
Abstract
In this work we studied the effect of adding MgO and/or Fe2O3 alone or simultaneously on in vitro bioactivity of a new sol-gel derived glass-ceramic in the system 56% SiO2 – (38 – x – y)% CaO – x% MgO – y% Fe2O3 – 6% P2O5 [(x,y) = (0,0); (6,0); (0,2); (6,2)] wt%. The in vitro bioactivity of the materials was assessed in a simulated body fluid (SBF). The materials characterization by Differential Scanning Calorimetry (DSC), BET analysis, FTIR spectroscopy, XRD analysis, ICP-OES spectroscopy and SEM-EDS, before and after soaking in SBF, showed, an absence of changes in glass transition temperature (Tg) for Mg-doped materials, whereas we recorded an increase in Tg for the material doped with Fe only. A decrease in the ionic exchange kinetic between glass-ceramics and SBF was observed for all doped materials. For the Mg-doped materials, the hydroxyapatite (HA) formation rate was delayed contrary to the Mg-free materials. The detailed interpretation of the mechanism of the effect of Fe and Mg on the bioactivity of the glass-ceramics, with consideration of other explanations given in the literature, gives more understanding of how these chemical elements affect the physicochemical properties of the doped materials. We believe, this will help to define the optimal way to tailor the behavior of such biomaterials for biomedical applications.
Graphical Abstract

The magnesium incorporated in glass-ceramic retards the HA formation on its surface only if it is released in the physiological solution (SBF). The incorporation of the magnesium ions from SBF into the amorphous calcium phosphate (ACP) stabilizes it and inhibits its crystallization into HA. The formation rate of the HA layer is correlated to the concentration of magnesium ions in SBF.
Highlights
-
The glass transition temperature is not correlated to the in vitro bioactivity of the glass-ceramic.
-
The delay effect of magnesium on hydroxyapatite formation depends on whether the magnesium is released in SBF or not.
-
Hydroxyapatite formation rate on the glass-ceramic surface depends on the magnesium concentration in SBF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Bioactive glass and glass-ceramic are highly coveted biomaterials in the field of bone tissue engineering. Indeed, these materials have the ability, when in contact with a biological environment, to form on their surface a layer of hydroxy-carbonate apatite (HCA) that is close to the mineral component of the natural bone [1,2,3,4,5,6,7,8,9]. Thanks to the HCA layer and the products of their chemical reactivity, these materials bind to proteins and growth factors present in the biological environment, which allows the recruitment of cells at the implantation site, and thus the regeneration of a new bone tissue [10]. The physicochemical and biological behaviors of glass/glass-ceramic depend closely on their physicochemical characteristics and the medium they come into contact with. Several parameters are involved, including the pH of the solution [11], the glass/glass-ceramic composition [12,13,14,15,16,17], the thermal treatment [18,19,20,21] as well as the textural properties such as the surface area, the shape, and the size of the pores [11, 22,23,24]. The tailoring of the physicochemical and biological properties is governed by the qualitative and quantitative composition of the glass [25, 26], the synthesis method [27, 28], as well as the synthesis parameters [29, 30]. A plethora of different metal oxide compositions have been studied in previous works to evaluate the physicochemical and biological properties acquired through the introduction of different chemical elements [1, 16, 17, 31,32,33,34,35,36,37,38,39,40,41,42].
One of the most investigated elements is magnesium (Mg). It is a trace alkali-earth metal naturally present in the human body, it represents the 2nd intracellular cation and the 4th most abundant metallic cation in the human body [43]. In total, 50–60% of Mg content of an average human body is stored in bone tissues, [44] and is essential for human metabolism [45, 46]. It is known that Mg stimulates bone regrowth [47,48,49,50], promotes the adhesion of osteoblasts [51] and stimulates their proliferation [13, 52,53,54]. A lack of Mg appears to lead to a halt in bone growth due to decreased osteoblastic and osteoclastic activity [16, 52, 55, 56].
It was reported in a previous study that Mg incorporation results in an increase in glass dissolution due to disruption of the connectivity of the glass network and delays the formation of the HA layer. The intensity of these effects depends on the MgO wt% content, which must be greater than or equal to 1.2% [40, 57]. In another study, it was shown that the delay effect on HA formation, occurs only at a level greater than or equal to 7% molar of MgO [13]. This shows that the effect of the addition of MgO studied in several works has led to various interpretations. It was also demonstrated that the effect of MgO on the formation of HA is related to the glass system and the MgO content, which resulted in either an improvement [58, 59], a reduction [60], or a no effect on the formation rate of HA [61].
Another chemical element of biological interest, is iron (Fe), it is considered as an enzymatic cofactor in the control of bone metabolism [62, 63]. Its deficiency leads to a decrease in bone mineralization [64]. Furthermore, a high concentration of Fe can result in osteopenia [65]. This shows that the protective or destructive effect of Fe depends closely on its concentration. Thus, an optimal Fe content improves bone metabolism by acting on proliferation, osteoblast differentiation, and calcification [66] In addition, the incorporation of Fe in the glass matrix can confer ferromagnetic properties on glass, allowing them to be used in controlled and targeted therapy [67]. Other studies have shown that adding Fe2O3 confers to the glass antibacterial properties [42]. Glass/glass-ceramic containing Fe have good mechanical properties [68,69,70]. The chemical durability of glass/glass-ceramic has been shown to be highly influenced by the structure, composition and nature of ions present in the glass network [71]. Gupta et al., demonstrated that incorporating Fe2O3 strengthens the connectivity of the glass network [42]. It was shown that the increase in chemical durability leads to a decrease in bioactivity of glass [41, 72,73,74]. Paradoxically, another study has shown that the addition of Fe improves bioactivity [75].
The interesting set of biological properties of chemical elements such Mg and Fe make them suitable to be incorporated in glasses and glass-ceramics for bone tissue engineering.
For this aim we decide to study the Fe and Mg effects after their incorporation, on the physicochemical properties of a new glass-ceramic composition synthesized via sol-gel route. This method represents a good alternative to the synthesis by fusion, in fact, the glass/glass-ceramic is synthesized at low temperature contrary to the conventional route, which allows to control the composition and consequently the physicochemical properties of the material [76, 77]. The low temperature favors surfaces with a large number of silanol groups which can act as nucleation sites for the crystallization of HA [76,77,78]. In addition, this method leads to materials of high porosity, purity, and homogeneity which are essential characteristics for the biological response of glass/glass-ceramic [79, 80].
A serie of new compositions was synthesized in the system 56% SiO2 – (38 – x – y)% CaO – x% MgO – y% Fe2O3 – 6% P2O5 [(x,y) = (0,0); (6,0); (0,2); (6,2)] wt%. The results obtained in this work are discussed in detail to explain the mechanism of the effect of Fe and Mg on the in vitro bioactivity of glass-ceramic with a consideration of various explanations and controversies reported in the literature.
2 Materials and methods
2.1 Glass synthesis reactants
The different reactants for the materials synthesis are summarized in Table 1.
2.2 Preparation of the tested samples
The different theoretical compositions in weight % of the studied materials are summarized in Table 2.
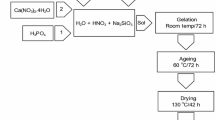
The materials were synthesized according to a procedure described elsewhere [81]. The hydrolysis of TEOS and TEP was performed using nitric acid HNO3 (1 M) as a catalyst with a molecular ratio of (HNO3 + H2O)/(TEOS + TEP) = 8. The mixture was made in a Teflon container that remains closed throughout the synthesis. After the addition of each reactant at an interval of 1 h, the solution was agitated for 1 h. The container was kept closed at room temperature for 3 days to allow hydrolysis and polycondensation of the gel. The formed gel then matured in an oven set at 70 °C for 3 days and was then dried at 150 °C for 3 days. After drying, the samples were calcined at 700 °C for 6 h at a heating rate of 10 °C per minute. All samples were grounded and sieved to obtain a particle size between 40 and 63 µm.
2.3 Materials characterization
Thermal analysis was performed on a Labsys evo 1600 TG-DTA/DSC (Setaram) with a heating rate of 10 K.min−1 in nitrogen. This technique allows to verify if the phenomenon of glass transition occured or not.
The verification of the presence of crystalline phases and their identification were accomplished by XRD analysis using a PANalytical X’Pert Pro diffractometer (Cu‑L2,L3 radiation, λ = 1.5418 Å, 40 kV,40 mA, PIXcel 1D detector), with 0.026° step size and a counting time 400 ms per step, over a range of 10° <2θ < 60°, at room temperature.
The identification of the different functional groups within the material chemical structure was achieved by FTIR spectroscopy carried out at room temperature on KBr pellets containing the sample by Alpha Bruker spectrometer between 4000 cm−1 and 400 cm−1 in transmittance mode with a resolution of 1 cm−1.
Specific surface area and porosity features were analyzed at 77 K using Nitrogen adsorption isotherms by a Gemini VII volumetric adsorption analyzer (Micromeritics, USA). Prior to measurements, the samples were outgassed during one night at ambient temperature in a VacPrep 061 degas system. Specific surface areas were derived from the isotherms using the BET equation and a set of 10 experimental points of the linear range of the BET plot (0.05 < P/P0 < 0.3).
2.4 In vitro bioactivity test
To evaluate the in vitro bioactivity, all materials, in a powder form, were immersed in a simulated body fluid. This later is a physiological solution with an ionic composition close to that of the human plasma. The SBF solution was synthesized as described by Kokubo and collaborators [82, 83]. The elemental composition of the SBF is given in Table 3. In bref, 25 mg of each material were immersed in 50 mL of SBF. The recipients were then placed in an incubator set at 37 °C with a rotation speed of 60 rpm for different immersion times {1, 5, 10 and 15 days}. The procedure was realized in triplicate.
2.5 Materials characterization before and after soaking in SBF
The glass ceramics surface changes were characterized by FTIR spectroscopy, XRD analysis before and after immersion in SBF. Scanning electron microscopy coupled to Energy dispersive X-ray spectroscopy (SEM-EDS) was also performed by a JEOL JSM 7100 F EDS EBSD (Oxford instrument) to examine the materials surfaces before and after different immersion times. After each time of immersion, the supernatant of SBF was collected and analyzed by ICP-OES to quantify the ionic exchange between the materials surfaces and SBF. The analysis was performed by Thermo Scientific iCAP 7000 series ICP-AES spectrometer.
3 Results
3.1 Determination of the materials structure state by X-ray diffraction
The different XRD patterns (Fig. 1) show a scattering halo between 2θ = 25° and 2θ = 35° due to the presence of an amorphous phase [84]. The diffraction peaks, recorded for the different materials, reveal the presence of a crystalline phase (dicalcium silicate Ca2(SiO4): ICSD#01-086-0399) for all materials. In addition, we noticed for 56S6 and 56S6-Fe the presence of two other crystalline phases that might be attributed to hydroxyapatite (ICSD#96-230-0274) for 56S6, and calcium phosphate silicate Ca5(PO4)2(SiO4)6 (ICSD#00-049-1674) for 56S6-Fe.

XRD patterns of the different glass-ceramics. * ICSD#01-086-0399. ** ICSD#96-230-0274. *** ICSD#00-049-1674
3.2 Identification of the different functional groups by infrared spectroscopy
The different infrared spectra (Fig. 2) show characteristic bands of stretching vibrations of the Si-O-Si (bridging oxygen) at about 1040 cm−1 [85] and bending vibrations of O-Si-O (bridging oxygen and non-bridging oxygen) bonds between 473 cm−1 and 501 cm−1 [85]. This confirms that all materials are made of the same silica glass network interrupted by network modifiers. The band at 788 cm−1 is attributed to the symmetric stretching vibrations of the Si-O-Si bonds [86,87,88,89]. The shoulder at approximately 940 cm−1 is attributed to the asymmetric stretching vibrations of the Si-O bonds (NBO) within the tetrahedrons [87, 90], the shoulder is larger for the doped materials. The band recorded at 606 cm−1 for 56S6 and 603 cm−1 for 56S6-Fe is attributed to the bending vibrations of the O-P-O bonds of the group PO43- in the crystalline form [91], this band disappears for Mg-doped materials. The band at approximately 1040 cm−1 is attributed to the asymmetric stretching vibrations of the Si-O-Si bonds within the tetrahedrons [87, 89, 92]. The large shoulder at 1235 cm−1 is assigned to the vibrations of the PO43- group present in the glass matrix. Indeed, P2O5 can be incorporated both as network former and network modifier in the form of PO43- [93, 94].

