
Abstract
The radiochronometric model age is an important signature in nuclear forensic analysis. Recent studies have illustrated the need for controlled experiments on the behavior of decay products during uranium metal casting to provide a foundation for interpretation of discordant model ages. A variety of uranium metal and alloy samples cast under known conditions were analysed by three laboratories. This work is the first multi-laboratory study of its kind to explore how these progeny isotopes are chemically fractionated from uranium metal during casting. The intercomparison allowed for capability demonstration and method development on samples and provided data to increase our understanding of the behavior of decay progeny in these complex systems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nuclear forensics is the analysis of measurable parameters in nuclear or other radioactive material to support law enforcement investigations and nuclear security applications [1, 2]. Radiochronometry is a useful technique for constraining production timelines for an unknown sample [3]. The measured model ages can be used to define model sample purification dates which can then be compared to any available records or other samples. To maximize the benefit of radiochronometry, it is essential that the community understands the behavior of radionuclides throughout key processes within the nuclear fuel cycle. This understanding is crucial for underpinning confidence in the use of radiochronometry as a tool to answer specific investigatory questions and to understand radiochronometry data when applied samples with a potentially complex production history. As uranium is the most common nuclear material found outside of regulatory control, work on understanding forensic signatures in uranium-based material types is important [1].
In radioactive materials, measurements of decay progeny-parent pairs are used to calculate a model age that can be used for nuclear forensics. In the case of uranium, 230Th/234U and 231 Pa/235U are the most commonly used progeny-parent ratios [4, 5]. These decay progeny pairs are useful as they have sufficiently high atomic ratios, concentrations, and half-lives to allow for accurate measurements from a wide range of ages, material types, and enrichments we may encounter. Measurement of decay progeny-parent pairs in actinide sample matrices by a variety of analytical techniques can be used to produce model ages for interpretation within specific timescales, accuracy, and precision requirements [6,7,8]. These measurements of ultra-trace nuclides (e.g. 231 Pa/235U <1 × 10-8) can require considerable effort [9]. The most precise and accurate results are currently obtained by isotope dilution mass spectrometry methods. All ages obtained through radiochronometry are model ages based on several assumptions reported in previous studies [3]. If these assumptions are accurate, then the model age reflects a relevant purification date in the history of the material.
A variety of challenges remain to improve our ability to make rapid and robust radiochronometry measurements over the wide range of potential sample types required for nuclear forensics. Recent advances in certified reference standards underpin measurement traceability for radiochronometry and continue to improve accuracy and precision [10,11,12].
Laboratory intercomparisons on samples from the nuclear fuel cycle offer an excellent opportunity to test established analytical capabilities and to develop new and improved methodologies when needed. Analysis of certified reference standards does not always allow for true validation of methodologies. Such reference standards have typically been produced under highly controlled conditions for specific purposes and in some cases do not give a suitable test of laboratory method robustness for potential unknown sample matrix interferences. There are few of these materials available and they are also limited in terms of age and material composition relative to the wide range of material types that could be encountered.
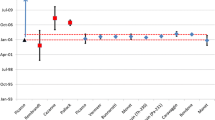
The measurement of multiple chronometer pairs and agreement within measurement uncertainty (termed concordance) adds confidence to our ability to interpret the likely significance of the model age in question [14]. Concordant model ages are observed in many U and Pu reference materials that are commonly analysed for validation of analytical methods. Concordance is also observed in materials produced in specific production processes [15]. As an example, for U fuel pellets, the model age might reflect the date of UO2 production, but not the sintering or thermomechanical processing of said material occurring later in production [16]. Experimental data to study these processes may be most pragmatically obtained by application of radiochronometry to samples of known provenance (Fig. 1).

Example of concordant chronometer pairs from the analysis of UO2 fuel pellets by multiple labs (orange and blue) in the Collaborative Materials eXchange (CMX) 5 exercise [16]
If any of the model age assumptions are not met, we may measure discordant chronometer pairs which are more challenging to interpret [17, 18]. Discordant chronometer pairs are typically caused by differential chemical fractionation of decay progeny during a material’s processing history. This can be important forensic information. For example, discordant model ages may be used to bound the maximum age of the sample, allowing comparison with relevant knowledge on process history. However, data on materials that have been sourced from common uranium production processes is limited. Consequently, the complex effects of nuclear fuel cycle processes on radiochronometry data can be poorly understood. There are multiple published examples where progeny have not been completely purified during material processing [15, 19, 20]. Constraining the mechanisms that lead to discordance of chronometric pairs will enhance the diagnostic value of radiochronometric signatures.
