
Abstract
In this study, we report on the synthesis of a bioactive glass powder with the original 45S5 composition (Bioglass®) by means of an acetic acid-assisted sol–gel route. A glassy material was obtained after the gels underwent a thermal stabilization treatment at 600 °C for 3 h. Above this temperature, the heat-treated gels crystallized partially, forming a sodium-calcium-silicate Na2CaSi2O6 phase. Even after crystallization, this material showed in vitro bioactivity in simulated body fluid after 12 h, when the formation of hydroxycarbonate apatite on the material surface was identified by X-ray diffraction. Not surprisingly, microbiological assays revealed that these gel-derived materials appear to have an antibacterial effect against Pseudomonas aeruginosa (ATCC 27853)—a Gram-negative bacterium that is noted for its environmental survival versatility, ability to produce biofilm and resistance to some antibiotics. Thus, using common precursors that are widely available, relatively cheap, simple to use, and which result in gels with low stabilization temperature, it was possible to explore the versatility of sol–gel processing to obtain the golden standard 45S5 bioglass.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Bioactive glasses are able to interact rapidly and positively with hard and soft tissues [1, 2]. The first bioactive glass (Bioglass®) was developed by Prof. L. L. Hench at the University of Florida in 1969 [3]. It was obtained by the traditional melt-quenching route, and its original composition (45S5) 46.1SiO2−26.9CaO−24.4Na2O−2.6P2O5 (mol%) is still one of the bioactive glasses most widely studied for different purposes [1,2,3].
The use of a sol–gel method to obtain bioactive glasses was first proposed by Li and colleagues in 1991 [4], comprising compositions in the SiO2−CaO−P2O5 ternary system. These authors proposed a potential processing technique for the molecular and textural tailoring of microstructure and biological behavior, leading to a new class of bioactive materials [3, 5] and facilitating the development of scaffolds suitable for tissue-engineering applications [6, 7]. However, despite these advantages and the possibility of exploring new glass compositions and systems in milder laboratory conditions, the study of the original 45S5 composition containing sodium via sol–gel has only recently been investigated. Sodium oxide (Na2O) is a network-modifier component that enables the development of glasses that are more soluble in physiological media and also the formation of certain crystalline phases (resulting in glass-ceramics), which improve mechanical properties without adversely affecting bioactivity [8].
In 2011, we proposed the synthesis of gel-glasses close to the 45S5 composition [9], with tetraethoxysilane (Si(OC2H5)4), calcium nitrate tetrahydrate (Ca(NO3)2 • 4H2O), sodium nitrate (NaNO3) and both triethyl phosphate (OP(OC2H5)3) and phosphoric acid (H3PO4) as precursors for SiO2, CaO, Na2O, and P2O5, respectively. At that time, it was not possible to obtain fully glassy materials since the temperature (~680 °C) used for gel stabilization—a condition necessary for the complete removal of counter ions, especially nitrate from the NaNO3—coincides with the temperature range (~600–700 °C) at which the material easily crystallizes. Thus, glass-ceramics containing Na2Ca2Si3O9 (normally attributed as the main phase) are more readily obtained with the aforementioned precursors, as first reported by Chen and co-authors, in the form of powder [10] and foam [11], and by other authors [9, 12,13,14,15,16,17] in powder form.
It is interesting to note that although only a relatively small amount of P2O5 is present in silicate bioactive glasses, the use of different precursors in some chemical routes is sufficient to affect their resulting materials [9, 18, 19], especially in terms of their mineralization behavior and physical properties. In 2012, Cacciotti et al. [20] demonstrated that it is possible to obtain gel-glasses with 45S5 composition using triethyl phosphite (P(C2H5O)3) as precursor for P2O5—the disadvantage of P(C2H5O)3 is its hazardous classification and very strict purchase procedures. Upon testing diammonium hydrogen phosphate ((NH4)2HPO4), Shankhwar et al. [21] obtained a gelatinous powder by a distinct sol–gel route involving freeze drying at −40 °C followed by heat treatment at 70 °C for 3 days. However, further discussions about this interesting method are needed to find an explanation for the mechanism whereby unreacted substances and counter ions (mainly nitrate) are removed at a mere 70 °C.