FTIR spectra of the different glass-ceramics
3.3 Thermal behavior of the different materials
The thermal analysis of the different materials (Fig. 3) show a glass transition for all tested materials, This is consistent with the results of XRD analysis which show a scattering halo between 2θ = 25° and 2θ = 35° due to the presence of an amorphous phase [84]. The results show also that the incorporation of Mg alone or simultaneously with Fe does not affect the glass transition temperature (Tg). Whereas, the incorporation of Fe alone increases the Tg slightly.

DSC curves for the different materials
3.4 Textural properties by nitrogen sorption measurements
All obtained adsorption/desorption isotherms (Fig. 4) are of type IV isotherm indicating that all materials are mesoporous [95]. The textural parameters: specific surface area (SBET), porous volume (Vp), and pore diameter (Dp), are listed in Table 4. The substitution by Fe alone seems to decrease the different textural parameters; this is in agreement with the results obtained by Gupta et al. [42]. Substitution by Mg alone or simultaneously with Fe does not seem to vary the surface area but decreases the porous volume and the diameter of the pores with a higher degree for simultaneous substitution of Ca by both Mg and Fe. On the other hand, when Fe is incorporated alone it decreases the surface area.

Nitrogen adsorption/desorption isotherms for the different glass-ceramics
3.5 In vitro bioactivity tests
3.5.1 Surface characterization by X-ray diffraction
The XRD patterns of the different glass-ceramics (Fig. 5) before and after immersion show, for all materials, a disappearance of the peaks specific to the crystalline phases present before immersion from the first day of immersion. This is may be due to the dissolution of these crystalline phases due to ionic exchanges between glass-ceramics and SBF. We also recorded an appearance of new peaks attributed to two different crystalline phases. Peaks recorded at 23.0°, 29.4°, 36.0°, 39.4°, 43.2°, 47.2°, 48.4° and 57.5° which are assigned to calcite (COD#9000967), and peaks recorded at 25.9°, 31.9°, 39.5°, 46.8°, 49.5° and 53.2°, which correspond to (002), (211), (310), (222), (213) and (004) reflection plans of hydroxyapatite (COD#9003554) respectively. From the first day of immersion, we observe the presence of calcite for all glass-ceramics. This is due to the strong release, by the materials, of the Ca2+ ions which combine with the carbonate ions (CO32-) present in the SBF [96], the calcium carbonate formed precipitates in the form of calcite on the surface of the glass-ceramics [97,98,99]. For 56S6 and 56S6-Fe, we observe a concomitant formation of calcite and HA. This is often the case for glasses and glass-ceramics products which are highly bioactive [96, 100]. The calcite formed remains present throughout the immersion period because of its low water solubility [101]. By comparing the different XRD spectra, we notice that HA is formed simultaneously with calcite on the surface of the 56S6 and 56S6-Fe on the first day of immersion. Whereas, for the Mg-doped glass-ceramics, only calcite is observed. It is only, from day 5, that we observe the appearance of HA peaks on the surface of Mg-doped compositions. The formation of HA on the surface of the different glass-ceramics, confirms their bioactive character.

XRD patterns of the different glass-ceramics surfaces before and after immersion in SBF solution at different immersion times *Calcium hydroxide phosphate, AR powder (Alfa Aesar, Zeppelinstraße 7, 76185 Karlsruhe, Cat. # 304288, Lot#1386600). ** ICSD#96-900-0968
3.5.2 Surface characterization by infrared spectroscopy
The infrared spectra of the different glass-ceramics (Fig. 6), before and after immersion, show differences in the evolution of the spectral bands only between the compositions containing Mg (56S6-Mg and 56S6-MgFe) and those free of Mg (56S6 and 56S6-Fe). On day 1 of immersion, for Mg-doped glass-ceramics, the band centered at about 500 cm−1, which includes the shoulder at about 605 cm−1 is transformed into two bands, one at 468 cm−1 attributed to the Si-O-Si rocking vibration [102], and a more or less broad band between 530 cm−1 and 630 cm−1 attributed to asymmetrical deformation vibrations of P-O bonds in ACP [103,104,105,106]. For Mg-free glass-ceramics, the same band is observed at 468 cm−1, however, the small band at 606 cm−1 (56S6) and 603 cm−1 (56S6-Fe) increases in intensity, in parallel, with the appearance of a new band at 564 cm−1. This dual band is due to the vibrations of the crystalline phosphate groups present in HA [107,108,109]. For 56S6-Mg and 56S6-MgFe, the broad band between 1000 and 1200 cm−1 is attributed to asymmetric stretching vibrations of P-O bonds in ACP [103,104,105]. For the 56S6 and 56S6-Fe, the band, ranging from 1000 cm−1 to 1100 cm−1, has spikes at 1034 cm−1 and 1093 cm−1 that correspond to the asymmetric stretching vibrations of the P-O bonds in the PO43- groups present in HA [110]. The weak band (for 56S6) and the shoulder (for 56S6-Fe, 56S6-Mg and 56S6-MgFe) at 788 cm−1 (Fig. 2), are both assigned to the symmetric stretching vibrations of the Si-O-Si bonds [86,87,88,89], this band/shoulder increases in intensity and becomes centered at 800 cm−1. This change is due to the formation of the silica gel layer on the surface of the glass-ceramics [111,112,113]. On day 1, we recorded, for all materials, an appearance of a broad band between 1400 cm−1 and 1500 cm−1, and another band at 874 cm−1, these bands are attributed to the vibrations of the C-O and C = O bonds in carbonate groups. The band at 1456 cm−1 is characteristic of the crystalline carbonates [114], while the other bands are common to both crystalline and amorphous carbonates [115]. This is in a good agreement with the XRD patterns of calcite, which precipitates on the surface the 1st day of immersion for all tested glass-ceramics. After the first day of immersion, no change is recorded in the spectra of Mg-free glass-ceramics. On the other hand, for Mg-doped glass-ceramics, on the 5th day, the band between 530 cm−1 and 630 cm−1 takes a shape of a dual-band at 603 cm−1 and 564 cm−1. This is due to the crystallization of ACP in HA [106]. This crystallization is also confirmed by the transformation of the shape of the broad band between 1000 cm−1 and 1200 cm−1. Indeed, like the spectral bands recorded on the 1st day, in the same region for the 56S6 and 56S6-Fe, this band takes the form of the spectral bands attributed to asymmetric stretching vibrations of the P-O bonds of the phosphate groups in HA [106, 110]. Beyond the 5th day of immersion, no other changes in the spectra are observed. The presence of phosphates and carbonates together may indicate that the HA formed on the surface is carbonated [116]. The FTIR results are consistent with the XRD results. Indeed, the spectral bands of the carbonates of calcite as well as the carbonates and phosphates present in the hydroxycarbonate apatite (HCA), evolve in perfect correlation with the XRD patterns of calcite and HA. By comparing the different FTIR spectra and XRD patterns, the results show that the incorporation of Fe does not affect the formation rate of HCA, while the incorporation of Mg delays it.

FTIR spectra of the different glass-ceramics surfaces before and after immersion in SBF solution at different soaking times. *Calcium hydroxide phosphate, AR powder (Alfa Aesar, Zeppelinstraße 7, 76185 Karlsruhe, Cat. # 304288, Lot#1386600)
3.5.3 ICP-OES analysis of SBF
Obtained results of ICP analysis (Fig. 7) show the evolution of the different elements concentrations in SBF versus immersion time.

Evolution of (Ca), (Mg), (P) and (Si) concentrations in SBF versus immersion time
Ca and P concentrations
On day 1 of immersion, we recorded for all glass-ceramics, an increase in Ca concentration in parallel with a decrease in P concentration which is slower for Mg-doped materials. Between day 1 and day 5, the Ca concentration decreases for 56S6-MgFe, while it continues to increase for 56S6 and 56S6-Fe. For 56S6-Mg, the Ca concentration remains unchanged. The P concentration continues to decrease for all materials. Beyond the 5th day, the concentration of Ca decreases with the same trend for all glass-ceramics. The P concentration continues to decrease until depletion, as noted from the 5th day for 56S6, 56S6-Fe and 56S6-Mg, and from the 10th day for 56S6-MgFe.
Mg concentration
The Mg concentration follows the same pattern for all materials except for 56S6-MgFe. For 56S6, 56S6-Fe and 56S6-Mg, starting on day 1, there is a decrease in the concentration. Between day 1 and day 5, the concentration increases. After day 5, the concentration decreases again but slightly. For 56S6-MgFe, the concentration increases from the first day and then, remains unchanged until the 5th day. Beyond the 5th day, the evolution of the concentration follows the same trend as the rest of glass-ceramics.
Fe concentration
ICP-Fe results (not presented here) show no release of Fe3+ in SBF.
Si concentration
The Si concentration increases rapidly from the first day of immersion for all glass-ceramics. Between Day 1 and Day 5, a slight increase was recorded. Beyond day 5, the concentration remains relatively unchanged during the remaining immersion time.
3.5.4 SEM-EDS micrographs
The SEM micrographs (Fig. 8) of the different materials, before immersion in the SBF, show irregular surfaces with a granular appearance (Fig. 8a, d, g, j). After 1 day of immersion, rods appear on the surfaces of the 56S6 (Fig. 8b) and 56S6-Fe (Fig. 8e), while for 56S6-Mg (Fig. 8h) and 56S6-MgFe (Fig. 8k), the surface remains unchanged. After 15 days of immersion, rods similar to those observed on the 1st day on the surfaces of 56S6 and 56S6-Fe, appear on the surfaces of 56S6-Mg (Fig. 8i) and 56S6-MgFe (Fig. 8l). at this stage of immersion, a more or less smooth layer appears on the rods, For 56S6 (Fig. 8c) and 56S6-Fe (Fig. 8f). The chronology of the appearance of the rods recorded on the SEM micrographs, coincides perfectly with the chronology of the appearance of the FTIR spectral bands and the XRD patterns attributed to the HCA.

SEM micrographs of the different material surfaces before and after 1 day and 15 days of immersion in SBF. a, d, g, j Before immersion for 56S6, 56S6-Fe, 56S6-Mg, and 56S6-MgFe, respectively; b, e, h, k day 1 of immersion for 56S6, 56S6-Fe, 56S6-Mg, and 56S6-MgFe, respectively; c, f, i, l day 15 of immersion for 56S6, 56S6-Fe, 56S6-Mg, and 56S6-MgFe, respectively
This result allows us to conclude that the rods that appeared on the glass-ceramics surfaces correspond to the HCA crystals formed from the 1st day, for 56S6 and 56S6-Fe, and that we only observed from the 5th day, for 56S6-Mg and 56S6-MgFe. The smooth layer that covers the rods after 15 days of immersion for 56S6 and 56S6-Fe, corresponds, probably, to a silica gel layer that forms as a result of the Ca leaching that continued throughout the immersion period.
The SEM results confirm the delayed formation of HA on the surface of Mg-doped glass-ceramics in comparison to Mg-free ones.
4 Discussion
4.1 Thermal analysis
The results obtained in the thermal analysis needs to be dressed meticulously. Previous studies [117, 118] have shown that Tg increases as the content of MgO increases. Paradoxically, De Araujo Bastos Santana et al. [119] have shown the opposite result, and they attributed these changes to the fact that the Mg-O bond strength is lower than the Ca-O bond based on the bond dissociation energies (Mg-O: 394 kJ.mol−1; Ca-O: 464 kJ.mol−1), which leads to a weakening of the glass network. Contrary to this explanation, Lee et al. [117], and Labbilta et al., [118] attributed the increase of Tg, following the incorporation of Mg, to the fact that the Mg-O ionic bond is stronger than the Ca-O ionic bond, which made the glass network more rigid. They reported that the strength of a cation-oxygen ionic bond is affected by the cation field strength (CFS), the more important it is, the stronger the bond. They reported that, the CFS (Mg2+) is higher than CFS (Ca2+) (0.46 for Mg2+ and 0.36 for Ca2+), which explains the network reinforcement after the addition of MgO, and consequently the increase of the Tg.
