchanger regulatory protein (NHERF3) through the PDZ domain and that expression of NHERF3 increases the amount of EAAC1 at the plasma membrane. NHERF3 is not detected in human or mouse brain (https://www.proteinatlas.org/ENSG00000174827-PDZK1/tissue; http://www.informatics.jax.org/expression.shtml). This prompts a relatively simple and testable hypothesis that the differential effects of PKC on EAAC1 trafficking in these two systems is related to the differential expression of NHERF3. It is also reasonable to hypothesize that PKC regulates the interaction of an unidentified protein with this PDZ domain to slow endocytosis as observed in C6 glioma [199].
In a more recent study, Su and colleagues used a database to identify proteins that contain the YNxxF[Y] motif because they had developed evidence that this motif was important for clathrin-dependent endocytosis that utilizes the clathrin-associated sorting protein, Numb [224]. They demonstrate a direct interaction between Numb and this motif. They also found that EAAC1 surface expression increased with knock-down of Numb. It is interesting to note that PKCα was identified as containing the YNxxF[Y] consensus sequence. Perhaps activation of PKCα slows EAAC1 endocytosis by preventing Numb binding to EAAC1, but this has not been tested. A very recent study identified SorSC2 as a protein that controls intracellular EAAC1 sorting and delivery to the cell surface [225]. Perhaps analyses of SorSC2 interactions with EAAC1 will lead to an understanding of how PDGF and PKC increase the rate of delivery of EAAC1 to the plasma membrane [199].
In one other study, the same YVN region has also been implicated in amphetamine-induced regulation of EAAC1 [226]. In this study, the group found that amphetamine decreases EAAC1 cell surface expression in dopaminergic neurons and in HEK293 cells. The effect in HEK cells was dependent upon co-expression of the dopamine transporter, suggesting that intracellular dopamine somehow triggers this response. They also showed the mutation of VN and the next three amino acids, GGF, eliminated this response. A cell permeable peptide corresponding to these five amino acids also blocks the effects of amphetamine. These same investigators have found that amphetamine can trigger internalization of EAAC1 in noradrenergic neurons as well [227].
In summary, several different studies have identified specific amino acids or domains within the carboxy terminus of EAAC1 that are required for constitutive and regulated trafficking, but there are several questions that remain. For example, are all of the effects of PKC on EAAC1 dependent upon phosphorylation of serine-465? How do PKC and PDGF/PI3K converge to increase delivery of EAAC1 to the plasma membrane? What are the signals that underlie the amphetamine-induced internalization of EAAC1? What receptors trigger these effects in neurons?
Although EAAC1 is far less abundant than either GLT-1 or GLAST [54], it has critical roles in brain function. As mentioned earlier, glutamate is one of the three amino acids incorporated into glutathione. Both EAAC1 and GLT-1 also transport cysteine, one of the other amino acids required for glutathione synthesis [228]. In fact, mice deleted of EAAC1 display an age-dependent neurodegenerative phenotype that is associated with decreased glutathione and increased oxidant levels [229]. These effects were attenuated using N-acetylcysteine, a membrane permeant form of cysteine that does not require a transporter. In several of the studies described above, the regulation of EAAC1 has been associated with changes in glutathione levels. For example, there is a tight linkage between the effects of GTRAP3-18 on EAAC1 and changes in glutathione levels [197, 213, 230]. Similarly, deletion of SorCS2 causes decreased cysteine uptake and increased neuronal sensitivity to oxidant insults [225]. Regulation of EAAC1 has also been linked to the control of excitatory signaling. For example, the amphetamine-induced regulation of EAAC1 is associated with increased activation of NMDA receptors [226, 231]. Together, these studies provide a strong rationale for developing a clearer understanding of the mechanisms that regulate EAAC1 with the distinct possibility that regulation could represent a novel target for neurogenerative disease.
Regulation of EAAT4 and EAAT5 by PKC
As indicated in the introduction, EAAT4 and EAAT5 are relatively slow transporters compared to the other members of the family, and they are coupled to a larger Cl− current than that observed with the other members of the family [59, 67, 232, 233]. We have found no studies examining the effect of PKC activation on EAAT5. The effects of acute activation of PKC on EAAT4 have been examined after expression in Xenopus oocytes [234]. This group found that activation of PKC increases the glutamate-activated Cl− current, but had no effect on glutamate uptake, suggesting that the Cl− conductance and glutamate uptake can be independently regulated. In a follow-up study, some of these same scientists showed that ethanol increases EAAT4-mediated currents, and this effect is blocked by inhibitors of PKC or phosphatidylinositol 3-kinase [235]. In this later study, they also showed that phorbol esters increase EAAT4-mediated currents. EAAT4 is enriched on perisynaptic processes of Purkinje cell neurons and overlaps with the metabotropic glutamate receptor, mGluR1, that couples to activation of phospholipase C and should activate PKC. Although there is strong evidence that EAAT4 regulates activation of mGluR1, there is no evidence that mGluR1 regulates EAAT4 in this system [236, 237]. To summarize, there is some evidence that the EAAT4-mediated Cl− current can be regulated by activation of PKC in oocytes, but it is not clear if this occurs in a native system. The mechanism by which PKC regulates this conductance has not been examined.
Glutamate Transport-Dependent Regulation of Glutamate Transport
Several studies have examined the effects of glutamate transporter substrates on glutamate transport activity; both increases and decreases have been observed. Duan et al. demonstrated that pre-incubation with glutamate transporter substrates increases Na+-dependent glutamate uptake in cortical astrocytes [238]. This effect is relatively rapid (occurs with 15 min), is caused by an increase in Vmax, is not blocked by PKC antagonists, is blocked by non-substrate inhibitors of transport, and is associated with an increase in cell surface expression of GLAST. They also demonstrated that the total intracellular glutamate accumulation increases using high performance liquid chromatography (HPLC), eliminating the possibility that this increase is due to exchange (see introduction). Munir and colleagues made a nearly identical set of observations using a variety of primary cultures [239]. They found that the rate of total accumulation of non-radioactive glutamate increases using HPLC, but they found no evidence of changes in cell surface expression of either GLAST or GLT-1, even though both contribute to the increase in uptake. The opposite effect on glutamate uptake has also been observed by more than one group. González and colleagues showed that glutamate transport substrates increase the Km for glutamate transport in cerebellar Bergman glial cultures [240]. This effect was blocked by a PKC inhibitor. Nakagawa and colleagues demonstrated that glutamate transport substrates induce clustering and internalization of GLT-1 [241]. These studies were conducted with a variant of GLT-1 with green fluorescent protein fused to the carboxy terminus. In this same paper, the authors mention that they observed a glutamate-dependent increase in uptake and GLAST surface expression as unpublished data. Ibánez et al. showed that glutamate transporter substrates also cause internalization and ubiquitination of GLT-1 in primary cultures and transfected HEK293 cells [242]. In a follow-up study, they showed that this effect is dependent on transport-dependent activation of reversed Na+/Ca2+ exchange [243]. Two different studies have shown that glutamate triggers clusters of GLT-1 to dissociate in the plasma membrane and increases surface diffusion of GLT-1 in astrocytes and brain slices [81, 244]. In the later study, they show that this effect is dependent on glutamate interaction with the transporter [244]. In the former study, they show that dis-aggregating the clusters of GLT-1 speeds the clearance of glutamate [81].
Together these studies strongly suggest that glutamate transport activity can bi-directionally control GLT-1- and GLAST-mediated activity. It seems clear that the mechanisms of these effects need to be understood. Without this information, it will be impossible to determine when or if this regulation occurs under physiologic or pathologic conditions.
Regulation of Glutamate Transport by Nitric Oxide/Nitrosylation
Nitric oxide (NO) is an important physiological regulator of biological functions in multiple tissues and was recognized as the molecule of the year in 1992 [245]. NO mediates several signals that control neurotransmission and cerebrovascular coupling in the CNS. NO is enzymatically synthesized by three different isoforms of nitric oxide synthase (NOS), including neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). nNOS and eNOS are the dominant isoforms in the CNS, but all isoforms have various roles in the brain [246,247,248,249,250,251,252,253]. The biological effects of NO are carried out by two principal mechanisms: activation of soluble guanylate cyclase or by direct protein nitrosylation. Both of these mechanisms have been implicated in the regulation of the glial glutamate transporters.
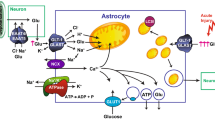
Using cerebellar Bergmann glia, Balderas and colleagues showed that the NO donor, sodium nitroprusside, or dbcGMP cause an acute increase in glutamate uptake [254]. This effect is associated with both an increase in Vmax and an increase in Km. They showed that an inhibitor of protein kinase G or an inhibitor of the reversed-operation of Na+/Ca2+-exchangers blocked the effect of sodium nitroprusside. They also used a binding assay to measure transporters on the plasma membrane in intact cells and observed an increase in the number of binding sites. As indicated above, this group has consistently found that these cells only express the GLAST subtype of glutamate transporter. These studies indicate that activation of the NO/guanylate cyclase pathway stimulates protein kinase G, that protein kinase G activates reversed operation of the Na+/Ca2+ exchanger causing an increase in intracellular Ca2+, and that the increase in Ca2+ is somehow linked to an increase in GLAST in the plasma membrane.