Strategies using other CaO and Na2O precursors containing easily removed counter ions (at low temperatures) could also be considered, as previously suggested [9]. According to Catteaux et al. [22], gel-glasses can be obtained using dimethoxy calcium (Ca(OCH3)2) and sodium acetate (CH3COONa), respectively, with thermal treatment at 700 °C to stabilize the gel. Rezabeigi et al. [23] also obtained a gel-glass using calcium l-lactate pentahydrate ((CH3CH(OH)COO)2Ca·5H2O) and a sodium dl-lactate solution (CH3CH(OH)COONa), employing a heat treatment at 550 °C for 3 h. But this route required approximately 50 days for the complete synthesis of the final materials.
In this paper, we report an alternative synthesis procedure based on the low decomposition temperature of the precursors to obtain fully glassy material with the 45S5 composition by a sol–gel route, using acetic acid as catalyst. This strategy proved to be convenient since the precursors used are relatively inexpensive, easy to acquire and handle. In addition, they enable a significant reduction in the gel stabilization temperature compared to that required in the synthesis procedure using calcium and sodium nitrates [9, 20]. Hence, this expanded the range of possible treatments for stabilizing the gel without concomitant crystallization. Finally, the resulting materials in glassy and crystalline forms were subjected to in vitro bioactivity test, and their antibacterial effect against Pseudomonas aeruginosa (ATCC 27853) was evaluated.
2 Materials and methods
2.1 Preparation of the glass powder
To obtain 10 g of a 46.1SiO2−26.9CaO−24.4Na2O−2.6P2O5 (mol%) glass, the relative proportions of each chemical in the batch were calculated using the freely available software tool Glass Panacea [24]. The preparation of gels involved hydrolysis and polycondensation reactions by mixing 16.88 mL tetraethoxysilane (TEOS, Si(OC2H5)4 99%), 1.44 mL triethyl phosphate (TEP, OP(OC2H5)3 99.8%), 4.42 g calcium (CaCO3 99%) and 4.24 g sodium (Na2CO3 99%) carbonates pre-solubilized in a diluted acetic acid solution—all the chemicals provided by Sigma-Aldrich. Starting with the hydrolysis of TEOS, catalyzed by a solution of acetic acid (pH = 2), the other chemicals were sequentially added to the reaction mixture at 60 min intervals while the mixture was stirred constantly under pH < 4. The sol was stored at room temperature for 7 days in a polytetrafluoroethylene mold, followed by aging at 60 °C for 3 days and drying for 1 day at 120 °C. The gels thus formed were then ground in an agate mortar before stabilization at 600 °C (heating rate <2 °C/min) in an oxidizing atmosphere for 3 h, in an electric furnace. Figure 1 outlines the procedures established for the preparation of the glass.

Flowchart of the steps involved in preparing the gel-glass powder
2.2 Characterization of the materials
Thermogravimetric analyses (TG/DTG) were performed in a Shimadzu DTG−60 differential thermal analyzer under air atmosphere. Typical analyses involved ~10 mg of gel particles smaller than 150 μm and heating from room temperature to 1000 °C at a heating rate of 10 °C/min to identify an adequate thermal treatment to stabilize the gels and obtain the glass. The glass was analyzed by Fourier transform infrared (FTIR) spectroscopy using a PerkinElmer Spectrum GX spectrometer operating in transmission mode employing the KBr pellet technique with a spectral resolution of 4 cm−1 from 4000 to 400 cm−1. Spectra were collected as the means of 30 scans and plotted as adsorption. The crystallization behavior of the gel-derived glass was analyzed by X-ray diffraction (XRD) using a Rigaku Ultima IV X-ray diffractometer operating with CuKα radiation (λ = 0.15418 nm). The diffraction patterns were obtained for values of 2θ ranging from 10 to 70° in continuous scan mode at 1°/min. The surface areas of the materials were verified using a Micromeritics Gemini VII surface area analyzer by measuring the nitrogen adsorption/desorption isotherms at 77 K with the Brunauer–Emmett–Teller (BET) equation [25]. Finally, the chemical composition of the synthesized powders was analyzed by X-ray fluorescence spectrometry (XRF). Samples with a particle size <50 μm were selected and converted into glassy disks by melting before the measurements, which were carried out using a Philips PW2404 sequential X-ray fluorescence spectrometer.