The second explanation seems to be more appropriate. In fact, Mg2+ and Ca2+ are bound to non-bridging oxygen (NBO) by ionic bonds. As explained before, the higher CFS of Mg2+ results in a Mg-O bond that is shorter and stronger than the Ca-O bond. In addition, the dissociation energy of a bond is the energy that must be provided for a homolytic rupture of a bond at 25 °C [120], the fact that the Mg is more electronegative than Ca, the electronic doublet in the Mg-O bond will be less shifted from the center to the oxygen atom than it will be in the Ca-O bond, thus, the energy required for a homolytic rupture of the (Cation2+-O2-) bond will be more important for the Ca-O bond than the Mg-O bond. Consequently, this type of energy cannot be used to interpret the changes in glass transition in this case.
In the present study, the absence of changes in Tg following the incorporation of Mg alone can be explained as follows, the molar mass of Ca is approximately 1.67 times greater than that of Mg, and since substitution occurs in wt%, this results in a number of substituting Mg2+ ions 1.67 times greater than the number of substituted Ca2+ ions. This leads to an increase in the number of NBO in the glass network. Although the Mg-O ionic bond is stronger than the Ca-O ionic bond, the increase in the number of NBO leads to more breaking of Si-O-Si bond, and since the Mg-O ionic bond is weaker than Si-O-Si bond, this causes the glass network to weaken [121], and thus, the strength of the new Mg-O bonds is compensated by the increase of broken Si-O-Si which explains the absence of effect on Tg.
The incorporation of Fe alone seems to increase the Tg. This result can be explained as follows, Fe is incorporated as Fe3+ ion, previous studies have shown that at this oxidation state, Fe3+ acts mainly as a network former because of its large ionic force field (CFS [Fe3+] = 0.76 ; CFS [Ca2+] = 0.36) [122,123,124,125,126,127,128,129]. The small increase can be explain by the fact that the higher electronegativity of Fe (χ (Fe) = 1.83) compared to Ca (χ (Ca) = 1) makes the bond Fe-O stronger than the bond Ca-O and consequently the network becomes more rigid and less easier to break. On the other hand, the number of substituting Fe3+ ions is lower than the number of substituted Ca2+ ions n(Fe3+) = 0.71×n(Ca2+), and since the Fe3+ ions are likely present in the tetrahedral form in the glass network [130], the number of the Fe-O bond is greater than that of the substituted Ca-O bond n(Fe-O) = 1.43 × n(Ca-O), which causes more breaking of Si-O-Si bonds. Knowing that the electronegativity of Fe (χ (Fe) = 1.83) is slightly lower than that of silicon (χ (Si) = 1.9), this leads to a slightly weak Fe-O bond compared to the Si-O, which explains the compensator effect on the bonds energies between Fe and Si, and the difference results in the small increase recorded.
The unchanged Tg for the composition where the Mg2+ and Fe3+ are incorporated simultaneously can be explained by the same approach mentioned above, this result is likely due to an equilibrium between the energies of the new created bonds Si-O-Mg and Si-O-Fe, and the replaced bond Si-O-Ca and/or the broken bonds Si-O-Si.
4.2 Textural properties
The decrease recorded in the size of the pores is due to a combination of two phenomena, the ions diffusion occurring during calcination [131, 132], and to the higher electronegativity of the substituting elements compared to that of Ca, the descending order of the electronegativity is as follows: χ(Fe) > χ (Mg) > χ (Ca). The sol-gel synthesis of our materials occurs in different steps [131, 132], first the polycondensation of Si-O-Si and P-O-P bonds leads to gel formation which consists of SiO2-P2O5 primary particles. The condensation of silanol groups at the surface allows the fusion of the primary particles to form secondary particles. Following this step, the removal of water by drying causes nitrates and calcium, and/or magnesium, and/or iron ions to recombine and deposit on the surface of secondary particles. During the calcination step which occurs at 700 °C, the nitrates are degraded and the cations diffuse from the edges to the core of secondary particles. The remaining ions at the surface act as a fuser of secondary particles to form tertiary particles. Simultaneously to the diffusion of modifier ions into the silica network, pores are created due to the formation of non-bridging oxygens related to the intercalation of ions into the silica network. Pore sizes are then directly related to the modifier ionic radius. The combination of more ions acting as smelting agents for secondary particles and a decrease in ionic radius induce the decrease of pore size. For our glass ceramics, knowing that the number of substituting Mg2+ ions is 1.67 times higher than the number of substituted Ca2+ ions, and the fact that Mg2+ ion is lighter and occupy less volume than Ca2+ ion, this leads to more and easy diffusion into more particles and causing their fusion. For Fe3+ ions, their diffusion causes, as discussed before, the creation of a number of Fe-O bond 1.43 times higher than that of the substituted Ca-O bond, and since Fe3+ is less voluminous than Ca2+, this leads also to a particles fusion that are tighter which explains the decrease of the pore size. At this stage and according to the difference of electronegativity of the different elements, the attraction between the NBO and the substituting elements is stronger than that of Ca. This results in Fe-O and Mg-O bonds that are stronger and shorter than the Ca-O bond [117, 118], leading to matrix compaction that results in reduced pore sizes [60, 133]. This also explains the reduction which is more important for the material substituted simultaneously by both Mg and Fe. The reduction in pore size is more important for the material substituted by Mg alone than that the one substituted by Fe alone, although Fe is more electronegative than Mg. This result is more likely related to the small incorporated amount of Fe compared to that of Mg as well as the ease of diffusion to the core of particles for Mg2+ ions.
The differences in the results regarding the specific surface between 56S6-Fe and the rest of glass-ceramics can be explained as follow, for 56S6-Fe, as explained above, the substitution of Ca2+ ions by Fe3+ ions leads to more breaking in Si-O-Si. This increases the porosity. On the other hand, as mentioned before the higher electronegativity of Fe compared to that of Ca leads to a more compact network which explains the decrease in all textural parameters.
For 56S6-Mg, as mentioned before, the number of substituting Mg2+ ions is 1.67 times higher than the number of substituted Ca2+ ions, this results in more breaking of the Si-O-Si bonds which causes more porosity in the glass-ceramic network. This should increase the specific surface area. However as mentioned before, -Si-O- --- Mg2+----O-Si- bonds are shorter and stronger than the -Si-O- ---- Ca2+-----O-Si- bonds, and Mg2+ ions occupy less volume than Ca2+. This explains the decrease in pores size and volume. Overall, the increase in porosity is compensated by the reduction of pores size, and this leads to an unchanged specific surface area.
For 56S6-MgFe, the concomitant substitution of Ca2+ by Mg2+ and Fe3+ ions causes much more breaking of Si-O-Si and this is likely to increase the porosity. This increase in porosity occurred alongside a decrease in pores size and volume as a result of the stronger new bonds namely Fe-O and Mg-O compared to the replaced Ca-O bonds. These changes in porosity and pores size may explain the unchanged specific surface for 56S6-MgFe.
4.3 X-ray diffraction
Before immersion in SBF, the two crystalline phases recorded by XRD analysis that contain phosphate groupes explain the absorption band at 606 cm−1 and 603 cm−1 recorded by FTIR spectroscopy for 56S6 and 56S6-Fe, respectively. As mentioned before, this band is attributed to the vibrations of the P-O bonds in the crystalline PO43- groups [91, 134]. Crystallization of dicalcium silicate for all materials is likely due to the high presence of residual –OH groups. These groups reduce viscosity and can act as nucleation sites for crystal growth. Following the heat treatment, the regions of the network with the most residual –OH, release SiO44- species that bind with Ca2+ present in the network and crystallize in Ca2(SiO4) [134, 135]. The presence of crystalline calcium phosphates is due to the presence of phosphorus in the network as a network modifier in the ionic form PO43- [94]. The network modifier ions, i.e Ca2+ and Mg2+, tend to act as charge compensators and preferably bind to orthophosphate [93, 94]. The resulting calcium phosphates or magnesium phosphates crystallize under the effect of heat treatment [134]. This explains the presence of HA crystals and calcium phosphates on the surface of 56S6 and 56S6-Fe respectively.
4.4 Infrared spectroscopy
Before immersion in SBF, the results show that the band at 788 cm−1 decreases in intensity and takes the form of a shoulder for the materials doped compared to the reference material (56S6). This is due to the rupture of the Si-O-Si bonds after the incorporation of Fe and Mg. The shoulder at 940 cm−1 is larger for the doped materials. These variations are due to the increase in the number of broken Si-O-Si bonds followed by the formation of Si-O bonds (NBO), as a result of the incorporation of the different elements namely Mg and Fe. Indeed, as explained before, for compositions doped with MgO, the number of substituting Mg2+ ions is 1.67 times greater than the number of substituted Ca2+ ions, which leads to an increase in the number of broken Si-O-Si bonds. For Fe-doped compositions, the number of substituting Fe3+ ions is lower than the number of substituted Ca2+ ions, however, the presence of Fe3+ in tetrahedral form [130] leads to an increase in broken Si-O-Si bonds as well.
The disappearance of the absorption band in the 600 cm−1 region for Mg-doped glass-ceramics is a result of the modification of the stability of the P-O bonds by the incorporation of Mg2+, this latter is more electronegative than Ca2+, and therefore the P-O(Mg) bond is more ionic than the P-O(Ca) bond [118].
4.5 ICP-OES analysis
4.5.1 Day 1of immersion
The ICP curves show that from the first moments of immersion, a cationic exchange occurs between the glass-ceramic and the SBF. Indeed, the modifier ions (Ca2+ and Mg2+) leave the network in exchange for the H3O+ ions present in the SBF [136]. This explains the increase in Ca concentration recorded for all compositions. Following the fixation of H3O+ ions, silanol groups form and condense to form a silica gel layer [136]. The decrease in the concentration of H3O+ ions causes an increase in the concentration of OH- ions in SBF. These are responsible for the continuous hydrolysis of the Si-O-Si bonds, leading to the release of Si(OH)4 in the SBF. This explains the increase in the Si concentration from the 1st day of immersion [136].
The decrease in P concentration, for all glass-ceramics, is due to the concomitant migration of the Ca2+ and PO43- ions present in the SBF to the material surface. Although the Ca concentration seems to be just increasing on the 1st day of immersion, this does not prove the absence of migration of Ca2+ ions from SBF to the surface of the materials. Indeed, this can be explained by the fact that the release flow of these ions by the glass-ceramics is more important than their migration from the SBF to the surface of the material.
This concomitant migration of Ca2+ and PO43- ions to the surface of the material, leads to the formation of ACP layer [136], which crystallizes in HA, from the 1st day of immersion, for 56S6 and 56S6-Fe contrary to 56S6-Mg and 56S6-MgFe, where there is an absence of HA at this stage of immersion. This is confirmed by the ACP-specific FTIR spectral bands recorded on the 1st day of immersion for 56S6-Mg and 56S6-MgFe. For 56S6 and 56S6-Fe, the absence of ACP on their surface is due to its rapid transformation into HA.
The slopes of the phosphorus ICP curves show that the migration rate of the PO43- ions is slower for Mg-doped glass-ceramics. This result shows that the incorporation of Mg has a slowing effect on the migration of PO43- ions, and consequently on the formation of ACP and its crystallization in HA which is consistent with the literature [60, 137].
For Mg, the decrease in concentration, observed from the 1st day for all glass ceramics except for 56S6-MgFe, is probably due to the migration of the Mg2+ ions, which are incorporated and/or adsorbed into the ACP layer [138]. For 56S6-MgFe, the increase in the concentration confirms the release of Mg2+ by the material. However, the absence of decrease may be due to the absence of migration of the Mg2+ ions from SBF to the glass surface. This is probably the consequence of the delayed formation of the ACP layer compared to the other glass-ceramics.