In recent years, observations of discordant Th and Pa chronometer patterns have occurred in studies of cast U metal samples. As the final stage in U metal and alloy manufacturing that might separate decay progeny, casting is a key process that requires investigation. In addition, post-casting machining and thermomechanical processing is known to impact grain structure and mechanical properties. We hypothesize that such impacts to mechanical properties are unlikely to affect chronometer pairs at the spatial scale of a bulk analysis, however, experimental data are required to test this hypothesis. The production of U metal samples under controlled conditions for the sole purpose of investigating radiochronometry discordance can be prohibitively expensive and challenging to accomplish on a scale representative of commonly used production processes. Thus, analysis of ‘samples of opportunity’, i.e., materials of known provenance that are produced for some other purpose and can be utilized for radiochronometry studies offer a pragmatic approach for investigating radiochronometer behavior. In this study, we present radiochronometry measurements of several such U metal and alloy samples.
Several studies demonstrate that current knowledge of Th and Pa fractionation from U during metal casting is insufficient for accurate interpretations of 230Th-234U and 231 Pa-235U model ages [15] [21]. Metallic U and U alloys are important materials for many applications, especially in nuclear energy, which utilize low enriched U metallic fuel forms and new reactor fuel types, which require specific material properties or alloys [21, 22]. Alloy samples are typically produced by multi-stage casting processes and are therefore excellent candidates for study of chemical fractionation of trace actinides over multiple casting cycles in industrial scale casting processes such as Vacuum Induction Melting (VIM) and Vacuum Arc Remelting (VAR).
Hanlen et al. reported the first example of discordant chronometer pairs measured by one laboratory in cast U samples as part of the International Technical Working Group (ITWG) on nuclear forensics round robin exercise number three [22]. The samples analyzed for this exercise were subsamples of existing materials from Y-12 that had been produced by VIM processing of scrap metal. Following the completion of the exercise, Kayzar & Williams (2016) subsequently carried out a multiple U-series chronometer study on these materials [15]. They demonstrated that multiple decay progeny nuclides are fractionated from U to different extents during metal casting processes. The Th and Ac were almost entirely purified from U. They also showed that Ra and Pa are purified to a lesser extent in this example. The working hypothesis describing this behavior is based on the differing chemistry and physical properties of these elements upon melting and solidification in the casting media. As the casting media is key variable in a variety of casting processes, it is also a useful measurement for nuclear forensic signature discovery. Other studies suggest that Th and Ac may migrate to the ‘hot top’ of material in directional castings or can be fractionated between the top, sides, or even grains of a cast part [23,24,25,26]. Depending on outgassing of the cast and chemical reactivity, Ra may also be purified to a similar or lesser extent. The fate and reactivity of Pa in such systems is also poorly understood in the literature, but can be investigated using the 231 Pa/235U chronometer in cast U samples. Protactinium behaves similarly to Mo or Nb in uranium materials, and may not be segregated from uranium during casting under certain conditions [25, 26].
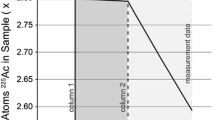
Other studies have also demonstrated repeatable discordance between Th and Pa chronometers in cast U samples [15] [18] [22]. Repeatable discordance is defined in this case such that the same relative bias in terms of discordance is observed in multiple different samples produced in the same way and that laboratories observe the same model age results between measurement uncertainty from repeated measurements of a given sample. Notably, one of the samples used in the sixth ITWG Collaborative Materials eXercise (CMX), was a VIM cast U metal sample that showed discordant 230Th/234U and 231 Pa/235U chronometers relative to the known casting date of less than a year [27]. This also adds weight that this impact must be considered and is measureable on recently produced samples. Both of these ITWG studies lack analytical data for the U feedstock material used to produce the cast items. However, these data are required in order to fully assess the fate of decay progeny during casting. It is therefore unclear if older discordant ages are representative of the most recent chemical purification step, or a composite age caused by minor spatial segregation of decay progeny in a material, or some other combination of processes (Fig. 2).