2.3 In vitro bioactivity test
The powder bioactivity was evaluated using an adaptation of the new TC04 method [26]. The solution employed in this test is known as simulated body fluid (SBF), and is acellular, protein-free and has a pH adjusted at 7.40 [27].
The powders were immersed in SBF using a ratio of 2 mg/mL. Different from the TC04 method, which prescribes a ratio of sample weight (mg) to SBF volume (mL) of 1.5:1, we opted to use that higher concentration because it greatly facilitated the powder characterization after the selected testing times. Each sample was cleaned ultrasonically for 10 s in acetone before its immersion in polyethylene bottles containing SBF. During the test, the powders were in contact with the solution for 12, 24, 48, 96, and 168 h, and the systems were kept under constant stirring at 37 °C on a shaker table. At the end of each test, the samples were removed from the bottles by filtration (particle retention > 3 μm) and washed with distilled water/acetone to eliminate the SBF and terminate any surface reaction. After drying, a set of samples was analyzed by XRD to check for the formation of a superficial hydroxycarbonate apatite (HCA) layer, and by scanning electron microscopy (SEM) to verify the surface modifications that occurred in the test, using a FEI Inspect S50 microscope coupled to an energy dispersive X-ray spectrometer (EDS), which allowed for qualitative chemical analysis. To finish, the filtered solution was collected to determine the variations in pH that occurred throughout the test due to the partial dissolution of the powder; each sample was processed in triplicate.
2.4 Antibacterial assay against Pseudomonas aeruginosa
The agar diffusion method was used to evaluate the inhibitory effect of the glass against Pseudomonas aeruginosa (ATCC 27853). Initially, a bacterial inoculum was standardized spectrophotometrically (λ = 625 nm and absorbance of 0.08–0.100) with 108 CFU/mL in physiological solution from fresh cultures at 37 °C for 24 h. Then, 0.5 mL of the inoculum was poured into Petri dishes (20 × 65 mm) containing 5 mL of Mueller-Hinton agar as culture medium. Samples previously pressed into disks (10 × 2.2 mm) by uniaxial pressing at 65 MPa were placed on the culture surface and after 2 h of acclimation, the Petri dishes were incubated at 37 °C for 24 h in aerobic conditions. After incubation, the bacterial interactions were examined based on the size of inhibition zone that were formed.
3 Results and discussion
3.1 Synthesis and characterization of the materials
The synthesized gels reached a gelation point, after which they lost their fluidity in approximately 5 h. They were optically homogeneous, translucent and colorless, as illustrated in Fig. 1. When pH is not controlled at a values <4 during synthesis, the gelation point is reached in ~30 min, and the reaction mixture reaches a values of pH higher than 5 after the addition of pre-solubilized carbonates. This occurred mainly when the soluble products from sodium carbonate, with a pH higher than 9, is added to the system. In addition, acetic acid, added as catalyst to prepare sol-gel silica systems, appears to reduce the gelation rates [28], a phenomenon that is highly sensitive to temperature.
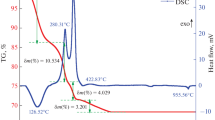
TG (Fig. 2) indicated that the dried gels underwent mass loss at approximately 65 °C, mostly due to water evaporation. The most intense mass loss occurred in the range of 300–500 °C, in which the maximum mass flow occurred from the solid to the vapor phase at 426 °C, corresponding to the removal of incompletely reacted precursors and mainly to the elimination of residual acetate (CH3COO−) ions. Above this temperature the gels became stable, allowing for thermal treatments between 500 and 600 °C to obtain the glass. Unlike the synthesis procedure using sodium and calcium nitrates [9, 20], in which the resulting gels apparently stabilized starting at 600 °C, in this study we reduced this point to 500 °C, significantly increasing the range of possible treatments for stabilizing the gel and obtaining fully glassy materials.

Thermogravimetric analyses of the gel
Choosing the final temperature of 600 °C, a slow heating rate (<2 °C/min) was employed to prevent the formation of residual carbon, as was observed in the TG. This condition was favorable, since the resulting sample did not exhibit the usual dark gray color produced by the presence or residual carbon, and its FTIR spectrum (Fig. 3) showed no traces of organic counter ions.