Indeed, the most plausible mechanism would be that 56S6-Mg would also release the Mg2+ ions, which is confirmed by the level of Mg concentration that is higher than those recorded for 56S6 and 56S6-Fe. Except that, the ACP formation on 56S6-Mg, which is faster compared to 56S6-MgFe, causes migration of Mg2+ ions and their incorporation or adsorption into the formed ACP layer. This phenomenon seems to be delayed for the 56S6-MgFe. Moreover, by comparing the (P) curve of 56S6-Mg to that of 56S6-MgFe, we noticed that the migration of PO43- ions is slower for the 56S6-MgFe. This shows that the incorporation of Fe in addition to Mg also seems to have a slowing effect on the migration of PO43- ions, and consequently, on the formation of the ACP layer.
4.5.2 Between day 1 and day 5 of immersion
Between day 1 and day 5, the Ca concentration continues to increase but with a lower rate for 56S6 and 56S6-Fe, this is due to the reduction of the release flow in relation to the migration flow. For 56S6-MgFe, the Ca concentration decreases significantly due to the significant migration of Ca2+ ions to the material surface. Finally, for 56S6-Mg, the concentration remains unchanged, which is probably due to an equilibrium between the release flow and the migration flow. This reinforces the hypothesis that the incorporation of Mg reduces ion exchange kinetics [60, 137].
As for the Ca concentration, the P concentration continues to decrease but with a lower rate, for the 56S6 and 56S6-Fe. This is due to the high concomitant migration of Ca2+ and PO43- ions to the surface of these materials at this stage of immersion, which contributes to the crystallization of ACP in HA. This crystallization is observed for all glass-ceramics after 5 days of immersion (see Figs. 5 and 6). At this stage, there is no PO43- in the SBF for 56S6, 56S6-Fe and 56S6-Mg due to its depletion, as a result of the continued migration of Ca2+ and PO43-. The Si concentration increases very slightly, with the same trend, for all glass-ceramics. This is due to the ionic exchange as explained above. This exchange continues at this stage but with a very low degree compared to the first hours of immersion as shown by the slopes of the different (Ca) ICP curves.
For Mg, there was an increase in the concentration for all glass-ceramics, between day 1 and day 5, except for 56S6-MgFe for which the concentration remained unchanged. This phenomenon is due to the release of the Mg2+ ions from the ACP layer which gradually dissolves in parallel with the continuous migration of the Ca2+ and PO43- ions [138]. This is confirmed by the crystallization of ACP in HA, as shown by the FTIR and DRX spectra on the 5th day of immersion (see Figs. 5 and 6). The constancy in the Mg concentration observed for 56S6-MgFe, is probably due to an equilibrium between the migration and the release flows of the Mg2+ ions by the material. This occurs at the same time as the intense and concomitant migration of Ca2+ and PO43- ions to the material surface, which is illustrated by the intense slopes of the ICP-Ca(56S6-MgFe) and ICP-P(56S6-MgFe) curves during this period of immersion.
4.5.3 Between day 5 and day 10 of immersion
After day 5 until day 10, the concentrations of Ca and Mg decrease, with the same trend, for all glass-ceramics. For 56S6-MgFe, the P concentration continues to decrease until depletion at day 10. For Si, the concentration remains unchanged.
Although there are no PO43- ions left in the SBF, for 56S6 and 56S6-Fe, the decrease in Ca concentration noted between day 5 and day 10 is due to the formation of Ca2+/CO32- ion pairs precipitating as calcite. This phenomenon is often observed when SBF is depleted in phosphate ions as described by Oyane et al. [139]. Indeed, the Ca2+ ions, present in high concentration, react with the HCO3- ions to form calcium carbonate following the reaction Ca2+ + HCO3- + H2O→ CaCO3 + H3O+. This explains the growth of calcite XRD peaks at this stage of immersion for 56S6 and 56S6-Fe.
The decrease in Mg concentration, between day 5 and day 10, may be due to the incorporation of Mg2+ ions into the HCA layer as suggested in another study by Chajri et al. [140].
4.5.4 Beyond day 10 of immersion
After the 10th day, the SBF depletion in PO43- is total for all materials. The plateau recorded for the Ca concentration is probably due to an equilibrium between the Ca2+ release flow and the precipitation flow of the Ca2+/CO32- pairs. The very slight decrease observed in the Mg concentration is probably due to their continued migration to the surface of the materials to be incorporated into the HCA layer.
4.6 Effect of Mg incorporation on the glass bioactivity
In this work, the results of the characterization of the surfaces of the different glass-ceramics, before and after immersion in SBF, revealed the delay in HA formation for Mg-doped glass-ceramics of the HCA. This is consistent with the results obtained in other studies [13, 57, 60, 133, 137].
HA formation involves several steps, step 1: the Ca2+ and PO43- ions, released from the material and/or present in SBF, precipitate to the surface as ACP. Step 2: the ACP gradually dissolves and releases the Ca2+ and PO43- ions [141]. At this stage, nucleation sites are formed, these are defined as the interfacial surfaces between the apatitic phase that begins to form and the solution. The Ca2+ and PO43- ions, released by ACP, are translocated and fixed on the apatitic nucleation sites. The repetition of this phenomenon allows the growth of HA crystals on the surface of the material [138, 142]. This description is consistent with the ionic exchange kinetics described in the literature for bioactive glasses/glass-ceramics [136], and which is observed for all the glass-ceramics studied in this work. However, the fact that the ACP is not observed for compositions free of Mg, can be related to the fact that the transformation would have occurred during the first hours of immersion as explained above. For Mg-doped glass-ceramics, this process was delayed.
Several studies have shown the complexity of the mechanism of the delayed transformation of ACP into HA, due to the presence of MgO. Some authors argue that the addition of MgO decreases the glass/glass-ceramic dissolution [60, 137]. Other authors have shown that the decrease in bioactivity was only due to the delay in crystallization of ACP in HA, while the dissolution kinetics of the Mg-doped glass/glass-ceramics was better than those of the bare glass/glass-ceramics [119]. This displays the controversies around the role that Mg2+ ions play within the glass network. Indeed, Mg2+ can be incorporated as a network modifier [143, 144], but can also, depending on the composition of the glass/glass ceramic and the conditions of synthesis, play an intermediate role by incorporating in a tetrahedral form (MgO4) [124, 145, 146].
Watts et al. suggested that Mg incorporated at 86% acts as a network modifier and that for the remaining 14%, it is present in a tetrahedral form (MgO4) [121]. This leads to the polymerization of the silicate network, which strengthens the network and therefore reduces its dissolution. In addition, although previous studies have shown a correlation between the MgO content and the retarding effect of HA formation [40, 57, 147], other authors have obtained results where there is no correlation between the MgO content and the observed effect on HA formation [58], which confirms the complexity of Mg’s role within the glass network.
4.6.1 Effect on dissolution kinetics
The results obtained in our study show that several parameters are involved in the delayed transformation of ACP into HA. The kinetic of ionic exchange appears to be slower for Mg-doped glass-ceramics, as advanced in the literature [60, 137]. This is confirmed by the slow release of Ca2+ ions as well as the slow migration of PO43- ions for compositions that contain Mg. Indeed, the 56S6-MgFe ICP curves show a high level of Ca concentration on Day 1 and a slow decrease in P concentration. For 56S6-Mg, on Day 1, the Ca concentration level is the same as for 56S6 and 56S6-Fe. However, the decrease in P concentration is slower. 56S6-Mg contains less CaO than 56S6 and 56S6-Fe, so if the release of Ca2+ had been as rapid as for Mg-free glass-ceramics, the Ca concentration of 56S6-Mg on Day 1 of immersion should be lower. However, the fact that it is at the same level as for the other compositions, confirms the slower release of Ca by 56S6-Mg. This is confirmed by the slow migration of PO43-ions, as shown in the 56S6-Mg ICP-P curve. In addition, between day 1 and day 5, the difference is clearly illustrated between the 56S6-Mg ICP-Ca curve and the 56S6 and 56S6-Fe ICP-Ca curves. Indeed, a plateau is recorded for the Ca concentration for the 56S6-Mg, contrary to the other two glass-ceramics where an increase in Ca concentration is recorded. Indeed, the slow release of Ca2+ ions, is likely in an equilibrium with their migration to the surface, which is at the origin of the transformation of the ACP into HA. For the 56S6 and 56S6-Fe the Ca release is faster than the migration of Ca2+ ions which explains the increase recorded.
For the evolution of Mg concentration, the curves show a migration of Mg2+ ions from SBF to the surface of the materials, to incorporate and/or adsorb in the layer and/or on the surface of ACP [138, 148,149,150,151], on the 1st day of immersion. A comparison of HA onset times of appearance at this stage of immersion, shows that the transformation from ACP to HA has been delayed for compositions containing Mg.
4.6.2 Mechanism of Mg effect on ACP delayed crystallization into HA
SBF contains Mg2+, the ICP results show its incorporation and/or adsorption into ACP for all glass-ceramics. However, the high Mg concentration due to the release of more Mg2+ ions in the solution by the Mg-doped glass-ceramics, accentuates the inhibitory effect of Mg. This finding corroborates what has been suggested in several studies, stipulating that the amount of Mg incorporated and/or adsorbed into ACP is related to its concentration in the SBF solution [138, 149,150,151].
The increase in Mg concentration between days 1 and 5 of immersion is due to the gradual dissolution of the ACP layer, which occurs simultaneously with its transformation into HA, as demonstrated by previous studies [138, 142]. The release of the incorporated Mg, suppresses its inhibitory effect, and the ACP begins its transformation into HA [142]. At this stage, interfacial surfaces are created between the new crystalline phase and the SBF. These surfaces act as HA nucleation sites. The Ca2+ and PO43- ions released after the dissolution of the ACP, migrate to the surface of the materials and deposit at the HA nucleation sites to form HA crystals [138], the repetition of this phenomenon leads to the growth of HA crystals. This corresponds to the formation of the HA layer observed on the 1st day for Mg-free glass-ceramics, and only from the 5th day for Mg-doped glass-ceramics.
The decrease in Mg concentration recorded for all glass-ceramics between day 5 and day 10 is due to the incorporation of Mg2+ ions in the HA layer as suggested by a previous study [140]. At this stage, Mg2+ ions adsorbed at the nucleation sites, is mainly responsible for delaying the growth of the HA layer. The degree of this delay depends on the adsorbed amount of Mg.
The results of our study corroborate and confirm the explanations and suggestions presented in the literature regarding the delay of HA formation rate caused by Mg. Researchers have suggested that the association of Mg with phosphates decreases their ionic activity, which would delay the induction time of crystallization of ACP in HA [152]. Others explain that this association of Mg with phosphates [13, 53, 60] leads to a decrease in the number of HA nucleation sites [153, 154]. Other studies [149,150,151, 155] argue that the presence of Mg in the ACP layer leads to a reduction in particle size and gives them an irregular shape rich in deficits, making their crystallization difficult.
Other researchers claim that the inhibition of the crystallization and growth of HA crystals, caused by Mg, is due to its adsorption at the HA nucleation sites [148,149,150, 156].
Ding et al. [138] demonstrated that the stabilization of ACP is achieved by both incorporated and adsorbed Mg, while inhibition of ACP crystallization in HA is primarily due to the adsorbed Mg. This proves that it is, indeed, the adsorbed Mg that invades the HA nucleation sites that inhibits the crystallization of ACP in HA. This result leads us to assert that the delay in the transformation of the ACP into HA is closely correlated with the amount of Mg present in the solution. If the Mg present in the glass-ceramic was not released into the solution, the delay in the transformation of ACP into HA would only be related to the indirect effects of Mg such as the decrease of the glass-ceramic dissolution in SBF, the degree of this effect would be directly related to the rate of this dissolution. However, since Mg-doped glass-ceramics release Mg2+, when in contact with SBF, these ions will incorporate into the ACP and reduce its dissolution. In addition, Mg2+ which subsequently adsorb at the nucleation sites, delay the transformation of the ACP into HA crystals, and the crystal growth. This is consistent with the results obtained in our study, which is in a good agreement with what is reported in the literature [40, 53, 57, 119, 157].