S-Nitrosylation is a reversible post-translational modification in which a NO group is covalently attached to cysteine thiols to form a S-nitrosothiol. At physiological concentrations of NO, S-nitrosylating enzymes (nitrosylases) and denitrosylating enzymes (denitrosylases) control the reaction. At higher concentrations the modification can occur independent of enzyme. In a hypothesis-generating exercise, proteomic analysis identified 136 brain proteins that undergo S-nitrosylation in wild-type (WT) mice [255]. Using transgenic mice that lack either eNOS−/− or nNOS−/−, they found that these two enzymes have different targets. In addition to several other proteins, they found that nNOS nitrosylates proteins that participate in the glutamate/glutamine cycle, including glutamine synthetase, glutamate dehydrogenase, and GLT-1. Glutamate uptake is higher in cortical synaptosomes from mice deleted of nNOS−/− compared to WT or eNOS−/− mice. GLT-1 has two sites of S-nitrosylation, Cys373 and Cys561. Using HEK-293T cells transfected with WT or mutant (C373S/C562S) GLT-1, they show that S-nitrosocysteine, a S-nitrosylating agent, causes nitrosylation of WT GLT-1 but not the double mutant. In parallel, they show that S-nitrosocysteine only reduces glutamate uptake mediated by WT GLT-1 and has no effect on the double mutant. S-Nitrosocysteine decreased both the Km and the Vmax. This effect of S-nitrosocysteine was independent of a change in cell surface GLT-1 measured using biotinylation. They also found that mice deleted of nNOS had higher levels of glutamate dehydrogenase activity. Together these studies suggest that activation of nNOS both decreases GLT-1 mediated activity and decreases glutamate oxidation.
Palmitoylation
Palmitoylation or s-acylation is a reversible post-translational modification that adds a palmitic acid to cysteine residues. This modification controls protein conformation, protein-protein interactions, and protein trafficking [256,257,258,259,260]. There are 23 different palmitoyltransferases (PATs), also referred to as Asp-His-His-Cys (DHHC) S-acyltransferases [261, 262]. Six different depalmitoylation enzymes have been identified, including acyl protein thioesterase 1 and 2 (APT1 and APT2) and α/β-hydrolase domain-containing proteins (ABH10, 17A, 17B and 17C) [263,264,265,266]. In a mass spectroscopic analysis of palymitoylated proteins, Kang and colleagues identified GLT-1, GLAST, and EAAC1, as targets of palmitoylation [267]. They used a secondary approach in which free cysteine residues are modified with N-ethylmaleimide, palymitoylate is cleaved from cysteine residues using hydroxylamine, and then these freshly reduced cysteine residues are modified with a biotin reagent that can be used for batch extraction and Western blot analysis of palmitoylated proteins. This approach, called acyl-biotin exchange (ABE), was used to determine if GLT-1 is palmitoylated. Although they demonstrated that GLT-1 is palmitoylated, the effect of palmitoylation of transport function was not examined.
In a subsequent study, Haung and colleagues used two different approaches (ABE and incorporation of radioactive palmitic acid) to confirm that GLT-1 is palmitylated [268]. They generated mutations at several different cysteine residues and identified Cys38 as the residue that is palmitoylated. They showed that 2-bromopalmitate (2BP), an inhibitor of palmitoylation, decreases glutamate uptake in COS-7 cells transfected with wild type GLT-1. This effect was associated with lower levels of palmitoylated GLT-1. They also showed that uptake mediated by Cys38Ser variant of GLT-1 was lower than that observed with wild type GLT-1. They observed no effect of palmitoylation on the cell surface levels of GLT-1 or on the levels of GLT-1 found in lipid rafts. These results suggest that palmitoylation regulates GLT-1-mediated activity. The mechanism has not been identified, and it is not known if palmitoylation affects GLT-1 in a native system.
Based on these analyses, we decided to determine if 2BP affects Na+-dependent glutamate uptake in crude rat cortical synaptosomes and if it affects the amount of palmitoylated GLT-1 in this same preparation. One would predict that the effects of 2BP should be time-dependent and proportional to the rate at which GLT-1 is constitutively depalmitoylated. We found the 2BP caused a nearly 40% decrease in uptake at 30 min, but it had a comparable effect essentially instantaneously (Fig. 4). This suggests that either the rate at which GLT-1 is palymitoylated and depalymitoylated in very fast (on the order of seconds to min) or 2BP has an off-target effect. To address these two possibilities, we measured the amount of palymitoylated GLT-1 using the ABE assay. We found that incubation for 30 min reduced the amount of palymitoylated GLT-1 by about 30%, but it had no effect on the amount of palmitoylated GLT-1 at 0 min (Fig. 5). Palmostatin B is a pan inhibitor of the depalmitoylation enzymes. As a complementary approach, we tested the effects of palmostatin B on the 2BP-induced inhibition of GLT-1. If the effects of 2BP are dependent on inhibition of a palmityltransferase, palmostatin B should block the effects of 2BP. We found that palmostatin B had no effect on the 2BP-induced inhibition of GLT-1 (Fig. 6). These studies provide additional evidence that GLT-1 is palmitoylated and that it is constitutively depalmitoylated. As indicated in the introduction, most of the GLT-1 protein found in synaptosomes is glial, but most of the uptake is mediated by neuronal pools of GLT-1. It is possible that glial pools of GLT-1 are regulated by palmitoylation and that neuronal pools of GLT-1 are inhibited by 2BP via a mechanism that is independent of palmitoylation.

Effects of 2 bromopalmitate (2BP) on Na+-dependent glutamate uptake. Crude synaptosomes (P2) were prepared from adult rat (male or female) cortical tissue as previously described [286]. After incubation with vehicle (0.05% ethanol in 0.32 M sucrose) or 2BP (50 µM) for the times indicated at 21 °C, Na+-dependent glutamate uptake (0.5 µM) was measured in duplicate replicates as previously described [286]. Each data point is an average of these technical replicates and represents an independent experiment. 2BP significantly reduced glutamate uptake at both 0 and 30 min, ****indicated p < 0.0001 compared to vehicle control, by a one-way ANOVA with Dunnett’s multiple comparisons test

Effect of 2BP on the amount of palmitoylated GLT-1. Crude synaptosomes were prepared and incubated with vehicle or 50 μm 2BP (see Fig. 4 legend). The amount of palymitoylated GLT-1 was determined as previously described [287]. In brief, synaptosomes were solubilized in RIPA buffer containing 50 mM N- ethylmaleimide (pH 7.5) for 1 h at 4 °C then incubated with anti-GLT-1 antibody [41] overnight at 4 °C. Protein agarose A beads were used to pull down anti-GLT-1 antibodies [288]. Samples were rinsed three times in RIPA buffer (pH 7.2), and split into two equal fractions. These two fractions are incubated for 1 h at 25 °C with hydroxylamine (1 M HAM) in RIPA buffer pH 7.2 or vehicle (RIPA, pH 7.2), and then they were rinsed three times with RIPA (pH 6.2). These fractions were then incubated with 2 μM EZ-Link™ BMCC-Biotin in RIPA (pH 6.2) for 1 h at 4 °C. These samples were then rinsed three times with RIPA (pH 7.5). The beads were then incubated with sample buffer (5% SDS, 5% glycerol, 125 mM Tris-HCl pH 6.8, 0.01% bromophenol blue) for 20 min at 37 °C. The resultant supernatants were incubated at 95 °C with 10 µL of β-mercaptoethanol (4%) for 5 min. The resultant supernatants were then resolved on polyacrylamide gels and probed with either streptavidin (palmitoylated proteins) or anti-GLT-1 antibody. Proteins were visualized LI-COR Odyssey Infrared Imaging system. The amount of palymitoylated GLT-1 was normalized to the amount of GLT-1 and expressed as a percent of that observed in vehicle treated synaptosomes. Data at 0 min are the results of two independent experiments. 2BP significantly reduces the amount of palymitoylated GLT-1 at 30 min. ****indicates a p < 0.0001 as determined by a one-sample T-test

Effect of 2BP and palmostatin B (PalmB), a general inhibitor of depalmitoylases, on Na+-dependent glutamate uptake. Rat cortical synaptosomes were prepared (see legend to Fig. 4) and incubated with vehicle, 2BP (50 µM), PalmB (10 µM), or a combination of 2BP with PalmB. PalmB had no effect on the 2-BP-induced decrease in glutamate uptake activity. ****indicates p < 0.0001 compared to vehicle by a one-way ANOVA with Dunnett’s multiple comparisons test
Summary/Future Directions
Given recent studies demonstrating that positive allosteric modulators of GLT-1 are neuroprotective [269,270,271], it seemed an auspicious time to review what is known about signals that regulate the activity of the transporters, to identify the gaps in knowledge, and discuss potential next steps for this field. While we focused on a subset of signals that have been linked to regulation of the glutamate transporters, calcium/calmodulin-dependent protein kinase II [272,273,274], protein kinase A [154, 156, 275], glycogen synthase kinase 3ß [276, 277], and arachidonic acid [278,279,280] have also been implicated in the regulation of one or more glutamate transporters by mechanisms that are likely independent of transcription/translation. Together these studies show that several different signals can relatively rapidly either increase or decrease the activity of these transporters. Throughout this review, we have identified several gaps, but there are others:
-
Non-native systems are useful to develop an understanding of mechanisms, but these studies will need to be extended to more native preparations. Astrocytes in culture are not polarized and are transcriptionally quite different from those in vivo [119]. Differences might be informative. For example, as discussed in the section on PKC-dependent regulation of EAAC1, it seems like a reasonable hypothesis that the differential expression of NHERF3 in MDCK cells and brain may contribute to differential regulation of EAAC1. Ultimately, it will be important to complement in vitro analyses with in vivo studies of glutamate clearance, changes in excitatory signaling, behavior, and pathology.