Example U metals analysis from the ITWG round robin 3 [22]
Many and varied factors contribute to potential complexity in the forensic signatures of cast U metal including economic constraints, U recycling, engineering or production specifications, geometry of a cast item, and material criticality. Additional chemical complexity may occur if certain elements are added to confer specific material properties. Radiochronometry studies of early fuel cycle materials have shown specific progeny pairs are purified to different extents during production of the feedstock used for metal production [28]. Kovarik et al. aimed to understand the fractionation of Th in such processes: Th fractionated in a variety of locations in bomb reduction and was shown to be associated with Fe, Al and Si in the slag [28]. Additional research is needed to understand how discordant ages are generated from casting processes with the additional complexity of feedstock blending (Fig. 3).

Example flow chart for U metal and alloy production, key = *feedstock preparation may include mining, recycle, dissolution, purification, enrichment and conversion. **Alloying stages will typically involve multi-stage castings and thermomechanical processing. ***Casting to a part or shape may also involve multiple casting stages. All stages have some potential to impart radiochronometric signatures
As a result of feedstock blending, various sources of U material may be combined in a single cast. The composite feed material may be cast multiple times to ensure material properties conferred by casting are consistent and reliable. The feed material to any one cast may contain new material, machined swarf/pieces from previous casts, recovered casting headers, or recycled massive scrap. Feedstock blending, therefore, results in progeny-parent relationships that reflect a complex purification history of the separate feed materials. Studies on these types of samples will be needed to understand the impact upon traditional nuclear forensic signatures.
Overviews of VIM and VAR are given in the literature [29,30,31]. In the case of the industry standard VIM method, molten U is transferred with release agents into a graphite crucible. Thermal transfer in the mould is efficient so the temperature profile is relatively uniform. The melt solidifies slowly and the mould for a shaped cast is designed such that the final region to solidify is the header region above a relevant part. This results in large grain size ranges (micron to mm-scale) in a given sample and preferential segregation of lower melting point trace impurities in the header region. The header region in the cast is expected to contain solid impurities, the largest grains, shrinkage defects and dissolved gaseous species [28].
This paper presents the first study to assess radiochronometry signatures in cast U metals with known production history and analysis of a feedstock to a casting. We explore the scenarios in which measured model ages reflect the time of sample casting for U metal and alloy samples with complex processing histories. Measuring these samples on an interlaboratory basis increases confidence in the accuracy of the measurements performed given the lack of relevant certified reference materials and challenging sample types. This inter-comparison provides laboratories the opportunity to improve methodologies and add information to add to our understanding of the nuclear fuel cycle [16].
This work is motivated by three main questions: (1) Can we explain and interpret the apparent model ages of cast U metals and alloys based on a known feedstock history?; (2) Does casting by VIM/VAR and other hybrids result in predictable discordance between 230Th/234U and 231 Pa/235U model ages in cast products?; and (3) What is the lab to lab variability in ultra-trace measurements of decay progeny in small samples from a bulk casting? To address these questions, we used depleted U (DU) samples from large scale casts that were previously prepared using multiple processing routes.
Experimental
Samples were size reduced for all labs from cut samples using end modified knipex snips, and 0.5-1 g subsamples were provided in Teflon bottles for all three laboratories.
Each laboratory used established and independent radiochemistry methods to prepare spikes for isotope dilution and to purify analytes for mass spectrometry. The methods for the U metals used currently published methods for the three laboratories [32,33,34]. New methodologies were developed for the DU-Nb alloy samples. All labs completed analyses gravimetrically via isotope dilution mass spectrometry (IDMS) and analysed samples in duplicate. All experimental details for the inter-comparison are detailed below for completeness. All laboratories used high-purity reagents and relevant certified reference standards.
AWE
The samples were dissolved either in Ultra High Purity (UHP) nitric acid (3 mL, Fisher, UK) for metals or aqua regia for alloys, followed by conc. UHP Sulphuric acid (Fisher, UK) and heating on a hotplate below the boiling point for 2 h. The resulting solutions were adjusted to HCl (Fisher, UK). Sub samples (0.2 g) were then taken for IDMS. The sub samples in duplicate were spiked with approximately 0.2 g 233U, 50 mg of 229Th and 0.2 g of 233Pa tracer solutions outlined below. The U630 (NIST, USA, 1 g of a 14 ppm solution) radiochronometry Certified Reference Material (CRM) was measured as an internal quality control sample.