4.7 Effect of Fe incorporation on the glass bioactivity
The results of this study show that the addition of Fe2O3 does not affect the formation of HA on the surface of the materials after contact with SBF. Some studies claim that the presence of Fe2O3 decreases this property [158, 159], while others confirm the opposite [75]. Indeed, Fe3+ ions have a stimulatory effect on the crystallization ACP into HA, as demonstrated in a previous study [160]. However, the fact that this ion is not released into the SBF explains the absence of incorporated Fe3+ ions in ACP layer, therefore there is no stimulation effect on its transformation into HA.
The ICP-OES results show that adding Fe2O3 appears to have a slight slowing effect on the kinetic of ionic exchange when added to 56S6. However, this effect remains insignificant because of the low Fe2O3 content. For the 56S6-MgFe, the slowing effect on ionic exchange kinetic is significant. This is probably due to the addition of the effects of both Fe2O3 and MgO. The slowing effect mechanism is detailed above for MgO. With regard to Fe2O3, it has been reported that the presence of Fe3+ in the glass network reduces the dissolution of the material and therefore the kinetic of the ionic exchange is slower [161, 162]. As mentioned above, Fe3+ ion act as a network former in tetrahedral form. This explains the absence of Fe3+ release in SBF as confirmed by ICP-OES (not presented here).
5 Conclusion
In this work we have shown that the Mg and Fe have many effects on the physicochemical properties of the glass/glass-ceramic. The incorporation of these chemical elements results in a decrease in the Mg-doped glass-ceramic bioactivity at different levels. Our results showed that when Mg is added, either alone or simultaneously with Fe, it delays the formation of HA at several levels, first by reducing the dissolution of the glass-ceramic in the SBF, then by stabilizing the ACP by decreasing its dissolution, and finally preventing its crystallization into HA by invading the HA nucleation sites. We also demonstrated that it is the free Mg2+ ions, previously present in the solution and/or released by the materials, that delays the formation of HA and not the Mg that remains incorporated in the matrix.
Fe3+ in solution, is known to have a stimulatory effect on HA crystallization from ACP. Surprisingly, there was no delay or acceleration observed on the HA formation rate, and this is due to either the low content of Fe2O3 or to the fact that Fe3+ was incorporated as a network former, leading to absence of its release in SBF. The results showed also that there is no systematic correlation between the glass transition temperature and the glass/glass-ceramic dissolution, indeed, although, the Tg was increased by incorporating Fe alone, the dissolution rate decreased less than for Mg-doped glass-ceramics, and where the Tg remains unchanged. Moreover, several parameters have an influence on the dissolution process, such as, composition of the solution, the overall glass-ceramic composition, the intrinsic physicochemical characteristics of the introduced elements, the behavior of each ion in the glass network whether as a network modifier or a network former, and also on how it is bonded to the rest of the network.
The findings and the detailed interpretation given in this study have shed light on the complexity of the mechanism by which Mg and Fe affect the physicochemical properties of the doped materials. Indeed, by comparing our results to other previous studies, we showed a non-correlated behavior between the glass transition and the bioactivity of the tested materials. These two physicochemical properties are influenced not only by the intrinsic physicochemical properties of the chemical elements but also by the exterior parameters that governed their behavior within the glass network, such as the overall glass or glass-ceramic composition, the conditions of synthesis, the composition of the physiological solution, etc. This complexity explains the different controversies and the contradictory interpretations found in the literature about these elements as mentioned above.
Based on these arguments, a profound studies are required to determine physically and mathematically the structure-activity relationship of the effect of Mg and Fe on glass/glass-ceramic physicochemical properties, in the aim to define the optimal way to tailor the behavior of such biomaterials for biomedical applications.
As mentioned before, Mg and Fe are elements with an interesting set of biological properties that can be beneficial for bone tissue engineering, therefore, the effect on biological properties, such as the antibacterial activity, and cytotoxicity test, will be studied in upcoming works to validate the biocompatibility of the doped materials and to evaluate their effect on antibacterial activity and bone regeneration.
References
Hench LL (1991) Bioceramics: from concept to clinic. J Am Ceram Soc 74:1487–1510. https://doi.org/10.1111/j.1151-2916.1991.tb07132.x
Jones JR (2013) Review of bioactive glass: from hench to hybrids. Acta Biomater 9:4457–4486. https://doi.org/10.1016/j.actbio.2012.08.023
Kitsugi T, Yamamuro T, Nakamura T, Kokubo T (1989) Bone bonding behavior of MgO-CaO-SiO2-P2O5-CaF2 glass (mother glass of A.W-glass-ceramics). J Biomed Mater Res 23:631–648. https://doi.org/10.1002/jbm.820230607
Ohura K, Nakamura T, Yamamuro T, Kokubo T, Ebisawa Y, Kotoura Y, Oka M (1991) Bone-bonding ability of P2O5-free CaO.SiO2 glasses. J Biomed Mater Res 25:357–365. https://doi.org/10.1002/jbm.820250307
Karlsson KH, Fröberg K, Ringbom T (1989) A structural approach to bone adhering of bioactive glasses. J Non-Crystalline Solids Proc Third Int Symp Glass 112:69–72. https://doi.org/10.1016/0022-3093(89)90495-X
Ohura K, Ikenaga M, Nakamura T, Yamamuro T, Ebisawa Y, Kokubo T, Kotoura Y, Oka M (1991) A heat-generating bioactive glass-ceramic for hyperthermia. J Appl Biomater 2:153–159. https://doi.org/10.1002/jab.770020303
Kitsugi T, Yamamuro T, Nakamura T, Higashi S, Kakutani Y, Hyakuna K, Ito S, Kokubo T, Takagi M, Shibuya T (1986) Bone bonding behavior of three kinds of apatite containing glass ceramics. J Biomed Mater Res 20:1295–1307. https://doi.org/10.1002/jbm.820200906
Kitsugi T, Nakamura T, Yamamura T, Kokubu T, Shibuya T, Takagi M (1987) SEM-EPMA observation of three types of apatite-containing glass-ceramics implanted in bone: the variance of a Ca-P-rich layer. J Biomed Mater Res 21:1255–1271. https://doi.org/10.1002/jbm.820211008
Ohtsuki C, Kushitani H, Kokubo T, Kotani S, Yamamuro T (1991) Apatite formation on the surface of Ceravital-type glass-ceramic in the body. J Biomed Mater Res 25:1363–1370. https://doi.org/10.1002/jbm.820251105
Valerio P, Pereira MM, Goes AM, Leite MF (2004) The effect of ionic products from bioactive glass dissolution on osteoblast proliferation and collagen production. Biomaterials 25:2941–2948. https://doi.org/10.1016/j.biomaterials.2003.09.086
Li P, Nakanishi K, Kokubo T, de Groot K (1993) Induction and morphology of hydroxyapatite, precipitated from metastable simulated body fluids on sol-gel prepared silica. Biomaterials 14:963–968. https://doi.org/10.1016/0142-9612(93)90186-6
Vallet-Regí M, Izquierdo-Barba I, Salinas AJ (1999) Influence of P2O5 on crystallinity of apatite formed in vitro on surface of bioactive glasses. J Biomed Mater Res 46:560–565. https://doi.org/10.1002/(sici)1097-4636(19990915)46:4<560::aid-jbm14>3.0.co;2-m
Vallet-Regí M, Salinas AJ, Román J, Gil M (1999) Effect of magnesium content on the in vitro bioactivity of CaO-MgO-SiO2-P2O5 sol-gel glasses. J Mater Chem 9:515–518. https://doi.org/10.1039/A808679F
Laczka M, Cholewa-Kowalska K, Laczka-Osyczka A, Tworzydlo M, Turyna B (2000) Gel-derived materials of a CaO-P2O5-SiO2 system modified by boron, sodium, magnesium, aluminum, and fluorine compounds. J Biomed Mater Res 52:601–612. https://doi.org/10.1002/1097-4636(20001215)52:4<601::aid-jbm4>3.0.co;2-i
Jokinen M, Rahiala H, Rosenholm JB, Peltola T, Kangasniemi I (1998) Relation Between Aggregation and Heterogeneity of Obtained Structure in Sol-Gel Derived CaO-P2O5-SiO2. J Sol-Gel Sci Technol 12:159–167. https://doi.org/10.1023/A:1008642117227
Hoppe A, Güldal NS, Boccaccini AR (2011) A review of the biological response to ionic dissolution products from bioactive glasses and glass-ceramics. Biomaterials 32:2757–2774. https://doi.org/10.1016/j.biomaterials.2011.01.004
Kargozar S, Baino F, Hamzehlou S, Hill RG, Mozafari M (2018) Bioactive glasses entering the mainstream. Drug Discov Today 23:1700–1704. https://doi.org/10.1016/j.drudis.2018.05.027
Letaief N, Lucas-Girot A, Oudadesse H, Meleard P, Pott T, Jelassi J, Dorbez-Sridi R (2014) Effect of aging temperature on the structure, pore morphology and bioactivity of new sol–gel synthesized bioglass. J Non-Crystalline Solids 402:194–199. https://doi.org/10.1016/j.jnoncrysol.2014.06.005
Yan H, Zhang K, Blanford CF, Francis LF, Stein A (2001) In Vitro Hydroxycarbonate Apatite Mineralization of CaO−SiO2 Sol−Gel Glasses with a Three-Dimensionally Ordered Macroporous Structure. Chem Mater 13:1374–1382. https://doi.org/10.1021/cm000895e
Arcos D, Greenspan DC, Vallet-Regí M (2002) Influence of the Stabilization Temperature on Textural and Structural Features and Ion Release in SiO2−CaO−P2O5 Sol−Gel Glasses. Chem Mater 14:1515–1522. https://doi.org/10.1021/cm011119p
Cho S-B, Nakanishi K, Kokubo T, Soga N, Ohtsuki C, Nakamura T, Kitsugi T, Yamamuro T (1995) Dependence of apatite formation on silica gel on its structure: effect of heat treatment. J Am Ceram Soc 78:1769–1774. https://doi.org/10.1111/j.1151-2916.1995.tb08887.x
Pereira MM, Clark AE, Hench LL (1995) Effect of texture on the rate of hydroxyapatite formation on gel-silica surface. J Am Ceram Soc 78:2463–2468. https://doi.org/10.1111/j.1151-2916.1995.tb08686.x
Aneb K, Oudadesse H, Khireddine H, Lefeuvre B, Merdrignac-Conanec O, Tessier F, Lucas A (2023) Study of the effect of ordered porosity and surface silanization on in vitro bioactivity of sol-gel-derived bioactive glasses. Mater Today Commun 34:104992. https://doi.org/10.1016/j.mtcomm.2022.104992
Rocton N, Oudadesse H, Mosbahi S, Bunetel L, Pellen-Mussi P, Lefeuvre B (2019) Study of nano bioactive glass for use as bone biomaterial comparison with micro bioactive glass behaviour. IOP Conf Ser: Mater Sci Eng 628:012005. https://doi.org/10.1088/1757-899X/628/1/012005
Brink M, Turunen T, Happonen RP, Yli-Urpo A (1997) Compositional dependence of bioactivity of glasses in the system Na2O-K2O-MgO-CaO-B2O3-P2O5-SiO2. J Biomed Mater Res 37:114–121. https://doi.org/10.1002/(sici)1097-4636(199710)37:1<114::aid-jbm14>3.0.co;2-g
Ogino M, Ohuchi F, Hench LL (1980) Compositional dependence of the formation of calcium phosphate films on bioglass. J Biomed Mater Res 14:55–64. https://doi.org/10.1002/jbm.820140107