-
It is clear that Nedd4.2 has effects on GLT-1 and GLAST. Under what circumstances is it activated? Does this occur under physiologic situations? Or is this only activated under pathologic conditions? What other proteins are targeted? Is the fate of internalized transporter regulated between recycling, degradation, and exocytosis?
-
It is interesting that both PKC and glutamate transporter substrates have bidirectional effects on GLT-1 and GLAST, and these effects are observed in several different studies. Both GLT-1 and GLAST are found in cholesterol-enriched microdomains called lipid rafts and are more active in these domains [188, 281, 282]. Flotillin serves as a scaffolding protein on the interior membrane of lipid rafts [283], and may contribute to internalization of GLT-1 under at least some circumstances [186]. Perhaps the PKC- and transporter substrate-dependent increases in activity are due to a transient redistribution of transporters to lipid rafts followed by internalization. One could imagine regulating the fate of transporters with a control that might delay or accelerate endocytosis. This type of regulation could also be dependent on cellular milieu with the variable expression of the regulatory proteins contributing to the diversity of effects observed. One of the reasons we had started exploring the effects of palmitoylation on GLT-1 is that flotillin is also a target of palmitoylation [284]. We had wanted to explore the possible interactions between these two pathways. As the field continues to evolve, it seems likely that there will be better tools to explore these types of questions.
-
Piniella and colleagues recently fused biotin ligase to the amino terminus of GLT-1 to identify proteins that co-assemble with GLT-1 in HT22 cells [285]. They identified several interesting proteins that likely contribute to regulation of GLT-1. In particular, they found that septin 2 regulates lateral mobility of GLT-1. They also identified Rac1 as a GLT-1 interacting protein that regulates GLT-1- and GLAST-mediated activity. These types of studies should help clarify the mechanisms by which transporter substrates regulate activity.
-
Several of these signals are not likely specifically targeting glutamate transporters, they presumably target groups of proteins that are regulated in some sort of coordinated fashion. With current technologies that make it relatively easy to edit the genome (e.g. CRISPR gene editing), it would be really useful to know the specific amino acids in a particular transporter that are required for regulation. This would make it much easier to determine how the specific regulation of one or more the glutamate transporters contribute to the control of glutamate signaling, to behavior, and to pathophysiology.
-
It seems counter-intuitive that glutamate transporter substrates can decrease glutamate transport. Presumably this doesn’t happen on a continuous basis during normal synaptic transmission because it would seemingly result in exaggerated excitatory transmission and/or excitotoxic activation of glutamate receptors. Perhaps this effect contributes to synaptic plasticity or maybe it only occurs under pathologic conditions when a loss of energy/ion gradients would allow the transporters to work in the reverse direction. This would also imply that there is a yet to be defined mechanism that functions to prevent this loss of transporter from the cell surface.
Data Availability
Raw data will be made available if requested.
Abbreviations
- CNS:
-
Central nervous system
- cAMP:
-
Cyclic adenosine monophosphate
- dbcAMP:
-
Dibutyryl cyclic adenosine monophosphate
- EAAC1:
-
Excitatory amino acid carrier 1
- EAAT:
-
Excitatory amino acid transporter
- GABA:
-
Gamma aminobutyric acid
- GLUT-4:
-
Glucose transporter type 4
- GLAST:
-
Glutamate aspartate transporter
- GLT-1:
-
Glutamate transporter 1
- iGluRs:
-
Ionotropic glutamate receptors
- MDCK:
-
Madin-Darby Canine Kidney
- mGluRs:
-
Metabotropic glutamate receptors
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MPP+ :
-
1-Methyl-4-phenylpyridinium
- NHERF3:
-
Na+/H+-exchanger regulatory protein
- PPARα:
-
Peroxisome proliferator-activated receptor-alpha
- PDGF:
-
Platelet-derived growth factor
- PDZ:
-
Post-synaptic density-95/Discs large/Zonula occludens
- PKC:
-
Protein kinase C
- WT:
-
Wild type
References
Schousboe A (1981) Transport and metabolism of glutamate and GABA in neurons are glial cells. Int Rev Neurobiol 22:1–45
Fonnum F (1984) Glutamate: a neurotransmitter in mammalian brain. J Neurochem 42:1–11
McDonald JW, Johnston MV (1990) Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15:41–70
Robinson MB, Coyle JT (1987) Glutamate and related acidic excitatory neurotransmitters: from basic science to clinical application. FASEB J 1:446–455
Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130:1007S–1015S
McKenna MC (2013) Glutamate pays its own way in astrocytes. Front Endocrinol 4:191
Schousboe A (2017) A Tribute to Mary C. McKenna: glutamate as energy substrate and neurotransmitter-functional interaction between neurons and astrocytes. Neurochem Res 42:4–9
Dienel GA (2013) Astrocytic energetics during excitatory neurotransmission: what are contributions of glutamate oxidation and glycolysis? Neurochem Int 63:244–258
Robinson MB, Jackson JG (2016) Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem Int 98:56–71
Kreft M, Bak LK, Waagepetersen HS, Schousboe A (2012) Aspects of astrocyte energy metabolism, amino acid neurotransmitter homoeostasis and metabolic compartmentation. ASN Neuro 4
Dringen R (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol 62:649–671
Meister A (1983) Selective modification of glutathione metabolism. Science 220:472–477
Hollman M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17:31–108
Nakanishi S (1994) Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron 13:1031–1037
Nakanishi S (1992) Molecular diversity of glutamate receptors and implications for brain function. Science 258:597–603
Schoepp DD, Conn PJ (1993) Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci 14:13–20
Conn PJ, Patel J (1994) The metabotropic glutamate receptors. Humana Press, Totowa
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7–61
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496
Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322
Tang CM, Dichter M, Morad M (1989) Quisqualate activates a rapidly inactivating high conductance ionic channel in hippocampal neurons. Science 243:1474–1477
Raman IM, Trussell LO (1992) The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron 9:173–186
Kleppe IC, Robinson HP (1999) Determining the activation time course of synaptic AMPA receptors from openings of colocalized NMDA receptors. Biophys J 77:1418–1427
Scimemi A, Beato M (2009) Determining the neurotransmitter concentration profile at active synapses. Mol Neurobiol 40:289–306
Vance KM, Simorowski N, Traynelis SF, Furukawa H (2011) Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat Commun 2:294
Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ, Traynelis SF (2018) Structure, function, and allosteric modulation of NMDA receptors. J Gen Physiol 150:1081–1105
Reiner A, Levitz J (2018) Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 98:1080–1098
Choi DW (1992) Excitotoxic cell death. J Neurobiol 23:1261–1276
Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695
Faden AI, Demediuk P, Panter SS, Vink R (1989) The role of excitatatory amino acids and NMDA receptors in traumatic brain injury. Science 244:798–800
Greene JG, Greenamyre JT (1996) Bioenergetics and glutamate excitotoxicity. Prog Neurobiol 48:613–634
Lewerenz J, Maher P (2015) Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence? Front NeuroSci 9:469
O’Donovan SM, Sullivan CR, McCullumsmith RE (2017) The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. NPJ Schizophr 3:32
Uno Y, Coyle JT (2019) Glutamate hypothesis in schizophrenia. Psychiatry Clin Neurosci 73:204–215
Butcher SP, Hamberger A (1987) In vivo studies on the extracellular, and veratrine-releasable, pools of endogenous amino acids in the rat striatum: effects of corticostriatal deafferentation and kainic acid lesion. J Neurochem 48:713–721
Butcher SP, Sandberg M, Hagberg H, Hamberger A (1987) Cellular origins of endogenous amino acids released into the extracellular fluid of the rat striatum during severe insulin-induced hypoglycemia. J Neurochem 48:722–728
van der Zeyden M, Oldenziel WH, Rea K, Cremers TI, Westerink BH (2008) Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav 90:135–147
Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741