FTIR spectrum of the gel treated at 600 °C
The FTIR spectrum of the stabilized gel was dominated by the presence of vibrational bands characteristic of silicate glasses, at around 1040 cm−1 for Si–O–Si stretching, 780 cm−1 for Si–O–Si bending and at 490 cm−1 for Si–O–Si rocking [29]. Stretching Si–NBO (non-bridging oxygen) vibration is visible at around 940 cm−1, due to the incorporation of sodium and calcium in the glass network. A comparison of the Si–NBO signal with that of the gel-glass obtained by means of a similar synthesis procedure [30], but containing approximately a half percent of Na2O, clearly shows that the difference in their intensities is a function of the amount of network modifiers present in their compositions. Despite this difference, the FTIR spectra are mostly in good agreement, and also with other spectra of glass compositions close to that of 45S5 [20, 22].
Asymmetric stretching C–O vibration associated with carbonate groups (CO3 2−) is also visible in the region of 1400 cm−1. The formation of carbonates in silicate-based glasses containing sodium and calcium is already known, and has been discussed in the literature for different systems [20, 31, 32]. It seems that an excess of both CO2 and water vapor are necessary to form CO3 2− ions through the dissolution of CO2 in a liquid-like water monolayer on the glass surface, and that these ions react with Na+ or Ca2+ cations, giving rise to ionic carbonates [31, 32]. As for the absence of signals associated with the phosphorus bonds, it should be noted that the amount of P2O5 (2.6 mol%) in the glass composition is relatively small. Thus, the intense bands assigned to silicate groups overlap the phosphate bands at around 500 cm−1 for PO4 3−, at 780 cm−1 for P−O−P bonds, from 900 to 1035 cm−1 for P−O- bond stretching, and at 1260 cm−1 for P=O double bond stretching [33, 34].
The glassy nature of the sample was confirmed by the XRD patterns (Fig. 4), which exhibited a broad halo centered at approximately 27° (2θ) that is typical of silicate glasses. Samples resulting from heat treatment tests with the same isothermal holding time of 3 h, but at higher temperatures (700 and 800 °C), underwent partial crystallization, forming a sodium calcium silicate phase (Na2CaSi2O6, PDF #77–2189). We attributed this phase to Na2CaSi2O6 rather than to Na2Ca2Si3O9, according a previous study involving the structural transformations of bioactive glass 45S5 with thermal treatments [35]. Solid solution is formed in a certain range of compositions in this system, including the 45S5, which we will discuss in another paper. With regard to increase in heat treatment temperature from 700 to 800 °C, the only noticeable alteration we observed was an increase in the diffraction peak intensities, which correspond to a greater crystallized fraction of the Na2CaSi2O6 phase as a function of temperature.

XRD patterns of the gel treated at different temperatures: * = sodium calcium silicate (Na2CaSi2O6)
Although our efforts to obtain glassy materials were successful, the process took approximately 12 days, which is a disadvantage of the method from the standpoint of research and production. Very recently, Ben-Arfa and colleagues [36] called attention to this point, proposing a new rapid method of sol–gel synthesis of 76SiO2−10CaO−8Na2O−6P2O5 (mol%) bioglass nanopowders—without an aging step and about 100-fold faster than our method and other traditional sol–gel routes. This new method is highly advantageous due to its speed and merits further investigation since it is an interesting alternative to obtain different gel-glass compositions.