Mezahi F-Z, Lucas-Girot A, Oudadesse H, Harabi A (2013) Reactivity kinetics of 52S4 glass in the quaternary system SiO2–CaO–Na2O–P2O5: Influence of the synthesis process: Melting versus sol–gel. J Non-Crystalline Solids 361:111–118. https://doi.org/10.1016/j.jnoncrysol.2012.10.013
Mabrouk M, Mostafa A, Oudadesse H, Wers E, Lucas-Girot A, El-Gohary MI (2014) Comparative Study of Nanobioactive Glass Quaternary System 46S6. Bioceramics Dev Appl 4:1–4. https://doi.org/10.4172/2090-5025.1000072
Letaief N, Lucas-Girot A, Oudadesse H, Dorbez-Sridi R (2014) Influence of Synthesis Parameters on the Structure, Pore Morphology and Bioactivity of a New Mesoporous Glass. J Biosci Med 2:57–63. https://doi.org/10.4236/jbm.2014.22009
Letaief N, Lucas-Girot A, Oudadesse H, Dorbez-Sridi R (2014) New 92S6 mesoporous glass: Influence of surfactant carbon chain length on the structure, pore morphology and bioactivity. Mater Res Bull 60:882–889. https://doi.org/10.1016/j.materresbull.2014.08.048
Wu C, Chang J (2014) Multifunctional mesoporous bioactive glasses for effective delivery of therapeutic ions and drug/growth factors. J Controlled Rel 193:282–295. https://doi.org/10.1016/j.jconrel.2014.04.026
Rocton N, Oudadesse H, Lefeuvre B (2018) Comparison of Zn and Sr effects on thermal properties and on the excess entropy of doped glasses for use in the biomedical field. Thermochim Acta 668:58–64. https://doi.org/10.1016/j.tca.2018.08.008
Oudadesse H, Dietrich E, Gal YL, Pellen P, Bureau B, Mostafa AA, Cathelineau G (2011) Apatite forming ability and cytocompatibility of pure and Zn-doped bioactive glasses. Biomed Mater 6:035006. https://doi.org/10.1088/1748-6041/6/3/035006
Miola M, Verné E, Vitale-Brovarone C, Baino F (2016) Antibacterial bioglass-derived scaffolds: innovative synthesis approach and characterization. Int J Appl Glass Sci 7:238–247. https://doi.org/10.1111/ijag.12209
Hoppe A, Mouriño V, Boccaccini AR (2013) Therapeutic inorganic ions in bioactive glasses to enhance bone formation and beyond. Biomater Sci 1:254–256. https://doi.org/10.1039/c2bm00116k
Mouriño V, Cattalini JP, Boccaccini AR (2012) Metallic ions as therapeutic agents in tissue engineering scaffolds: an overview of their biological applications and strategies for new developments. J R Soc Interface 9:401–419. https://doi.org/10.1098/rsif.2011.0611
Tripathi H, Rath C, Kumar AS, Manna PP, Singh SP (2019) Structural, physico-mechanical and in-vitro bioactivity studies on SiO2–CaO–P2O5–SrO–Al2O3 bioactive glasses. Mater Sci Eng C 94:279–290. https://doi.org/10.1016/j.msec.2018.09.041
Karakuzu-İkizler B, Terzioğlu P, Oduncu-Tekerek BS, Yücel S (2020) Effect of selenium incorporation on the structure and in vitro bioactivity of 45S5 bioglass. J Aust Ceram Soc 56:697–709. https://doi.org/10.1007/s41779-019-00388-6
Deliormanlı AM (2015) Synthesis and characterization of cerium- and gallium-containing borate bioactive glass scaffolds for bone tissue engineering. J Mater Sci: Mater Med 26:67. https://doi.org/10.1007/s10856-014-5368-0
Dietrich E, Oudadesse H, Lucas-Girot A, Le Gal Y, Jeanne S, Cathelineau G (2008) Effects of Mg and Zn on the surface of doped melt-derived glass for biomaterials applications. Appl Surf Sci First Int Symp Surf Interfaces Biomater 255:391–395. https://doi.org/10.1016/j.apsusc.2008.06.094
El-Batal FH, Khalil EM, Hamdy YM, Zidan HM, Aziz MS, Abdelghany AM (2010) FTIR spectral analysis of corrosion mechanisms in soda lime silica glasses doped with transition metal oxides. Silicon 2:41–47. https://doi.org/10.1007/s12633-010-9037-8
Gupta N, Santhiya D, Murugavel S, Kumar A, Aditya A, Ganguli M, Gupta S (2018) Effects of transition metal ion dopants (Ag, Cu and Fe) on the structural, mechanical and antibacterial properties of bioactive glass. Colloids Surf A: Physicochem Eng Asp 538:393–403. https://doi.org/10.1016/j.colsurfa.2017.11.023
Maguire ME, Cowan JA (2002) Magnesium chemistry and biochemistry. Biometals 15:203–210. https://doi.org/10.1023/A:1016058229972
Dermience M, Lognay G, Mathieu F, Goyens P (2015) Effects of thirty elements on bone metabolism. J Trace Elem Med Biol 32:86–106. https://doi.org/10.1016/j.jtemb.2015.06.005
Oudadesse H, Martin S, Derrien AC, Lucas-Girot A, Cathelineau G, Blondiaux G (2004) Determination of Ca, P, Sr and Mg in the synthetic biomaterial aragonite by NAA. J Radioanal Nucl Chem 262:479–483. https://doi.org/10.1023/B:JRNC.0000046781.15503.eb
Okuma T (2001) Magnesium and bone strength. Nutrition 17:679–680. https://doi.org/10.1016/s0899-9007(01)00551-2
Zreiqat H, Howlett CR, Zannettino A, Evans P, Schulze-Tanzil G, Knabe C, Shakibaei M (2002) Mechanisms of magnesium-stimulated adhesion of osteoblastic cells to commonly used orthopaedic implants. J Biomed Mater Res 62:175–184. https://doi.org/10.1002/jbm.10270
Yamasaki Y, Yoshida Y, Okazaki M, Shimazu A, Uchida T, Kubo T, Akagawa Y, Hamada Y, Takahashi J, Matsuura N (2002) Synthesis of functionally graded MgCO3 apatite accelerating osteoblast adhesion. J Biomed Mater Res 62:99–105. https://doi.org/10.1002/jbm.10220
Oliveira J, Correia RN, Fernandes M, Rocha J (2000) Influence of the CaO/MgO ratio on the structure of phase-separated glasses: A solid state29Si and31P MAS NMR study. J Non-Crystalline Solids 265:221–229. https://doi.org/10.1016/S0022-3093(99)00957-6
Wu C, Chang J, Wang J, Ni S, Zhai W (2005) Preparation and characteristics of a calcium magnesium silicate (bredigite) bioactive ceramic. Biomaterials 26:2925–2931. https://doi.org/10.1016/j.biomaterials.2004.09.019
Webster TJ, Ergun C, Doremus RH, Bizios R (2002) Hydroxylapatite with substituted magnesium, zinc, cadmium, and yttrium. II. Mechanisms of osteoblast adhesion. J Biomed Mater Res 59:312–317. https://doi.org/10.1002/jbm.1247
Liu CC, Yeh JK, Aloia JF (1988) Magnesium directly stimulates osteoblast proliferation. J Bone Miner Res 3:104
Saboori A, Rabiee M, Moztarzadeh F, Sheikhi M, Tahriri M, Karimi M (2009) Synthesis, characterization and in vitro bioactivity of sol-gel-derived SiO2–CaO–P2O5–MgO bioglass. Mater Sci Eng: C 29:335–340. https://doi.org/10.1016/j.msec.2008.07.004
Althoff J, Quint P, Krefting ER, Höhling HJ (1982) Morphological studies on the epiphyseal growth plate combined with biochemical and X-ray microprobe analyses. Histochemistry 74:541–552. https://doi.org/10.1007/BF00496668
Rude RK, Singer FR, Gruber HE (2009) Skeletal and hormonal effects of magnesium deficiency. J Am Coll Nutr 28:131–141. https://doi.org/10.1080/07315724.2009.10719764
Notomi T, Kuno M, Hiyama A, Nozaki T, Ohura K, Ezura Y, Noda M (2017) Role of lysosomal channel protein TPC2 in osteoclast differentiation and bone remodeling under normal and low-magnesium conditions. J Biol Chem 292:20998–21010. https://doi.org/10.1074/jbc.M117.780072
Dietrich E, Oudadesse H, Lucas-Girot A, Mami M (2009) In vitro bioactivity of melt-derived glass 46S6 doped with magnesium. J Biomed Mater Res A 88:1087–1096. https://doi.org/10.1002/jbm.a.31901
Moghanian A, Sedghi A, Ghorbanoghli A, Salari E (2018) The effect of magnesium content on in vitro bioactivity, biological behavior and antibacterial activity of sol–gel derived 58S bioactive glass. Ceram Int 44:9422–9432. https://doi.org/10.1016/j.ceramint.2018.02.159
Erol M, Özyuguran A, Çelebican Ö (2010) Synthesis, Characterization, and In Vitro Bioactivity of Sol-Gel-Derived Zn, Mg, and Zn-Mg Co-Doped Bioactive Glasses. Chem Eng Technol 33:1066–1074. https://doi.org/10.1002/ceat.200900495
Ma J, Chen CZ, Wang DG, Jiao Y, Shi JZ (2010) Effect of magnesia on the degradability and bioactivity of sol–gel derived SiO2–CaO–MgO–P2O5 system glasses. Colloids Surf B: Biointerfaces 81:87–95. https://doi.org/10.1016/j.colsurfb.2010.06.022
Moya JS, Tomsia AP, Pazo A, Santos C, Guitián F (1994) In vitro formation of hydroxylapatite layer in a MgO-containing glass. J Mater Sci: Mater Med 5:529–532. https://doi.org/10.1007/BF00124885
Medeiros DM, Stoecker B, Plattner A, Jennings D, Haub M (2004) Iron deficiency negatively affects vertebrae and femurs of rats independently of energy intake and body weight. J Nutr 134:3061–3067. https://doi.org/10.1093/jn/134.11.3061
Parelman M, Stoecker B, Baker A, Medeiros D (2006) Iron Restriction Negatively Affects Bone in Female Rats and Mineralization of hFOB Osteoblast Cells. Exp Biol Med (Maywood) 231:378–386. https://doi.org/10.1177/153537020623100403
Díaz-Castro J, López-Frías MR, Campos MS, López-Frías M, Alférez MJM, Nestares T, Ojeda ML, López-Aliaga I (2012) Severe nutritional iron-deficiency anaemia has a negative effect on some bone turnover biomarkers in rats. Eur J Nutr 51:241–247. https://doi.org/10.1007/s00394-011-0212-5
Guggenbuhl P, Deugnier Y, Boisdet JF, Rolland Y, Perdriger A, Pawlotsky Y, Chalès G (2005) Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos Int 16:1809–1814. https://doi.org/10.1007/s00198-005-1934-0
Yamasaki K, Hagiwara H (2009) Excess iron inhibits osteoblast metabolism. Toxicol Lett 191:211–215. https://doi.org/10.1016/j.toxlet.2009.08.023
Melchers S, Uesbeck T, Winter O, Eckert H, Eder D (2016) Effect of Aluminum Ion Incorporation on the Bioactivity and Structure in Mesoporous Bioactive Glasses. Chem Mater 28:3254–3264. https://doi.org/10.1021/acs.chemmater.5b04117
Hench LL (1998) Bioceramics. J Am Ceram Soc 81:1705–1728. https://doi.org/10.1111/j.1151-2916.1998.tb02540.x
Kokubo T, Ebisawa Y, Sugimoto Y, Kiyama M, Ohura K, Yamamuro T, Hiraoka M, Abe M (1992) Preparation of bioactive and ferromagnetic glass-ceramic for hyperthermia. Bioceramics 5:213–223
Da Li G, Zhou DL, Lin Y, Pan TH, Chen GS, Yin QD (2010) Synthesis and characterization of magnetic bioactive glass-ceramics containing Mg ferrite for hyperthermia. Mater Sci Eng: C 30:148–153. https://doi.org/10.1016/j.msec.2009.09.011
Martin RA, Twyman HL, Rees GJ, Smith JM, Barney ER, Smith ME, Hanna JV, Newport RJ (2012) A structural investigation of the alkali metal site distribution within bioactive glass using neutron diffraction and multinuclear solid state NMR. Phys Chem Chem Phys 14:12105–12113. https://doi.org/10.1039/C2CP41725A
Srivastava A, Pyare R, Singh S (2012) In vitro bioactivity and physical - mechanical properties of Fe2O3 substituted 45S5 Bioactive Glasses and Glass – ceramics. Int J Sci Eng Res 2:249–258
Sales BC, Boatner LA (1984) Lead phosphate glass as a stable medium for the immobilization and disposal of high-level nuclear waste. Mater Lett 2:301–304. https://doi.org/10.1016/0167-577X(84)90038-7
El-Meliegy E, Mabrouk M, El-Sayed SA, Abd El-Hady BM, Shehata MR, Hosny WM (2018) Novel Fe2O3-doped glass/chitosan scaffolds for bone tissue replacement. Ceram Int 44:9140–9151. https://doi.org/10.1016/j.ceramint.2018.02.122
Singh R, Kothiyal G, Srinivasan A (2009) In vitro evaluation of bioactivity of CaO–SiO2–P2O5–Na2O–Fe2O3 glasses. Appl Surf Sci 255:6827–6831. https://doi.org/10.1016/j.apsusc.2009.02.089
Li R, Clark AE, Hench LL In Chemical Processing of Advanced Materials, Hench L.L. and West J.K. eds John Wiley & Sons, New York, 1992, 627.