Moussawi K, Riegel A, Nair S, Kalivas PW (2011) Extracellular glutamate: functional compartments operate in different concentration ranges. Front Syst Neurosci 5:94
Herman MA, Nahir B, Jahr CE (2011) Distribution of extracellular glutamate in the neuropil of hippocampus. PloS one 6:e26501
Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13:713–725
Sims KD, Robinson MB (1999) Expression patterns and regulation of glutamate transporters in the developing and adult nervous system. Crit Rev Neurobiol 13:169–197
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Chaudhry FA, Lehre KP, Campagne MVL, Ottersen OP, Danbolt NC, Storm-Mathisen J (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720
Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC (1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15:1835–1853
Derouiche A, Rauen T (1995) Coincidence of L-glutamate/L-aspartate transporter (GLAST) and glutamine synthetase (GS) immunoreactions in retinal glia: evidence for coupling of GLAST and GS in transmitter clearance. J Neurosci Res 42:131–143
Pow DV, Barnett NL (1999) Changing patterns of spatial buffering of glutamate in developing rat retinae are mediated by the Muller cell glutamate transporter GLAST. Cell Tissue Res 297:57–66
Furuta A, Rothstein JD, Martin LJ (1997) Glutamate transporter protein subtypes are expressed differentially during rat central nervous system development. J Neurosci 17:8363–8375
Bar-Peled O, Ben-Hur H, Biegon A, Groner Y, Dewhurts S, Furata A, Rothstein JD (1997) Distribution of glutamate transporter subtypes during human brain development. J Neurochem 69:2571–2580
Conti F, DiBiasi S, Minelli A, Rothstein JD, Melone M (1998) EAAC1, a high-affinity glutamate transporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb Cortex 8:108–116
Kugler P, Schmitt A (1999) Glutamate transporter EAAC! is expressed in neurons and glial cells in the rat nervous system. Glia 27:129–142
Lane MC, Jackson JG, Krizman EN, Rothstein JD, Porter BE, Robinson MB (2014) Genetic deletion of the neuronal glutamate transporter, EAAC1, results in decreased neuronal death after pilocarpine-induced status epilepticus. Neurochem Int 73:152–158
Holmseth S, Dehnes Y, Bjornsen LP, Boulland JL, Furness DN, Bergles D, Danbolt NC (2005) Specificity of antibodies: unexpected cross-reactivity of antibodies directed against the excitatory amino acid transporter 3 (EAAT3). Neuroscience 136:649–660
Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE, Lehre KP, Danbolt NC (2012) The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci 32:6000–6013
Yamada K, Watanabe M, Shibata T, Tanaka K, Wada K, Inoue Y (1996) EAAT4 is a post-synaptic glutamate transporters in Purkinje cell synapses. Neuroreport 7:2013–2017
Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in the parts of the dendritic membrane facing astroglia. J Neurosci 18:3606–3610
Lin C-LG, Tzingounis AV, Jin L, Furuta A, Kavanaugh MP, Rothstein JD (1998) Molecular cloning and expression of the rat EAAT4 glutamate transporter subtype. Mol Brain Res 63:174–179
Dalet A, Bonsacquet J, Gaboyard-Niay S, Calin-Jageman I, Chidavaenzi RL, Venteo S, Desmadryl G, Goldberg JM, Lysakowski A, Chabbert C (2012) Glutamate transporters EAAT4 and EAAT5 are expressed in vestibular hair cells and calyx endings. PLoS ONE 7:e46261
Arriza JL, Eliasof S, Kavanaugh MP, Amara SG (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA 94:4155–4160
Eliasof S, Arriza JL, Leighton BH, Kavanaugh MP, Amara SG (1998) Excitatory amino acid transporters of the salamander retina: identification, localization, and function. J Neurosci 15:698–712
Pow DV, Barnett NL (2000) Developmental expression of excitatory amino acid transporter 5: a photoreceptor and bipolar cell glutamate transporter in rat retina. Neurosci Lett 280:21–24
Lee A, Balcar VJ, McCombe P, Pow DV (2020) Human brain neurons express a novel splice variant of excitatory amino acid transporter 5 (hEAAT5v). J Comp Neurol 528:3134–3142
Wersinger E, Schwab Y, Sahel JA, Rendon A, Pow DV, Picaud S, Roux MJ (2006) The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J Physiol 577:221–234
Zerangue N, Kavanaugh MP (1996) Flux coupling in a neuronal glutamate transporter. Nature 383:634–637
Levy LM, Warr O, Attwell D (1998) Stoichiometry of the glial glutamte transporter GLT-1 expressed inducibly in a chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J Neurosci 18:9620–9628
Owe SG, Marcaggi P, Attwell D (2006) The ionic stoichiometry of the GLAST glutamate transporter in salamander retinal glia. J Physiol 577:591–599
Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino acid transporter with properties of a ligand-gated chloride channel. Nature 375:599–603
Seal RP, Amara SG (1999) Excitatory amino acid transporters: a family in flux. Annu Rev Pharmacol Toxicol 39:431–456
Ryan RM, Vandenberg RJ (2005) A channel in a transporter. Clin Exp Pharmacol Physiol 32:1–6
Vandenberg RJ, Ryan RM (2013) Mechanisms of glutamate transport. Physiol Rev 93:1621–1657
Grewer C, Rauen T (2005) Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol 203:1–20
Tzingounis AV, Wadiche JI (2007) Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 8:935–947
Bergles DE, Jahr CE (1997) Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19:1297–1308
Gameiro A, Braams S, Rauen T, Grewer C (2011) The discovery of slowness: low-capacity transport and slow anion channel gating by the glutamate transporter EAAT5. Biophys J 100:2623–2632
Grewer C, Watzke N, Wiessner M, Rauen T (2000) Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci U S A 97:9706–9711
Wadiche JI, Kavanaugh MP (1998) Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci 18:7650–7661
Rosenberry TL (1975) Acetylcholinesterase. Adv Enzymol Relat Areas Mol Biol 43:103–218
Antosiewicz J, Gilson MK, Lee IH, McCammon JA (1995) Acetylcholinesterase: diffusional encounter rate constants for dumbbell models of ligand. Biophys J 68:62–68
Lehre KP, Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757
Tong G, Jahr CE (1994) Block of glutamate transporters potentiates postsynaptic excitation. Neuron 13:1195–1203
Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L, Oliet SH (2015) Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci 18:219–226
Conti F, Weinberg RJ (1999) Shaping excitation at glutamatergic synapses. Trends Neurosci 22:451–458
Rose CR, Felix L, Zeug A, Dietrich D, Reiner A, Henneberger C (2017) Astroglial glutamate signaling and uptake in the hippocampus. Front Mol Neurosci 10:451
Valtcheva S, Venance L (2019) Control of long-term plasticity by glutamate transporters. Front Synaptic Neurosci 11:10
Rossi DJ, Oshima T, Attwell D (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403:316–321
Kawahara K, Kosugi T, Tanaka M, Nakajima T, Yamada T (2005) Reversed operation of glutamate transporter GLT-1 is crucial to the development of preconditioning-induced ischemic tolerance of neurons in neuron/astrocyte co-cultures. Glia 49:349–359
Camacho A, Massieu L (2006) Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res 37:11–18
Zhang LN, Hao L, Guo YS, Wang HY, Li LL, Liu LZ, Li WB (2019) Are glutamate transporters neuroprotective or neurodegenerative during cerebral ischemia? J Mol Med (Berl) 97:281–289
Gegelashvili G, Schousboe A (1997) High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol 52:6–15
Robinson MB, Dowd LA (1997) Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Adv Pharmacol 37:69–115
Trotti D, Danbolt NC, Volterra A (1998) Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration. Trends Pharmacol Sci 19:328–334
Robinson MB (1999) The family of sodium-dependent glutamate transporters: A focus on the GLT-1/EAAT2 subtype. Neurochem Int 33:479–491
Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32:1–14
Gegelashgvili G, Robinson MB, Trotti D, Rauen T (2001) Regulation of glutamate transporters in health and disease. Prog Brain Res 132:267–286
Shigeri Y, Seal RP, Shimamoto K (2004) Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Rev 45:250–265
Dunlop J (2006) Glutamate-based therapeutic approaches: targeting the glutamate transport system. Curr Opin Pharmacol 6:103–107
Kanner BI (2006) Structure and function of sodium-coupled GABA and glutamate transporters. J Membr Biol 213:89–100
Sattler R, Rothstein JD (2006) Regulation and dysregulation of glutamate transporters. Handb Exp Pharmacol:277–303
Yi JH, Hazell AS (2006) Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int 48:394–403
Beart PM, O’Shea RD (2007) Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150:5–17
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51:333–355
Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, Reed JC, Stebbins JL, Pellecchia M, Sarkar D, Fisher PB (2011) Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. Journal of cellular physiology 226:2484–2493
Bridges R, Lutgen V, Lobner D, Baker DA (2012) Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev 64:780–802
Bridges RJ, Natale NR, Patel SA (2012) System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol 165:20–34
Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P (2013) The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18:522–555
Martinez-Lozada Z, Guillem AM, Robinson MB (2016) Transcriptional regulation of glutamate transporters: from extracellular signals to transcription factors. Adv Pharmacol 76:103–145
Murphy-Royal C, Dupuis J, Groc L, Oliet SHR (2017) Astroglial glutamate transporters in the brain: Regulating neurotransmitter homeostasis and synaptic transmission. J Neurosci Res 95:2140–2151
Magi S, Piccirillo S, Amoroso S, Lariccia V (2019) Excitatory amino acid transporters (EAATs): glutamate transport and beyond. Int J Mol Sci 20
Robinson MB, Lee ML, DaSilva S (2020) Glutamate transporters and mitochondria: signaling, co-compartmentalization, functional coupling, and future directions. Neurochem Res 45:526–540
Peterson AR, Binder DK (2019) Post-translational regulation of GLT-1 in neurological diseases and its potential as an effective therapeutic target. Front Mol Neurosci 12:164
Gegelashvili G, Danbolt NC, Schousboe A (1997) Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem 69:2612–2615
Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC (1997) Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci 17:932–940
Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB (1998) Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol 53:355–369
Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD (2009) Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron 61:880–894
Lee ML, Martinez-Lozada Z, Krizman EN, Robinson MB (2017) Brain endothelial cells induce astrocytic expression of the glutamate transporter GLT-1 by a Notch-dependent mechanism. J Neurochem 143:489–506
Martinez-Lozada Z, Robinson MB (2020) Reciprocal communication between astrocytes and endothelial cells is required for astrocytic glutamate transporter 1 (GLT-1) expression. Neurochem Int 139:104787
Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14:5559–5569
Benediktsson AM, Schachtele SJ, Green SH, Dailey ME (2005) Ballistic labeling and dynamic imaging of astrocytes in organotypic hippocampal slice cultures. J Neurosci Methods 141:41–53
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947
Henn FA, Anderson DJ, Rustad DG (1976) Glial contamination of synaptosomal fractions. Brain Res 101:341–344
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702
Divac I, Fonnum F, Storm-Mathisen J (1977) High affinity uptake of glutamate in terminals of corticostriatal axons. Nature 266:377–378
Storm-Mathisen J (1977) Glutamic acid and excitatory nerve endings: reduction of glutamaic acid uptake after axotomy. Brain Res 120:379–386
Taxt T, Storm-Mathisen J (1984) Uptake of D-aspartate and L-glutamate in excitatory axon terminals in hippocampus: autoradiographic and biochemical comparisons with gamma-aminobutyrate and other amino acids in normal rats and rats with lesions. Neuroscience 11:79–100
Storm-Mathisen J (1977) Localization of transmitter candidates in the brain: the hippocampal formation as a model. Prog Neurobiol 8:119–181
Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, Wojewodzic M, Zhou Y, Attwell D, Danbolt NC (2008) A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 157:80–94
Torp R, Danbolt NC, Babaie E, Bjoras M, Seeberg E, Storm-Mathisen J, Ottersen OP (1994) Differential expression of two glial glutamate transporters in the rat brain: An in situ hybridization study. Eur J Neurosci 6:936–942
Brooks-Kayal AR, Munir M, Jin H, Robinson MB (1998) The glutamate transporter, GLT-1, is expressed in cultured hippocampal neurons. Neurochem Int 33:95–100
Mennerick S, Dhond RP, Benz A, Xu W, Rothstein JD, Danbolt NC, Isenberg KE, Zorumski CF (1998) Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J Neurosci 18:4490–4499
Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA (2002) Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 22:2142–2152
Berger UV, DeSilva TM, Chen W, Rosenberg PA (2005) Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol 492:78–89
Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA (2015) Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 35:5187–5201
Zhou Y, Hassel B, Eid T, Danbolt NC (2019) Axon-terminals expressing EAAT2 (GLT-1; Slc1a2) are common in the forebrain and not limited to the hippocampus. Neurochem Int 123:101–113
Danbolt NC, Furness DN, Zhou Y (2016) Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem Int 98:29–45
Rimmele TS, Rosenberg PA (2016) GLT-1: The elusive presynaptic glutamate transporter. Neurochem Int 98:19–28
Dunlop J, Lou Z, Zhang Y, McIlvain HB (1999) Inducible expression and pharmacology of the human excitatory amino acid transporter 2 subtype of L-glutamate transporter. Br J Pharmacol 128:1485–1490
Tan J, Zelenaia O, Rothstein JD, Robinson MB (1999) Expression of the GLT-1 subtype of Na+-dependent glutamate transporter: Pharmacological characterization and lack of regulation by protein kinase C. J Pharmacol Exp Ther 289:1600–1610
Fontana ACK (2018) Protocols for Measuring Glutamate Uptake: Dose-Response and Kinetic Assays in In Vitro and Ex Vivo Systems. Curr Protoc Pharmacol 82:e45
Loder MK, Melikian HE (2003) The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J Biol Chem 278:22168–22174
Jabaudon D, Scanziani M, Gähwiler BH, Gerber U (2000) Acute decrease in net glutamate upatke during energy failure. Proc Natl Acad Sci USA 97:5610–5615
Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD (2007) Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27:11354–11365
Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ (2007) Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55:274–281
Zhou Y, Wang X, Tzingounis AV, Danbolt NC, Larsson HP (2014) EAAT2 (GLT-1; slc1a2) glutamate transporters reconstituted in liposomes argues against heteroexchange being substantially faster than net uptake. J Neurosci 34:13472–13485
Newton AC (2018) Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol 53:208–230
Brose N, Rosenmund C (2002) Move over protein kinase C, you’ve got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci 115:4399–4411
González MI, Ortega A (1997) Regulation of the Na+-dependent high affinity glutamate/aspartate transporter in cultured Bergmann glia by phorbol esters. J Neurosci Res 50:585–590
Bernabe A, Mendez JA, Hernandez-Kelly LC, Ortega A (2003) Regulation of the Na+-dependent glutamate/aspartate transporter in rodent cerebellar astrocytes. Neurochem Res 28:1843–1849
González MI, AM AML-C, Ortega A (1999) Sodium-dependent glutamate transport in Muller glial cells: regulation by phorbol esters. Brain Res 831:140–145
Wang Z, Li W, Mitchell CK, Carter-Dawson L (2003) Activation of protein kinase C reduces GLAST in the plasma membreane of rat Muller cells in primary culture. Vis Neurosci 20:611–619
Casado M, Zafra F, Aragón C, Giménez C (1991) Activation of high-affinity uptake of glutamate by phorbol esters in primary glial cell cultures. J Neurochem 57:1185–1190
Susarla BS, Seal RP, Zelenaia O, Watson DJ, Wolfe JH, Amara SG, Robinson MB (2004) Differential regulation of GLAST immunoreactivity and activity by protein kinase C: evidence for modification of amino and carboxy termini. J Neurochem 91:1151–1163
Sidoryk-Wegrzynowicz M, Lee E, Aschner M (2012) Mechanism of Mn(II)-mediated dysregulation of glutamine-glutamate cycle: focus on glutamate turnover. J Neurochem 122:856–867
Gosselin RD, Meylan P, Decosterd I (2013) Extracellular microvesicles from astrocytes contain functional glutamate transporters: regulation by protein kinase C and cell activation. Front Cell Neurosci 7:251
Adolph O, Koster S, Rath M, Georgieff M, Weigt HU, Engele J, Senftleben U, Fohr KJ (2007) Rapid increase of glial glutamate uptake via blockade of the protein kinase A pathway. Glia 55:1699–1707
Leonova J, Thorlin T, Eriksson NDA, Ronnback PS, Hansson L E (2001) Endothelin-1 decreases glutamate uptake in primary cultured rat astrocytes. American J Physiol Cell Physiol 281:C1495–C1503
Guillet BA, Velly LJ, Canolle B, Nieoullon FMM, Pisano AL P (2005) Differential regulation by protein kinases of activity and cell surface expression of glutamate transporters in neuron-enriched cultures. Neurochem Int 46:337–346
Conradt M, Stoffel W (1997) Inhibition of the high-affinity brain glutamate transporter GLAST-1 via direct phosphorylation. J Neurochem 68:1244–1251