Table 1 shows that the BET surface area of the synthesized materials decreased when the gels were stabilized at 600 (7.80 m2/g) and 800 °C (3.48 m2/g), respectively. Higher temperature favors the sintering process, which is driven by surface energy reduction, densifying the materials via viscous flow. This process was accompanied by the diminution of pore volume (~0.085–0.0040 cm3/g) and average pore diameter (~44–7 nm) of the samples during the treatment. A similar effect, but as a function of the time, was described in detail by Lin et al. [37] investigating the nanostructure evolution and calcium distribution in a sol–gel derived 70SiO2−30CaO (mol%) bioactive glass. The surface area of the gel-derived materials was higher than that of the melted glass (0.68 m2/g) synthesized for comparison, but their specific surface areas were smaller than those of the common gel-derived glasses. Furthermore, the use acetic acid as the catalyst in sol–gel synthesis favors the increase of surface area, as reported by Arenas et al. [28] for the silica xerogels, which reached a (huge) value of ~800 m2/g, and by Lei et al. [38] for a bioactive 60SiO2−36CaO−4P2O5 (mol%) gel-glass, which reached a value of ~190 m2/g. However, this effect was not observed in this study for bioactive materials of the SiO2−CaO−Na2O−P2O5 system. On the other hand, Rezabeigi et al. [23] also did not find a very high surface area (~12 m2/g) for their synthesized gel-glasses. They attributed that result to the combination of the starting chemicals, calcium and sodium lactate with lactic acid as the hydrolysis catalyst, and its slow gelation process (~45 days). Indeed, this can have an effect on the resulting materials; but, different researchers [12, 13, 16] have reported similar values (about 11 m2/g or smaller) for gel-derived materials with the 45S5 composition employing distinctly synthesis procedure and different precursors. Lucas-Girot et al. [30] found a surface area of ~80 m2/g for a 52.3SiO2−32.3CaO−13.7Na2O−1.7P2O5 (mol%) gel-glass composition synthesized by a procedure similar to that used here. Therefore, the concentration of sodium oxide in gel-glasses appears to have a more significant effect on their specific surface area, since by comparing the different materials there is a difference of almost half in the Na2O content (13.7 vs. 24.4% in the 45S5 composition) and practically one order of magnitude in the surface area (~80 vs. 7 m2/g).
The chemical composition of the gel heat treated at 600 °C and the melted glass are shown in Table 2. In general, the nominal composition established for the syntheses was in good agreement with the analyzed compositions, indicating that the sol–gel synthesis can be used as an alternative procedure for obtaining the 45S5 glass composition.
3.2 Bioactivity and antibacterial activity
The XRD patterns of the glass samples immersed in SBF (Fig. 5) showed two major peaks near 26 and 32° (2θ), corresponding to (002) and (121) atomic plane diffractions of the HCA-like phase (Ca5(PO4)3OH, PDF #84–1998). This association with HCA is made because the formation of pure hydroxyapatite on the glass surface is less likely to occur in SBF. This solution contains bicarbonate ions (HCO3 −) and the hydroxyapatite is saturated with respect to slightly carbonated apatite, where orthophosphates are generally replaced by carbonates in the crystal lattice [26, 39]. The main peaks increased with increasing testing time, and broad new peaks appeared at approximately 40, 44, 50, and 53° (2θ), corresponding, respectively, to the (310), (113), (123) and (004) atomic planes in the HCA lattice, which are correlated with the increase in crystallinity. The formation of calcium carbonate (calcite—CaCO3, PDF #5–586) was also observed after 24 h of testing. Previous studies have also reported this formation in gel-derived bioactive materials [10, 17, 26].

XRD patterns of the gels treated at 600 °C (a) and 800 °C (b) before and after immersion in SBF for different testing times: ● = hydroxyapatite (Ca5(PO4)3OH); ○ = calcite (CaCO3); * = sodium calcium silicate (Na2CaSi2O6)
The crystallized samples exhibited similar bioactive behavior, showing a significant decrease in the diffraction peaks of the Na2CaSi2O6 phase after contact with SBF. It is demonstrated in Fig. 5b with emphasis on the region from 20 to 40° (2θ), where the most intense peaks of the Na2CaSi2O6 and HCA formed are located. Amorphous phase became predominant at the surfaces of sample due to their partial dissolution in the test, with practically complete depletion of Na2CaSi2O6 peaks after 168 h.
The morphology of the samples’ surfaces (Fig. 6) changed significantly after various immersion times in SBF, resulting in a mixed polycrystalline layer of HCA. The formed HCA layer has a fair similarity to the mineral phase of human cortical bone [40] and this is what allows bone tissue to chemically bond to the bioactive glasses [1, 3]. During the test, the formation of HCA promoted the agglomeration of the powder, covering almost their entire surfaces. Consequently, a large compositional change was visible in the EDS spectra, with a predominance of Ca and P, which confirmed the formation of a HCA layer.

SEM micrograph and EDS spectrum of the gel treated at 600 °C before and after immersion in SBF for 168 h
As mentioned earlier, the initial pH of the solution used for evaluating the in vitro bioactivity was 7.4. During the test, all the samples presented a similar tendency for pH to vary (Fig. 7). The pH value increased very rapidly to approximately 8.30 in the first 24 h and maintained a slight upward tendency, reaching a value of ~60 in the last test of 168 h. The most abrupt change was found to occur in the first few hours, indicating that the samples were highly soluble until a substantial layer of HCA was formed. This change in pH reveals a direct relationship with cation (Na+ and Ca2+) exchange from the sample with H+ ions to the solution [1, 8, 18, 26], favoring the formation of HCA and confirming the antimicrobial effect observed here against Pseudomonas aeruginosa, an opportunistic pathogen and a frequent cause of healthcare-associated infections [41].