Li P, Ohtsuki C, Kokubo T, Nakanishi K, Soga N, Nakamura T, Yamamuro T (1992) Apatite formation induced by silica gel in a simulated body fluid. J Am Ceram Soc 75:2094–2097. https://doi.org/10.1111/j.1151-2916.1992.tb04470.x
Li P, Ohtsuki C, Kokubo T, Nakanishi K, Soga N, de Groot K (1994) The role of hydrated silica, titania, and alumina in inducing apatite on implants. J Biomed Mater Res 28:7–15. https://doi.org/10.1002/jbm.820280103
Jones RW (1989) Fundamental Principles of Sol–Gel Technology, first ed. The Institute of Metals, London
Brinker C, Scherer G (1990) Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing, first ed. Academic Press, San Diego
Balamurugan A, Balossier G, Michel J, Kannan S, Benhayoune H, Rebelo AHS, Ferreira JMF (2007) Sol gel derived SiO2-CaO-MgO-P2O5 bioglass system–preparation and in vitro characterization. J Biomed Mater Res – Part B: Appl Biomater 83:546–553. https://doi.org/10.1002/jbm.b.30827
Kokubo T, Kushitani H, Sakka S, Kitsugi T, Yamamuro T (1990) Solutions able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W. J Biomed Mater Res 24:721–734. https://doi.org/10.1002/jbm.820240607
Kokubo T, Takadama H (2006) How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27:2907–2915. https://doi.org/10.1016/j.biomaterials.2006.01.017
Siqueira RL, Costa LC, Schiavon MA, de Castro DT, dos Reis AC, Peitl O, Zanotto ED (2017) Bioglass® and resulting crystalline materials synthesized via an acetic acid-assisted sol–gel route. J Sol-Gel Sci Technol 83:165–173
Morsi, R., Ibrahim, S., Morsi, M., 2014. Electrical properties of silicate glasses of low level gadolinium oxide doping including dielectric and infrared measures. J Mater Sci: Mater Electr 26. https://doi.org/10.1007/s10854-014-2556-0
ElBatal HA, Azooz MA, Khalil EMA, Soltan Monem A, Hamdy YM (2003) Characterization of some bioglass–ceramics. Mater Chem Phys 80:599–609. https://doi.org/10.1016/S0254-0584(03)00082-8
Wang M, Cheng J, Li M, He F (2011) Structure and properties of soda lime silicate glass doped with rare earth. Phys B: Condens Matter 406:187–191. https://doi.org/10.1016/j.physb.2010.10.040
El-Alaily NA (2003) Study of some properties of lithium silicate glass and glass ceramics containing blast furnace slag. Glass Technol 44:30–35
Li Y, Liang K, Cao J, Xu B (2010) Spectroscopy and structural state of V4+ ions in lithium aluminosilicate glass and glass–ceramics. J Non-Crystalline Solids 356:502–508. https://doi.org/10.1016/j.jnoncrysol.2009.12.018
Annapurna K, Das M, Kundu P, Dwivedi RN, Buddhudu S (2005) Spectral properties of Eu3+: ZnO–B2O3–SiO2 glasses. J Mol Struct 741:53–60. https://doi.org/10.1016/j.molstruc.2005.01.062
Jastrzębski W, Sitarz M, Rokita M, Bułat K (2011) Infrared spectroscopy of different phosphates structures. Spectrochim Acta Part A: Mol Biomol Spectrosc, Xth Int Conf Mol Spectrosc 79:722–727. https://doi.org/10.1016/j.saa.2010.08.044
Feller S, Lodden G, Riley A, Edwards T, Croskrey J, Schue A, Liss D, Stentz D, Blair S, Kelley M, Smith G, Singleton S, Affatigato M, Holland D, Smith ME, Kamitsos EI, Varsamis CPE, Ioannou E (2010) A multispectroscopic structural study of lead silicate glasses over an extended range of compositions. J Non Crystalline Solids 356:304–313. https://doi.org/10.1016/j.jnoncrysol.2009.12.003
Elgayar I, Aliev A, Boccaccini A, Hill R (2005) Structural analysis of bioactive glasses. J Non-Crystalline Solids 351:173–183. https://doi.org/10.1016/j.jnoncrysol.2004.07.067
Lucas-Girot A, Mezahi F-Z, Mami M, Oudadesse H, Harabi A, Le Floch M (2011) Sol-gel synthesis of a new composition of bioactive glass in the quaternary system SiO2-CaO-Na2O-P2O5. J Non-Crystalline Solids 357:3322–3327. https://doi.org/10.1016/j.jnoncrysol.2011.06.002
Sing KSW (1985) Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl Chem 57:603–619. https://doi.org/10.1351/pac198557040603
Mami M, Lucas-Girot A, Oudadesse H, Dorbez-Sridi R, Mezahi F, Dietrich E (2008) Investigation of the surface reactivity of a sol–gel derived glass in the ternary system SiO2–CaO–P2O5. Appl Surf Sci 254:7386–7393. https://doi.org/10.1016/j.apsusc.2008.05.340
Gentleman E, Stevens MM, Hill RG, Brauer DS (2013) Surface properties and ion release from fluoride-containing bioactive glasses promote osteoblast differentiation and mineralization in vitro. Acta Biomaterialia 9:5771–5779. https://doi.org/10.1016/j.actbio.2012.10.043
Popescu RA, Magyari K, Vulpoi A, Trandafir DL, Licarete E, Todea M, Ştefan R, Voica C, Vodnar DC, Simon S, Papuc I, Baia L (2016) Bioactive and biocompatible copper containing glass-ceramics with remarkable antibacterial properties and high cell viability designed for future in vivo trials. Biomater Sci 4:1252–1265. https://doi.org/10.1039/c6bm00270f
Stan G, Pasuk I, Husanu M-A, Enculescu I, Pina S, Lemos A, Tulyaganov D, El Mabrouk K, Ferreira J (2011) Highly adherent bioactive glass thin films synthetized by magnetron sputtering at low temperature. J Mater Sci Mater Med 22:2693–710. https://doi.org/10.1007/s10856-011-4441-1
Mozafari M, Moztarzadeh F, Tahriri M (2010) Investigation of the physico-chemical reactivity of a mesoporous bioactive SiO2–CaO–P2O5 glass in simulated body fluid. J Non-Crystalline Solids 356:1470–1478. https://doi.org/10.1016/j.jnoncrysol.2010.04.040
Oyane A, Onuma K, Ito A, Kim H-M, Kokubo T, Nakamura T (2003) Formation and growth of clusters in conventional and new kinds of simulated body fluids. J Biomed Mater Res - Part A 64:339–348. https://doi.org/10.1002/jbm.a.10426
Jutarosaga T, Jeoung JS, Seraphin S (2005) Infrared spectroscopy of Si–O bonding in low-dose low-energy separation by implanted oxygen materials. Thin Solid Films 476:303–311. https://doi.org/10.1016/j.tsf.2004.10.006
Luginina M, Orru R, Cao G, Grossin D, Brouillet F, Chevallier G, Thouron C, Drouet C (2020) First successful stabilization of consolidated amorphous calcium phosphate (ACP) by cold sintering: toward highly-resorbable reactive bioceramics. J Mater Chem B 8:629–635. https://doi.org/10.1039/C9TB02121C
Combes C, Rey C (2010) Amorphous calcium phosphates: synthesis, properties and uses in biomaterials. Acta Biomater 6:3362–3378. https://doi.org/10.1016/j.actbio.2010.02.017
Vecstaudza J, Gasik M, Locs J (2019) Amorphous calcium phosphate materials: Formation, structure and thermal behaviour. J Eur Ceram Soc 39:1642–1649. https://doi.org/10.1016/j.jeurceramsoc.2018.11.003
Termine JD, Posner AS (1966) Infra-red determination of the percentage of crystallinity in apatitic calcium phosphates. Nature 211:268–270. https://doi.org/10.1038/211268a0
Ohtsuki C, Kokubo T, Yamamuro T (1992) Mechanism of apatite formation on CaO-SiO2-P2O5 glasses in a simulated body fluid. J Non-Crystalline Solids 143:84–92. https://doi.org/10.1016/S0022-3093(05)80556-3
LeGeros RZ (1991) Calcium phosphates in oral biology and medicine. Monogr Oral Sci 15:1–201
Li P, Ohtsuki C, Kokubo T, Nakanishi K, Soga N, Nakamura T, Yamamuro T (1993) Process of formation of bone-like apatite layer on silica gel. J Mater Sci: Mater Med 4:127–131. https://doi.org/10.1007/BF00120381
Manoj M, Mangalaraj D, Ponpandian N, Viswanathan C (2015) Core–shell hydroxyapatite/Mg nanostructures: surfactant free facile synthesis, characterization and their in vitro cell viability studies against leukaemia cancer cells (K562). RSC Adv 5:48705–48711. https://doi.org/10.1039/C5RA04663G
Crovace MC, Souza MT, Chinaglia CR, Peitl O, Zanotto ED (2016) Biosilicate® — A multipurpose, highly bioactive glass-ceramic. In vitro, in vivo and clinical trials. J Non-Crystalline Solids Glasses Healthc 432:90–110. https://doi.org/10.1016/j.jnoncrysol.2015.03.022
Peitl O, Zanotto E, Hench L (2001) Highly bioactive P2O5-Na2O-CaO-SiO2 glass-ceramics. J Non-Crystalline Solids 292:115–126. https://doi.org/10.1016/S0022-3093(01)00822-5
Hench LL, Polak JM (2002) Third-generation biomedical materials. Science 295:1014–1017. https://doi.org/10.1126/science.1067404
Wang X, Kong R, Pan X, Xu H, Xia D, Shan H, Lu JR (2009) Role of Ovalbumin in the Stabilization of Metastable Vaterite in Calcium Carbonate Biomineralization. J Phys Chem B 113:8975–8982. https://doi.org/10.1021/jp810281f
Andersen FA, Brecevic L (1991) Infrared spectra of amorphous and crystalline calcium carbonate. Acta Chem Scand 45:1018–1024
Lei B, Chen X, Wang Y, Zhao N, Du C, Fang L (2010) Influence of sintering temperature on pore structure and apatite formation of a sol-gel-derived bioactive glass. J Am Ceram Soc 93:32–35. https://doi.org/10.1111/j.1551-2916.2009.03358.x
Lee I-H, Shin S-H, Foroutan F, Lakhkar NJ, Gong M-S, Knowles JC (2013) Effects of magnesium content on the physical, chemical and degradation properties in a MgO−CaO−Na2O−P2O5 glass system. J Non-Crystalline Solids 363:57–63. https://doi.org/10.1016/j.jnoncrysol.2012.11.036
Labbilta T, Mesnaoui M, Aouad H, Abouliatim Y, Khouloud M, Abielaala L (2020) Study of the effect of calcium substitution by magnesium in the vitreous system 3P2O5-2K2O-(1 - x) CaO-x MgO. Mater (Basel) 13:2637. https://doi.org/10.3390/ma13112637
De Araujo Bastos Santana L, Oliveira Junior PH, Damia C, dos Santos Tavares D, dos Santos EA (2021) Bioactivity in SBF versus trace element effects: The isolated role of Mg2+ and Zn2+ in osteoblast behavior. Mater Sci Eng: C 118:111320. https://doi.org/10.1016/j.msec.2020.111320
Ouellette RJ, Rawn JD (2015) 3 - Introduction to Organic Reaction Mechanisms. In: Ouellette RJ, Rawn JD (Eds.) Organic Chemistry Study Guide. Elsevier, Boston, p 31–46. https://doi.org/10.1016/B978-0-12-801889-7.00003-0
Watts SJ, Hill RG, O’Donnell MD, Law RV (2010) Influence of magnesia on the structure and properties of bioactive glasses. J Non-Crystalline Solids 356:517–524. https://doi.org/10.1016/j.jnoncrysol.2009.04.074