Yan X, Yadav R, Gao M, Weng HR (2014) Interleukin-1 beta enhances endocytosis of glial glutamate transporters in the spinal dorsal horn through activating protein kinase C. Glia 62:1093–1109
Zhang Y, He X, Meng X, Wu X, Tong H, Zhang X, Qu S (2017) Regulation of glutamate transporter trafficking by Nedd4-2 in a Parkinson’s disease model. Cell Death Dis 8:e2574
Ma S, Zheng X, Zheng T, Huang F, Jiang J, Luo H, Guo Q, Hu B (2019) Amitriptyline influences the mechanical withdrawal threshold in bone cancer pain rats by regulating glutamate transporter GLAST. Mol Pain 15:1744806919855834
Garcia-Tardon N, Gonzalez-Gonzalez IM, Martinez-Villarreal J, Fernandez-Sanchez E, Gimenez C, Zafra F (2012) Protein kinase C (PKC)-promoted endocytosis of glutamate transporter GLT-1 requires ubiquitin ligase Nedd4-2-dependent ubiquitination but not phosphorylation. J Biol Chem 287:19177–19187
Boehmer C, Henke G, Schniepp R, Palmada M, Rothsetin JD, Broer S, Lang F (2003) Regulation of the glutamate transporter EAAT1 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid-inducible kinase isoforms SGK1/3 and protein kinase B. J Neurochem 86:1181–1188
Sun XL, Zeng XN, Zhou F, Dai CP, Ding JH, Hu G (2008) KATP channel openers facilitate glutamate uptake by GluTs in rat primary cultured astrocytes. Neuropsychopharmacology 33:1336–1342
Vermeiren C, Najimi M, Vanhoutte N, Tilleux S, Hemptinne Id, Maloteauz J-M, Hermans E (2005) Acute up-regulation of glutamate uptake mediated by mGluR5a in reactive astrocytes. J Neurochem 94:405–416
Vermeiren C, Hemptinne I, Vanhoutte N, Tilleux S, Maloteaux JM, Hermans E (2006) Loss of metabotropic glutamate receptor-mediated regulation of glutamate transport in chemically activated astrocytes in a rat model of amyotrophic lateral sclerosis. J Neurochem 96:719–731
Vergouts M, Doyen PJ, Peeters M, Opsomer R, Hermans E (2018) Constitutive downregulation protein kinase C epsilon in hSOD1(G93A) astrocytes influences mGluR5 signaling and the regulation of glutamate uptake. Glia 66:749–761
Pisano P, Samuel D, Nieoullon A, Goff LK-L (1996) Activation of the adenylate cyclase-dependent protein kinase pathway increases high affinity glutamate uptake into rat striatal synaptosomes. Neuropharmacology 35:541–547
Lu YM, Lu BF, Zhao FQ, Yan YL, Ho XP (1993) Accumulation of glutamate is regulated by calcium and protein kinase C in rat hippocampal slices exposed to ischemic states. Hippocampus 3:221–227
Daniels KK, Vickroy TW (1999) Reversible activation of glutamate transport in rat brain glia by protein kinase C and an okadaic acid-sensitive phosphoprotein phosphatase. Neurochem Res 24:1017–1025
Hoogland G, Bos IW, Kupper F, van Willigen G, Spierenburg HA, van Nieuwenhuizen O, de Graan PN (2005) Thrombin-stimulated glutamate uptake in human platelets is predominantly mediated by the glial glutamate transporter EAAT2. Neurochem Int 47:499–506
Chen T, Tanaka M, Wang Y, Sha S, Furuya K, Chen L, Sokabe M (2017) Neurosteroid dehydroepiandrosterone enhances activity and trafficking of astrocytic GLT-1 via sigma1 receptor-mediated PKC activation in the hippocampal dentate gyrus of rats. Glia 65:1491–1503
Ganel R, Crosson CE (1998) Modulation of human glutamate transporter activity by phorbol ester. J Neurochem 70:993–1000
Kalandadze A, Wu Y, Robinson MB (2002) Protein kinase C activation decreases cell surface expression of the GLT-1 subtype of glutamate transporter. Requirement of a carboxyl-terminal domain and partial dependence on serine 486. J Biol Chem 277:45741–45750
Fang H, Huang Y, Zuo Z (2002) The different responses of rat glutamate transporter type 2 and its mutant (tyrosine 403 to histidine) activity to volatile anesthetics and activation of protein kinase C. Brain Res 953:255–264
Najimi M, Stephenne X, Sempoux C, Sokal E (2014) Regulation of hepatic EAAT-2 glutamate transporter expression in human liver cholestasis. World J Gastroenterol 20:1554–1564
Wang Y, Lu S, Qu Z, Wu L, Wang Y (2017) Sonic hedgehog induces GLT-1 degradation via PKC delta to suppress its transporter activities. Neuroscience 365:217–225
Zhou J, Sutherland ML (2004) Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J Neurosci 24:6301–6306
González MI, Susarla BTS, Robinson MB (2005) Evidence that protein kinase Ca interacts with an regulates the glial glutamate transporter GLT-1. J Neurochem 94:1180–1188
Gonzalez-Gonzalez IM, Garcia-Tardon N, Gimenez C, Zafra F (2008) PKC-dependent endocytosis of the GLT1 glutamate transporter depends on ubiquitylation of lysines located in a C-terminal cluster. Glia 56:963–974
Sheldon AL, Gonzalez MI, Krizman-Genda EN, Susarla BT, Robinson MB (2008) Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem Int 53:296–308
Boehmer C, Palmanda M, Rajamanickam J, Schneipp R, Amara S, Lang F (2006) Post-translational regulation of EAAT2 function by co-expressed ubiquitin ligase Nedd4-2 is impacted by SGK kinases. J Neurochem 97:911–921
Martinez-Villarreal J, Garcia Tardon N, Ibanez I, Gimenez C, Zafra F (2012) Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia 60:1356–1365
Perez-Jimenez E, Viana R, Munoz-Ballester C, Vendrell-Tornero C, Moll-Diaz R, Garcia-Gimeno MA, Sanz P (2020) Endocytosis of the glutamate transporter 1 is regulated by laforin and malin: Implications in Lafora disease. Glia
Susarla BT, Robinson MB (2008) Internalization and degradation of the glutamate transporter GLT-1 in response to phorbol ester. Neurochem Int 52:709–722
Guo S, Zhang X, Zheng M, Zhang X, Min C, Wang Z, Cheon SH, Oak MH, Nah SY, Kim KM (2015) Selectivity of commonly used inhibitors of clathrin-mediated and caveolae-dependent endocytosis of G protein-coupled receptors. Biochim Biophys Acta 1848:2101–2110
Cremona ML, Matthies HJ, Pau K, Bowton E, Speed N, Lute BJ, Anderson M, Sen N, Robertson SD, Vaughan RA, Rothman JE, Galli A, Javitch JA, Yamamoto A (2011) Flotillin-1 is essential for PKC-triggered endocytosis and membrane microdomain localization of DAT. Nat Neurosci 14:469–477
Sorkina T, Caltagarone J, Sorkin A (2013) Flotillins regulate membrane mobility of the dopamine transporter but are not required for its protein kinase C dependent endocytosis. Traffic 14:709–724
Butchbach ME, Tian G, Guo H, Lin CL (2004) Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for EAAT localization and function. J Biol Chem
Huang HT, Liao CK, Chiu WT, Tzeng SF (2017) Ligands of peroxisome proliferator-activated receptor-alpha promote glutamate transporter-1 endocytosis in astrocytes. Int J Biochem Cell Biol 86:42–53
Casado M, Bendahan A, Zafra F, Danbolt NC, Gimenez C, Kanner BI (1993) Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem 268:27313–27317
Dowd LA, Coyle AJ, Rothstein JD, Pritchett DB, Robinson MB (1996) Comparison of Na+-dependent glutamate transport activity in synaptosomes, C6 glioma, and Xenopus Oocytes expressing excitatory amino acid carrier 1 (EAAC1). Mol Pharmacol 49:465–473
Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB (1998) Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci 18:2475–2485
Dowd LA, Robinson MB (1996) Rapid stimulation of EAAC1-mediated Na+-dependent L-glutamate transport activity in C6 glioma by phorbol ester. J Neurochem 67:508–516
Bianchi MG, Rotoli BM, Dall’Asta V, Gazzola GC, Gatti R, Bussolati O (2006) PKC-dependent stimulation of EAAT3 glutamate transporter does not require the integrity of actin cytoskeleton. Neurochem Int 48:341–349
Sheldon AL, Gonzalez MI, Robinson MB (2006) A carboxyl-terminal determinant of the neuronal glutamate transporter, EAAC1, is required for platelet-derived growth factor-dependent trafficking. J Biol Chem 281:4876–4886
Murphy A, Vines A, McBean GJ (2009) Stimulation of EAAC1 in C6 glioma cells by store-operated calcium influx. Biochim Biophys Acta 1788:551–558
Watabe M, Aoyama K, Nakaki T (2007) Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3–18 (GTRAP3-18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol Pharmacol 72:1103–1110
Foster DJ, Heacock AM, Fisher SK (2010) Muscarinic receptor stimulation of D-aspartate uptake into human SH-SY5Y neuroblastoma cells is attenuated by hypoosmolarity. J Pharmacol Exp Ther 333:297–309
Fournier KM, González MI, Robinson MB (2004) Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J Biol Chem 279:34505–34513
Huang Y, Zuo Z (2005) Isoflurane induces a protein kinase Ca-dependent increase in cell surface protein level and activity of glutamate transporter type 3. Mol Pharmacol 67:1522–1533
Sims KD, Straff DJ, Robinson MB (2000) Platelet-derived growth factor rapidly increases activity and cell surface expression of the EAAC1 subtype of glutamate transporters through activation of phosphatidylinositol 3-kinase. J Biol Chem 274:5228–5327