Variations in pH as a function of the immersion time of samples in SBF
For the antibacterial assay, the pH of the agar medium should vary from 7.2 to 7.4 at room temperature [42], because the optimum pH range for most bacteria to grow is normally around 7. Thus, the zone of inhibition induced by the glass disk exemplified in Fig. 8, with an average size of 17 mm, in principle can be explained by the increased osmotic pressure and a local change in pH (around the disks) due to the release of ions from the sample to the medium—probably reaching a final pH of approximately 8.30 (or higher) in 24 h of incubation based on the pH reached in the bioactivity test during the same testing time. In this case, it is important to note that the SBF is a buffer solution and that its buffering capacity differs from that of the Mueller-Hinton agar used as culture medium. In addition, increases in pH during the glass leaching and dissolution processes can promote an antibacterial effect, which is in agreement with other studies [43,44,45]. However, the zone of inhibition found in our case study did not necessarily indicate that bacteria were killed, but simply that they were prevented from growing, so further tests are required to reach more definitive conclusions.

Representative image of the zone of inhibition produced by the gel treated at 600 °C against Pseudomonas aeruginosa
4 Conclusions
We prepared a bioactive glass powder with a composition very close to that of the standard 45S5 (Bioglass®) by means of an alternative sol–gel route using acetic acid as a catalyst. The material was obtained after a cycle of approximately 12 days (vs. ~50 days for the lactic acid route), without the need for special apparatuses, using common precursors and in milder laboratory conditions than most previously proposed routes. This enabled us to explore the most widely studied bioactive glass composition associated with the intrinsic beneficial characteristics offered by the sol–gel processing.
Although the dried gels subjected to stabilization treatments above 600 °C underwent crystallization (formation of the Na2CaSi2O6 phase), all the glassy and crystalline samples were bioactive, showing HCA formation after 12 h of in vitro testing with no negative influence from crystallization. A preliminary microbiological assay revealed that the glassy and resulting crystalline materials have an antibacterial effect against Pseudomonas aeruginosa, indicating the feasibility of testing their properties to prevent skin and soft tissue infections—with the concomitant benefit of inducing tissue regeneration.
References
Jones JR (2013) Review of bioactive glass: from Hench to hybrids. Acta Biomater 9:4457–4486
Miguez-Pacheco V, Hench LL, Boccaccini AR (2015) Bioactive glasses beyond bone and teeth: emerging applications in contact with soft tissues. Acta Biomater 13:1–15
Hench LL (2006) The story of Bioglass®. J Mater Sci Mater Med 17:967–978
Li R, Clark AE, Hench LL (1991) An investigation of bioactive glass powders by sol–gel processing. J App Biomater 2:231–239
Hench LL (1998) Biomaterials: a forecast for the future. Biomaterials 19:1419–1423
Jones JR, Ehrenfried LM, Hench LL (2006) Optimising bioactive glass scaffolds for bone tissue engineering. Biomaterials 27:964–973
Jones JR (2009) New trends in bioactive scaffolds: the importance of nanostructure. J Eur Ceram Soc 29:1275–1281
Peitl O, Zanotto ED, Hench LL (2001) Highly bioactive P2O5–Na2O–CaO–SiO2 glass-ceramics. J Non-Cryst Solids 292:115–126
Siqueira RL, Peitl O, Zanotto ED (2011) Gel-derived SiO2–CaO–Na2O–P2O5 bioactive powders: synthesis and in vitro bioactivity. Mater Sci Eng C 31:983–991