Arslan G, Karasu B, Dolekcekic E, Kaya G, Gunkaya G (2009) Effect of transition metal oxide additions on the chemical durability of SrO-MgO-ZrO2 SiO2 glasses. European. J Glass Sci Technol Part a Glass Technol 50:17–24
Cicconi MR, Giuli G, Ertel-Ingrisch W, Paris E, Dingwell DB (2015) The effect of the [Na/(Na+K)] ratio on Fe speciation in phonolitic glasses. Am Mineralogist 100:1610–1619. https://doi.org/10.2138/am-2015-5155
Kuryaeva RG (2009) The state of magnesium in silicate glasses and melts. Glass Phys Chem 35:378–383. https://doi.org/10.1134/S1087659609040051
Burkhard D (2000) Iron-bearing silicate glasses at ambient conditions. J Non-Crystalline Solids 275:175–188. https://doi.org/10.1016/S0022-3093(00)00252-0
Farges F, Lefrère Y, Rossano S, Berthereau A, Calas G, Brown Jr G-E (2004) The effect of redox state on the local structural environment of iron in silicate glasses: a combined XAFS spectroscopy, molecular dynamics, and bond valence study. J Non-Crystalline Solids 344:176–188. https://doi.org/10.1016/j.jnoncrysol.2004.07.050
Mysen BO, Seifert F, Virgo D (1980) Structure and redox equilibria of iron-bearing silicate melts. Am Mineralogist 65:867–884
Virgo D, Mysen BO (1985) The structural state of iron in oxidized vs. reduced glasses at 1 atm: A57Fe Mössbauer study. Phys Chem Miner 12:65–76. https://doi.org/10.1007/BF01046829
Holland D, Mekki A, Gee IA, McConville CF, Johnson JA, Johnson CE, Appleyard P, Thomas M (1999) The structure of sodium iron silicate glass – a multi-technique approach. J Non-Crystalline Solids 253:192–202. https://doi.org/10.1016/S0022-3093(99)00353-1
Wang Z, Ni W, Jia Y, Zhu L, Huang X (2010) Crystallization behavior of glass ceramics prepared from the mixture of nickel slag, blast furnace slag and quartz sand. J Non Crystalline Solids 356:1554–1558. https://doi.org/10.1016/j.jnoncrysol.2010.05.063
Lin S, Ionescu C, Pike KJ, Smith ME, Jones JR (2009) Nanostructure evolution and calcium distribution in sol–gel derived bioactive glass. J Mater Chem 19:1276–1282. https://doi.org/10.1039/B814292K
Borges R, Oliveira JSS, Queiroz AP, Zambanini T, Hanashiro AM, Lima NB, Schneider JF, Marchi J (2023) On the structure of Ag-containing sol-gel bioactive glasses: A surface crystal growth of metallic silver removes its network modifier role in the glass structure. Open Ceram 16:100449. https://doi.org/10.1016/j.oceram.2023.100449
Ma J, Chen CZ, Wang DG, Meng XG, Shi JZ (2010) In vitro degradability and bioactivity of mesoporous CaO-MgO-P2O5-SiO2 glasses synthesized by sol–gel method. J Sol-Gel Sci Technol 1:69–76. https://doi.org/10.1007/s10971-010-2159-z
Cacciotti I, Lombardi M, Bianco A, Ravaglioli A, Montanaro L (2012) Sol-gel derived 45S5 bioglass: synthesis, microstructural evolution and thermal behaviour. J Mater Sci: Mater Med 23:1849–1866. https://doi.org/10.1007/s10856-012-4667-6
Nawaz Q, de Pablos-Martín A, Martins de Souza e Silva J, Hurle K, Jaimes ATC, Brauer DS, Boccaccini AR (2020) New insights into the crystallization process of sol-gel–derived 45S5 bioactive glass. J Am Ceram Soc 103:4234–4247. https://doi.org/10.1111/jace.17124
Cao W, Hench LL (1996) Bioactive materials. Ceram Int 22:493–507. https://doi.org/10.1016/0272-8842(95)00126-3
Soulié J, Nedelec JM, Jallot E (2009) Influence of Mg doping on the early steps of physico-chemical reactivity of sol–gel derived bioactive glasses in biological medium. Phys Chem Chem Phys 11:10473–10483. https://doi.org/10.1039/B913771H
Ding H, Pan H, Xu X, Tang R (2014) Toward a detailed understanding of magnesium ions on hydroxyapatite crystallization inhibition. Cryst Growth Des 14:763–769. https://doi.org/10.1021/cg401619s
Oyane A, Kim H-M, Furuya T, Kokubo T, Miyazaki T, Nakamura T (2003) Preparation and assessment of revised simulated body fluids. J Biomed Mater Res Part A 65:188–195. https://doi.org/10.1002/jbm.a.10482
Chajri S, Bouhazma S, Adouar I, Herradi S, Khaldi M, El Bali B, Lachkar M (2019) Synthesis, characterization and evaluation of bioactivity of glasses in the CaO-SiO2 -P2O5 -MgO system with different CaO/MgO ratios. J Phys: Conf Ser 1292:012013. https://doi.org/10.1088/1742-6596/1292/1/012013
Blumenthal NC, Posner AS (1973) Hydroxyapatite: Mechanism of formation and properties. Calcif Tissue Res 13:235–243. https://doi.org/10.1007/BF02015413
Eanes ED, Gillessen IH, Posner AS (1965) Intermediate States in the Precipitation of Hydroxyapatite. Nature 208:365–367. https://doi.org/10.1038/208365a0
Oliveira JM, Correia RN, Fernandes MH (2002) Effects of Si speciation on the in vitro bioactivity of glasses. Biomaterials 23:371–379. https://doi.org/10.1016/S0142-9612(01)00115-6
George AM, Stebbins JF (1998) Structure and dynamics of magnesium in silicate melts: A high-temperature 25Mg NMR study. Am Mineralogist 83:1022–1029. https://doi.org/10.2138/am-1998-9-1010
Shimoda K, Tobu Y, Hatakeyama M, Nemoto T, Saito K (2007) Structural investigation of Mg local environments in silicate glasses by ultra-high field Mg-25 3QMAS NMR spectroscopy. Am Mineralogist 92:695–698. https://doi.org/10.2138/am.2007.2535
Zhao Y, Song M, Liu J (2008) Characteristics of bioactive glass coatings obtained by pulsed laser deposition. Surf Interface Anal 40:1463–1468. https://doi.org/10.1002/sia.2925
Tabia Z, El Mabrouk K, Bricha M, Nouneh K (2019) Mesoporous bioactive glass nanoparticles doped with magnesium: drug delivery and acellular in vitro bioactivity. RSC Adv 9:12232–12246. https://doi.org/10.1039/c9ra01133a
Boskey AL, Posner AS (1974) Magnesium stabilization of amorphous calcium phosphate: A kinetic study. Mater Res Bull 9:907–916. https://doi.org/10.1016/0025-5408(74)90169-X
Abbona F, Franchini-Angela M (1990) Crystallization of calcium and magnesium phosphates from solutions of low concentration. J Cryst Growth 104:661–671. https://doi.org/10.1016/0022-0248(90)90009-A
Cao X, Harris W (2008) Carbonate and magnesium interactive effect on calcium phosphate precipitation. Environ Sci Technol 42:436–442. https://doi.org/10.1021/es0716709
Bigi A, Falini G, Foresti E, Ripamonti A, Gazzano M, Roveri N (1993) Magnesium influence on hydroxyapatite crystallization. J Inorg Biochem 49:69–78. https://doi.org/10.1016/0162-0134(93)80049-F
Yang X, Xie B, Wang L, Qin Y, Henneman Z, Nancollas G (2011) Influence of magnesium ions and amino acids on the nucleation and growth of hydroxyapatite. CrystEngComm 13:1153–1158. https://doi.org/10.1039/C0CE00470G
Vernè E, Bretcanu O, Balagna C, Bianchi C, Cannas M, Gatti S, Vitale-Brovarone C (2008) Early stage reactivity and in vitro behavior of silica-based bioactive glasses and glass-ceramics. J Mater Sci Mater Med 20:75–87. https://doi.org/10.1007/s10856-008-3537-8
Pérez-Pariente J, Balas F, Vallet-Regí M (2000) Surface and chemical study of SiO2·P2O5·CaO·(MgO) bioactive glasses. Chem Mater 12:750–755. https://doi.org/10.1021/cm9911114
Abbona F, Baronnet A (1996) A XRD and TEM study on the transformation of amorphous calcium phosphate in the presence of magnesium. J Cryst Growth 165:98–105. https://doi.org/10.1016/0022-0248(96)00156-X
Jallot E, Moretto PH (2006) Characterisation, by the PIXE method, of trace elements during physicochemical reactions at the periphery of bioactive glass pastilles in contact with biological fluids. Instrum Sci Technol 34:405–416. https://doi.org/10.1080/10739140600648803
Kibalczyc W, Christoffersen J, Christoffersen MR, Zielenkiewicz A, Zielenkiewicz W (1990) The effect of magnesium ions on the precipitation of calcium phosphates. J Cryst Growth 106:355–366. https://doi.org/10.1016/0022-0248(90)90080-5
Ebisawa Y, Sugimoto Y, Hayashi T, Kokubo T, Ohura K, Yamamuro T (1991) Crystallization of (FeO, Fe2O3)-CaO-SiO2 glasses and magnetic properties of their crystallized products. J Ceram Soc Jpn 99:7–13. https://doi.org/10.2109/jcersj.99.7
Kawashita M, Tanaka M, Kokubo T, Inoue Y, Yao T, Hamada S, Shinjo T (2005) Preparation of ferrimagnetic magnetite microspheres for in situ hyperthermic treatment of cancer. Biomaterials 26:2231–2238. https://doi.org/10.1016/j.biomaterials.2004.07.014
Okamoto Y, Hidaka S (1994) Studies on calcium phosphate precipitation: effects of metal ions used in dental materials. J Biomed Mater Res 28:1403–1410. https://doi.org/10.1002/jbm.820281204
Ebisawa Y, Kokubo T, Ohura K, Yamamuro T (1990) Bioactivity of CaO·SiO2-based glasses:in vitro evaluation. J Mater Sci: Mater Med 1:239–244. https://doi.org/10.1007/BF00701083
Krajewski A, Ravaglioli A, Fabbri B, Azzoni CB (1987) Doping influence on the interaction between a bioactive glass and a simulated physiological solution: Chemical and EPR tests. J Mater Sci 22:1228–1234. https://doi.org/10.1007/BF01233113
Acknowledgements
We would like to acknowledge Loïc Joanny (CMEBA) for SEM-EDS analysis, Odile Merdrignac-Conanec (ICSR, Laboratory “Glasses and Ceramics”) for BET analysis and Christophe Calers (ICSR, Laboratory “Glasses and Ceramics”) for XRD analysis at the University of Rennes.
Author contributions
All authors have participated in the conception and design, or analysis and interpretation of the data; drafting the article or revising it critically for important intellectual content, and approval of the final version.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aneb, K., Oudadesse, H., Khireddine, H. et al. Effect of magnesium and iron incorporation on the physicochemical properties of a new sol-gel derived glass-ceramic. J Sol-Gel Sci Technol 109, 502–522 (2024). https://doi.org/10.1007/s10971-023-06290-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-023-06290-9