Najimi M, Maloteaux JM, Hermans E (2002) Cytoskeleton-related trafficking of the EAAC1 glutamate transporter after activation of the G(q/11)-coupled neurotensin receptor NTS1. FEBS Lett 523:224–228
Najimi M, Maloteaux J-M, Hermans E (2005) Pertussis toxin-sensitive modulation of glutamate transport by endothelin-1 type A receptors in glioma cells. Biochem Biophys Acta 1668:195–202
Yu YX, Shen L, Xia P, Tang YW, Bao L, Pei G (2006) Syntaxin 1A promotes the endocytic sorting of EAAC1 leading to inhibition of glutamate transport. J Cell Sci 119:3776–3787
Shepherd JD, Huganir RL (2007) The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol 23:613–643
Evans AJ, Gurung S, Henley JM, Nakamura Y, Wilkinson KA (2019) Exciting times: new advances towards understanding the regulation and roles of kainate receptors. Neurochem Res 44:572–584
Bryant NJ, Govers R, James DE (2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3:267–277
Watson RT, Kanzaki M, Pessin JE (2004) Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev 25:177–204
Dugani CB, Klip A (2005) Glucose transporter 4: cycling, compartments and controversies. EMBO Rep 6:1137–1142
González MI, Kazanietz MG, Robinson MB (2002) Regulation of the neuronal glutamate transporter excitatory amino acid carrier-1 (EAAC1) by different protein kinase C subtypes. Mol Pharmacol 62:901–910
Lin C-LG, Orlov I, Ruggiero AM, Dykes-Hoberg M, Lee A, Jackson M, Rothstein JD (2001) Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3-18. Nature 410:84–88
Butchbach ME, Lai L, Lin CL (2002) Molecular cloning, gene structure, expression profile and functional characterization of the mouse glutamate transporter (EAAT3) interacting protein GTRAP3-18. Gene 292:81–90
Watabe M, Aoyama K, Nakaki T (2008) A dominant role of GTRAP3-18 in neuronal glutathione synthesis. J Neurosci 28:9404–9413
Ikemoto MJ, Inoue K, Akiduki S, Osugi T, Imamura T, Ishida N, Ohtomi M (2002) Identification of addicsin/GTRAP3-18 as a chronic morphine-augmented gene in amygdala. Neuroreport 13:2079–2084
Akiduki S, Ikemoto MJ (2008) Modulation of the neural glutamate transporter EAAC1 by the addicsin-interacting protein ARL6IP1. J Biol Chem 283:31323–31332
González MI, Bannerman PG, Robinson MB (2003) Phorbol myristate acetate-dependent interaction of protein kinase Ca and the neuronal glutamate transporter EAAC1. J Neurosci 23:5589–5593
Huang Y, Feng X, Sando JJ, Zuo Z (2006) Critical role of serine 465 in isoflurane-induced increase of cell-surface redistribution and activity of glutamate transporter type 3. J Biol Chem 281:38133–38138
Baik HJ, Huang Y, Washington JM, Zuo Z (2009) Critical role of s465 in protein kinase C-increased rat glutamate transporter type 3 activity. Int J Neurosci 119:1419–1428
Huang Y, Li L, Washington JM, Xu X, Sando JJ, Lin D, Zuo Z (2011) Inhibition of isoflurane-induced increase of cell-surface redistribution and activity of glutamate transporter type 3 by serine 465 sequence-specific peptides. Eur J Pharmacol 655:16–22
Trotti D, Peng J-B, Dunlop J, Hediger MA (2001) Inhibition of the glutamate transporter EAAC1 expressed in Xenopus oocytes by phorbol esters. Brain Res 914:196–203
Padovano V, Massari S, Mazzucchelli S, Pietrini G (2009) PKC induces internalization and retention of the EAAC1 glutamate transporter in recycling endosomes of MDCK cells. Am J Physiol Cell Physiol 297:C835–C844
D’Amico A, Soragna A, Di Cairano E, Panzeri N, Anzai N, Vellea Sacchi F, Perego C (2010) The surface density of the glutamate transporter EAAC1 is controlled by interactions with PDZK1 and AP2 adaptor complexes. Traffic 11:1455–1470
Cheng C, Glover G, Banker G, Amara SG (2002) A novel sorting motif in the glutamate transporter excitatory amino acid transporter 3 directs its targeting in Madin-Darby canine kidney cells and hippocampal neurons. J Neurosci 22:10643–10652
Su JF, Wei J, Li PS, Miao HH, Ma YC, Qu YX, Xu J, Qin J, Li BL, Song BL, Xu ZP, Luo J (2016) Numb directs the subcellular localization of EAAT3 through binding the YxNxxF motif. J Cell Sci 129:3104–3114
Malik AR, Szydlowska K, Nizinska K, Asaro A, van Vliet EA, Popp O, Dittmar G, Fritsche-Guenther R, Kirwan JA, Nykjaer A, Lukasiuk K, Aronica E, Willnow TE (2019) SorCS2 controls functional expression of amino acid transporter EAAT3 and protects neurons from oxidative stress and epilepsy-induced pathology. Cell Rep 26:2792–2804 (e2796)
Underhill SM, Wheeler DS, Li M, Watts SD, Ingram SL, Amara SG (2014) Amphetamine modulates excitatory neurotransmission through endocytosis of the glutamate transporter EAAT3 in dopamine neurons. Neuron 83:404–416
Underhill SM, Colt MS, Amara SG (2020) Amphetamine Stimulates Endocytosis of the Norepinephrine and Neuronal Glutamate Transporters in Cultured Locus Coeruleus Neurons. Neurochem Res 45:1410–1419
Chen Y, Swanson RA (2003) The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem 84:1332–1339
Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA (2006) Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci 9:119–126
Aoyama K, Watabe M, Nakaki T (2012) Modulation of neuronal glutathione synthesis by EAAC1 and its interacting protein GTRAP3-18. Amino Acids 42:163–169
Li MH, Underhill SM, Reed C, Phillips TJ, Amara SG, Ingram SL (2017) Amphetamine and Methamphetamine Increase NMDAR-GluN2B Synaptic Currents in Midbrain Dopamine Neurons. Neuropsychopharmacology 42:1539–1547
Palmer MJ, Taschenberger H, Hull C, Tremere L, von Gersdorff H (2003) Synaptic activation of presynaptic glutamate transporter currents in nerve terminals. J Neurosci 23:4831–4841
Mim C, Balani P, Rauen T, Grewer C (2005) The glutamate transporter subtypes EAAT4 and EAATs 1–3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J Gen Physiol 126:571–589
Fang H, Huang Y, Zuo Z (2006) Enhancement of substrate-gated Cl- currents via rat glutamate transporter EAAT4 by PMA. Am J Physiol Cell Physiol 290:C1334–C1340
Park HY, Kim JH, Zuo Z, Do SH (2008) Ethanol increases the activity of rat excitatory amino acid transporter type 4 expressed in Xenopus oocytes: role of protein kinase C and phosphatidylinositol 3-kinase. Alcohol Clin Exp Res 32:348–354
Brasnjo G, Otis TS (2001) Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron 31:607–616
Otis TS, Brasnjo G, Dzubay JA, Pratap M (2004) Interactions between glutamate transporters and metabotropic glutamate receptors at excitatory synapses in the cerebellar cortex. Neurochem Int 45:537–544
Duan S, Anderson CM, Stein BA, Swanson RA (1999) Glutamate induces rapid upregulation of glutamate transport and cell-surface expression of GLAST. J Neurosci 19:10193–10200
Munir M, Correale DM, Robinson MB (2000) Substrate-induced up-regulation of Na+-dependent glutamate transport activity. Neurochem Int 37:147–162
González MI, Ortega A (2000) Regulation of high-affinity glutamate uptake activity in Bergmann glia cells by glutamate. Brain Res 866:73–81
Nakagawa T, Otsubo Y, Yatani Y, Shirakawa H, Kaneko S (2008) Mechanisms of substrate transport-induced clustering of a glial glutamate transporter GLT-1 in astroglial-neuronal cultures. Eur J Neurosci 28:1719–1730
Ibanez I, Diez-Guerra FJ, Gimenez C, Zafra F (2016) Activity dependent internalization of the glutamate transporter GLT-1 mediated by beta-arrestin 1 and ubiquitination. Neuropharmacology 107:376–386
Ibanez I, Bartolome-Martin D, Piniella D, Gimenez C, Zafra F (2019) Activity dependent internalization of the glutamate transporter GLT-1 requires calcium entry through the NCX sodium/calcium exchanger. Neurochem Int 123:125–132
Al Awabdh S, Gupta-Agarwal S, Sheehan DF, Muir J, Norkett R, Twelvetrees AE, Griffin LD, Kittler JT (2016) Neuronal activity mediated regulation of glutamate transporter GLT-1 surface diffusion in rat astrocytes in dissociated and slice cultures. Glia 64:1252–1264
Wang Y, Yun BW, Kwon E, Hong JK, Yoon J, Loake GJ (2006) S-nitrosylation: an emerging redox-based post-translational modification in plants. J Exp Bot 57:1777–1784
Stamler JS, Lamas S, Fang FC (2001) Nitrosylation. the prototypic redox-based signaling mechanism. Cell 106:675–683
Foster MW, McMahon TJ, Stamler JS (2003) S-nitrosylation in health and disease. Trends Mol Med 9:160–168
Stomberski CT, Hess DT, Stamler JS (2019) Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid Redox Signal 30:1331–1351
Yin CY, Huang SY, Gao L, Lin YH, Chang L, Wu HY, Zhu DY, Luo CX (2021) Neuronal nitric oxide synthase in nucleus accumbens specifically mediates susceptibility to social defeat stress through cyclin-dependent kinase 5. J Neurosci
McLeod F, Boyle K, Marzo A, Martin-Flores N, Moe TZ, Palomer E, Gibb AJ, Salinas PC (2020) Wnt Signaling Through Nitric Oxide Synthase Promotes the Formation of Multi-Innervated Spines. Front Synaptic Neurosci 12:575863