Chen Q-Z, Li Y, Jin L-Y, Quinn JMW, Komesaroff PA (2010) A new sol–gel process for producing Na2O-containing bioactive glass ceramics. Acta Biomater 6:4143–4153
Chen Q-Z, Thouas GA (2011) Fabrication and characterization of sol–gel derived 45S5 Bioglass®-ceramic scaffolds. Acta Biomater 7:3616–3626
Bahniuk MS, Pirayesh H, Singh HD, Nychka JA, Unsworth LD (2012) Bioactive glass 45S5 powders: effect of synthesis route and resultant surface chemistry and crystallinity on protein adsorption from human plasma. Biointerphases 7:1–15
Pirayesh H, Nychka JA (2013) Sol-gel synthesis of bioactive glass-ceramic 45S5 and its in vitro dissolution and mineralization behavior. J Am Ceram Soc 96:1643–1650
Li HC, Wang DG, Hu JH, Chen CZ (2013) Crystallization, mechanical properties and in vitro bioactivity of sol–gel derived Na2O–CaO–SiO2–P2O5 glass-ceramics by partial substitution of CaF2 for CaO. J Sol Gel Sci Technol 67:56–65
Zheng K, Solodovnyk A, Li W, Goudouri O-M, Stähli C, Nazhat SN, Boccaccini AR (2015) Aging time and temperature effects on the structure and bioactivity of gel-derived 45S5 glass-ceramics. J Am Ceram Soc 98:30–38
Faure J, Drevet R, Lemelle A, Jaber NB, Tara A, Btaouri HE, Benhayoune H (2015) A new sol–gel synthesis of 45S5 bioactive glass using an organic acid as catalyst. Mater Sci Eng C 47:407–412
Thomas A, Bera J (2016) Sol–gel synthesis and in vitro bioactivity of glass-ceramics in SiO2–CaO–Na2O–P2O5 system. J Sol Gel Sci Technol 80:411–416
Siqueira RL, Zanotto ED (2013) The influence of phosphorus precursors on the synthesis and bioactivity of SiO2–CaO–P2O5 sol–gel glasses and glass-ceramics. J Mater Sci Mater Med 24:365–379
Shah AT, Ain Q, Chaudhry AA, Khan AF, Iqbal B, Ahmad S, Siddiqi SA, ur Rehman I (2015) A study of the effect of precursors on physical and biological properties of mesoporous bioactive glass. J Mater Sci 50:1794–1804
Cacciotti I, Lombardi M, Bianco A, Ravaglioli A, Montanaro L (2012) Sol–gel derived 45S5 bioglass: synthesis, microstructural evolution and thermal behaviour. J Mater Sci Mater Med 23:1849–1866
Shankhwar N, Kothiyal GP, Srinivasan A (2015) Influence of phosphate precursors on the structure, crystallization behaviour and bioactivity of sol–gel derived 45S5 bioglass. RSC Adv 5:100762–100768
Catteaux R, Grattepanche-Lebecq I, Désanglois F, Chai F, Hornez J-C, Hampshire S, Follet-Houttemane C (2013) Synthesis, characterization and bioactivity of bioglasses in the Na2O–CaO–P2O5–SiO2 system prepared via sol gel processing. Chem Eng Res Des 91:2420–2426
Rezabeigi E, Wood-Adams PM, Drew RAL (2014) Synthesis of 45S5 Bioglass® via a straightforward organic, nitrate-free sol–gel process. Mater Sci Eng C 40:248–252
Siqueira RL, Alano JH, Peitl O, Zanotto ED (2017) GlassPanacea: a user-friendly free software tool for the formulation of glasses, glass-ceramics, and ceramics. Am Ceram Soc Bull 96:48–49
Brunauer S, Emmett PH, Teller E (1938) Adsorption of gases in multimolecular layers. J Am Chem Soc 60:309–319
Maçon ALB, Kim TB, Valliant EM, Goetschius K, Brow RK, Day DE, Hoppe A, Boccaccini AR, Kim IY, Ohtsuki C, Kokubo T, Osaka A, Vallet-Regí M, Arcos D, Fraile L, Salinas AJ, Teixeira AV, Vueva Y, Almeida RM, Miola M, Vitale-Brovarone C, Verné E, Höland W, Jones JR (2015) A unified in vitro evaluation for apatite-forming ability of bioactive glasses and their variants. J Mater Sci Mater Med 26:115–125
Kokubo T, Takadama H (2006) How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27:2907–2915
Arenas LT, Simm CW, Gushikem Y, Dias SLP, Moro CC, Costa TMH, Benvenutti EV (2007) Synthesis of silica xerogels with high surface area using acetic acid as catalyst. J Braz Chem Soc 18:886–890
Aguiar H, Serra J, González P, León B (2009) Structural study of sol–gel silicate glasses by IR and Raman spectroscopies. J Non-Cryst Solids 355:475–480
Lucas-Girot A, Mezahi FZ, Mami M, Oudadesse H, Harabi A, Floch ML (2011) Sol–gel synthesis of a new composition of bioactive glass in the quaternary system SiO2–CaO–Na2O–P2O5: comparison with melting method. J Non-Cryst Solids 357:3322–3327
Cerruti M, Morterra C (2004) Carbonate formation on bioactive glasses. Langmuir 20:6382–6388
Perardi A, Cerrruti M, Morterra C (2005) Carbonate formation on sol–gel bioactive glass 58S and on Bioglass® 45S5. Stud Surf Sci Catal 155:461–469
Langille KB, Nguyen D, Bernt JO, Veinot DE, Murthy MK (1993) Constitution and properties of phosphosilicate coatings. Part I: influence of sodium phosphates on the constitution of sodium silicate coatings. J Mater Sci 28:4175–4182
Khawaja EE, Durrani SMA, Al-Adel FF, Salim MA, Hussain MS (1995) X-ray photoelectron spectroscopy and Fourier transform-infrared studies of transition metal phosphate glasses. J Mater Sci 30:225–234
Lefebvre L, Chevalier J, Gremillard L, Zenati R, Thollet G, Bernache-Assolant D, Govin A (2007) Structural transformations of bioactive glass 45S5 with thermal treatments. Acta Mater 55:3305–3313
BAE Ben-Arfa, IMM Salvado, JMF Ferreira, RC Pullar (2016) A hundred times faster: novel, rapid sol–gel synthesis of bioglass nanopowders (Si–Na–Ca–P system, Ca:P = 1.67) without aging. Int. J Appl Glass Sci doi:10.1111/ijag.12255.1-7
Lin S, Ionescu C, Pike KJ, Smith ME, Jones JR (2009) Nanostructure evolution and calcium distribution in sol-gel derived bioactive glass. J Mater Chem 19:1276–1282
Lei B, Chen X, Wang Y, Zhao N, Du C, Zhang L (2019) Acetic acid derived mesoporous bioactive glasses with an enhanced in vitro bioactivity. J Non-Cryst Solids 355:2583–2587
Šupová M (2015) Substituted hydroxyapatites for biomedical applications: a review. Ceram Int 41:9203–9231
Rehman I, Hench LL, Bonfield W, Smith R (1994) Analysis of surface layers on bioactive glasses. Biomaterials 15:865–870
Streeter K, Katouli M (2016) Pseudomonas aeruginosa: a review of their pathogenesis and prevalence in clinical settings and the environment. Infect. Epidemiol Med 2:25–32
Clinical and Laboratory Standards Institute (CLSI) (2012) Performance standards for antimicrobial disk susceptibility tests. Approved standard M02-A11. Clinical and Laboratory Standards Institute, Wayne, PA
Munukka E, Leppäranta O, Korkeamäki M, Vaahtio M, Peltola T, Zhang D, Hupa L, Ylänen H, Salonen JI, Viljanen MK, Eerola E (2008) Bactericidal effects of bioactive glasses on clinically important aerobic bacteria. J Mater Sci Mater Med 19:27–32
Hu S, Chang J, Liu M, Ning C (2009) Study on antibacterial effect of 45S5 Bioglass®. J Mater Sci Mater Med 20:281–286
Zhang D, Leppäranta O, Munukka E, Ylänen HMK, Eerola E, Hupa M, Hupa L (2010) Antibacterial effects and dissolution behavior of six bioactive glasses. J Biomed Mater Res A 93:475–483
Acknowledgements
We are grateful to the following Brazilian research funding agencies: FAPESP—São Paulo Research Foundation (CEPID—project no. 2013/07793-6) for its generous funding, and CNPq—Conselho Nacional de Desenvolvimento Científico e Tecnológico (Project no. 140516/2013-1) for granting a scholarship to R. L. Siqueira.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Siqueira, R.L., Costa, L.C., Schiavon, M.A. et al. Bioglass® and resulting crystalline materials synthesized via an acetic acid-assisted sol–gel route. J Sol-Gel Sci Technol 83, 165–173 (2017). https://doi.org/10.1007/s10971-017-4402-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-017-4402-3