Lenz IJ, Plesnila N, Terpolilli NA (2020) Role of endothelial nitric oxide synthase for early brain injury after subarachnoid hemorrhage in mice. J Cereb Blood Flow Metab:271678 × 20973787
Sun Y, Zhao Z, Zhang H, Li J, Chen J, Luan X, Min W, He Y (2020) The interaction of lead exposure and CCM3 defect plays an important role in regulating angiogenesis through eNOS/NO pathway. Environ Toxicol Pharmacol 79:103407
Zhou Q, Tu T, Tai S, Tang L, Yang H, Zhu Z (2021) Endothelial specific deletion of HMGB1 increases blood pressure and retards ischemia recovery through eNOS and ROS pathway in mice. Redox Biol 41:101890
Balderas A, Guillem AM, Martinez-Lozada Z, Hernandez-Kelly LC, Aguilera J, Ortega A (2014) GLAST/EAAT1 regulation in cultured Bergmann glia cells: role of the NO/cGMP signaling pathway. Neurochem Int 73:139–145
Raju K, Doulias PT, Evans P, Krizman EN, Jackson JG, Horyn O, Daikhin Y, Nissim I, Yudkoff M, Nissim I, Sharp KA, Robinson MB, Ischiropoulos H (2015) Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci Signal 8:ra68
Greaves J, Chamberlain LH (2007) Palmitoylation-dependent protein sorting. J Cell Biol 176:249–254
Greaves J, Prescott GR, Gorleku OA, Chamberlain LH (2009) The fat controller: roles of palmitoylation in intracellular protein trafficking and targeting to membrane microdomains (Review). Mol Membr Biol 26:67–79
Fukata Y, Fukata M (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci 11:161–175
Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M (2013) Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J Cell Biol 202:145–161
Blaskovic S, Adibekian A, Blanc M, van der Goot GF (2014) Mechanistic effects of protein palmitoylation and the cellular consequences thereof. Chem Phys Lipids 180:44–52
Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS (2004) Identification of PSD-95 palmitoylating enzymes. Neuron 44:987–996
Korycka J, Lach A, Heger E, Boguslawska DM, Wolny M, Toporkiewicz M, Augoff K, Korzeniewski J, Sikorski AF (2012) Human DHHC proteins: a spotlight on the hidden player of palmitoylation. Eur J Cell Biol 91:107–117
Cao Y, Qiu T, Kathayat RS, Azizi SA, Thorne AK, Ahn D, Fukata Y, Fukata M, Rice PA, Dickinson BC (2019) ABHD10 is an S-depalmitoylase affecting redox homeostasis through peroxiredoxin-5. Nat Chem Biol 15:1232–1240
Hirano T, Kishi M, Sugimoto H, Taguchi R, Obinata H, Ohshima N, Tatei K, Izumi T (2009) Thioesterase activity and subcellular localization of acylprotein thioesterase 1/lysophospholipase 1. Biochim Biophys Acta 1791:797–805
Lin DT, Conibear E (2015) ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. eLife 4:e11306
Lin DT, Conibear E (2015) Enzymatic protein depalmitoylation by acyl protein thioesterases. Biochem Soc Trans 43:193–198
Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR 3rd, Davis NG, El-Husseini A (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456:904–909
Huang K, Kang MH, Askew C, Kang R, Sanders SS, Wan J, Davis NG, Hayden MR (2010) Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol Dis 40:207–215
Fontana AC, Fox DP, Zoubroulis A, Mortensen OV, Raghupathi R (2016) Neuroprotective Effects of the Glutamate Transporter Activator (R)-(-)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following Traumatic Brain Injury in the Adult Rat. J Neurotrauma 33:1073–1083
Kortagere S, Mortensen OV, Xia J, Lester W, Fang Y, Srikanth Y, Salvino JM, Fontana ACK (2018) Identification of novel allosteric modulators of glutamate transporter EAAT2. ACS Chem Neurosci 9:522–534
Falcucci RM, Wertz R, Green JL, Meucci O, Salvino J, Fontana ACK (2019) Novel positive allosteric modulators of glutamate transport have neuroprotective properties in an in vitro excitotoxic model. ACS Chem Neurosci 10:3437–3453
Zhu BG, Chen YZ, Xing BR (1999) Effect of calcium on the uptake of glutamate by synaptosomes: possible involvement of two different mechanisms. J Neural Transm (Vienna) 106:257–264
Chawla AR, Johnson DE, Zybura AS, Leeds BP, Nelson RM, Hudmon A (2017) Constitutive regulation of the glutamate/aspartate transporter EAAT1 by Calcium-Calmodulin-Dependent Protein Kinase II. J Neurochem 140:421–434
Ashpole NM, Chawla AR, Martin MP, Brustovetsky T, Brustovetsky N, Hudmon A (2013) Loss of calcium/calmodulin-dependent protein kinase II activity in cortical astrocytes decreases glutamate uptake and induces neurotoxic release of ATP. J Biol Chem 288:14599–14611
Hughes EG, Maguire JL, McMinn MT, Scholz RE, Sutherland ML (2004) Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA-induced EAAT2 cell surface trafficking. Brain Research Molecular Brain Research 124:114–123
Abousaab A, Lang F (2016) Up-Regulation of Excitatory Amino Acid Transporters EAAT3 and EAAT4 by Lithium Sensitive Glycogen Synthase Kinase GSK3ss. Cell Physiol Biochem 40:1252–1260
Jimenez E, Nunez E, Ibanez I, Draffin JE, Zafra F, Gimenez C (2014) Differential regulation of the glutamate transporters GLT-1 and GLAST by GSK3beta. Neurochem Int 79:33–43
Trotti D, Volterra A, Lehre KP, Rossi D, Gjesdal O, Racagni G, Danbolt NC (1995) Arachidonic acid inhibits a purified and reconsititued glutamate transporter directly from the water phase and not via the phospholipid membrane. J Biol Chem 270:9890–9895
Volterra A, Trotti D, Cassutti P, Tromba C, Salvaggio A, Melcangi RC, Racagi G (1992) High sensitivity of glutamate uptake to extracellular free arachidonic acid levels in rat cortical synaptosomes and astrocytes. J Neurochem 59:600–606
Volterra A, Trotti D, Racagni G (1994) Glutamate uptake is inhibited by arachidonic acid and oxygen radicals via two distinct and additive mechanisms. Mol Pharmacol 46:986–992
Escartin C, Brouillet E, Gubellini P, Trioulier Y, Jacquard C, Smadja C, Knott GW, Kerkerian-Le Goff L, Deglon N, Hantraye P, Bonvento G (2006) Ciliary neurotrophic factor activates astrocytes, redistributes their glutamate transporters GLAST and GLT-1 to raft microdomains, and improves glutamate handling in vivo. J Neurosci 26:5978–5989
Raunser S, Haase W, Franke C, Eckert GP, Muller WE, Kuhlbrandt W (2006) Heterologously expressed GLT-1 associates in approximately 200-nm protein-lipid islands. Biophys J 91:3718–3726
Otto GP, Nichols BJ (2011) The roles of flotillin microdomains–endocytosis and beyond. J Cell Sci 124:3933–3940
Jang D, Kwon H, Jeong K, Lee J, Pak Y (2015) Essential role of flotillin-1 palmitoylation in the intracellular localization and signaling function of IGF-1 receptor. J Cell Sci 128:2179–2190
Piniella D, Martinez-Blanco E, Ibanez I, Bartolome-Martin D, Porlan E, Diez-Guerra J, Gimenez C, Zafra F (2018) Identification of novel regulatory partners of the glutamate transporter GLT-1. Glia 66:2737–2755
Robinson MB (1998) Examination of glutamate transporter heterogeneity using synaptosomal preprations. Methods Enzymol 296:189–202
Brigidi GS, Bamji SX (2013) Detection of protein palmitoylation in cultured hippocampal neurons by immunoprecipitation and acyl-biotin exchange (ABE). J Vis Exp
Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, Spruce LA, Xiao R, Guo W, Putt M, Seeholzer S, Ischiropoulos H, Robinson MB (2011) Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 31:18275–18288
Acknowledgements
The authors would like to thank Dr. Zila Martinez-Lozada for her thoughtful comments and suggestions for improving this review article. The authors were supported by a grant from the National Institutes of Neurologic Disease and Stroke (R01 NS106693).
Funding
The authors were supported by a Grant from the National Institutes of Neurologic Disease and Stroke (R01 NS106693).
Author information
Authors and Affiliations
Contributions
The idea for the topic of this review was developed by AMG and MBR. AMG and MBR gathered literature, and they each drafted parts of the manuscript. AMG, MBR, and ENK all planned the experiments described. AMG and ENK conducted the experiments and performed the data analyses. AMG, MBR, and ENK all contributed to editing and critical revision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
The work with rats was reviewed and approved by the Children’s Hospital of Philadelphia animal care and use committee and was conducted in accordance with NIH policy.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Special issue: In Honor of Baruch Kanner.
Rights and permissions
About this article

Cite this article
Guillem, A.M., Krizman, E.N. & Robinson, M.B. Rapid Regulation of Glutamate Transport: Where Do We Go from Here?. Neurochem Res 47, 61–84 (2022). https://doi.org/10.1007/s11064-021-03329-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03329-7