A 233U tracer (103.57 ± 0.31 Bq/g,) and a 229Th tracer (100.53 ± 1.20 Bq/g) certified radiometrically by the National Physical Laboratory (UK) was used for isotope dilution. A 233Pa tracer (1.07 × 1010 ± 5.5 × 107 atoms/g) was standardized using the CIEMAN-NIST method, produced internally from an activity certified 237Np [32]. The 233Pa prepared by chemical separation from 1 mg 237Np using TK400 (Triskem, France) followed by ZR extraction chromatography resin (Triskem, France). A separation factor of around 1 × 107 of neptunium from Pa is typically achieved [32]. The 233Pa solution was also measured by gamma spectrometry as an additional check on the CIEMAN-NIST assay.
For alloy samples a modification of our previously published method was used [32]. This allows for novel, no-dry down steps in chromatography, single methodology for Nb-U alloy samples for Th, Pa and U radiochronometry measurements on a vacuum box (Triskem, Fr). A sample was taken for purification in a Teflon beaker. The above 233Pa, 229Th and 233U tracers were added and the sample was evaporated to near dryness. The samples were conditioned with 3 × 1 mL c.HCl, evaporating to near dryness between additions. The samples were re-dissolved in 1 mL c.HCl. A stacked resin setup was prepared. A 1 mL resin column of TK400 and Zr Resin and a 2 mL column of AG1-X8 and UTEVA (Triskem, Fr) were prepared in Rockbourne R1010 medium columns. These columns ensure reduced adsorption to the frit of Np and Pa. The TK400 (top, Pa/Nb absorbed) AG1-X8 (middle, U absorbed) UTEVA (bottom, Th absorbed) triple stacked column. The sample is loaded with 1 mL of 11 M HCl, followed by 4 × 1 mL of 11 M HCl. The three columns are then separated. To the AG1-X8 column, the U can be rinsed with 5 mL of 18.2 MΩ cm (Elga) deionized water. The TK400 column should be washed with 4 × 5 mL of 11 M HCl (to waste). The TK400 column should then be stacked on top of a Zr resin column (Pa). The double stack should then be washed with 3 × 5 mL of 1 M HCl. The Zr column should then be separated. To remove the ingrown 233U, perform the next step as close to measurement time as possible. Wash the Zr resin column with 4 × 5 mL of 5.5 M HCl. The Zr resin column can then be stacked onto a 2 mL AG1-X8 column previously conditioned with 5 mL of c.HCl. The Pa fraction can then be eluted using 2 × 5 mL of 5.5 M HCl/0.1 M HF, with no dry down stages, into a pre-weighed LSC vial for HRGS to determine chemical recovery. For Th, the UTEVA column is washed with 5 mL of 11 M HCl (waste). The column is then stacked on top of an AG1-X8 column conditioned with 5 mL c.HCl. The Th is eluted with 2 × 5 mL of 5.5 M HCl into a 20 mL Liquid Scintillation Vial (LSV).
A ThermoScientific Neptune Plus MC-ICP-MS was used for all ID-MS measurements. For U, approximately 0.1 ng of U was consumed for isotope ratio measurements. For U ID-MS, samples were diluted by a factor of approx. 200, then approx. 50 mg of diluted sample (8 × 1014 atoms 238U) was spiked with 0.2 g of 233U tracer (4.5 × 1014 atoms). This was diluted to approx. 10 ppt for measurement by mass spec. Measurements were corrected for mass bias and blank subtraction. An Ion Counter (IC)/Faraday gain correction was performed for 234U/238U. Static multi-collection where 233U, 235U and 238U were measured using Faraday cups equipped with 1013 Ohm amplifiers. 234U was measured on the centre IC equipped with a Retarding Potential Quadrupole (RPQ). An integration time of 8 s run for 40 cycles was used. U010, U050 and U630 CRMs were used for mass bias correction.
In the case of the Th and Pa, 5 mL column elution volumes, approx. 0.1–1 pg was taken for measurement. Th measurements were performed by peak-jumping on the center IC. Integration of 2 s for 100 cycles was completed. For Th, mass bias correction was quantified by U010 and U050. Three internal QC standards were produced from a mix of 229Th and 230Th certified concentration solutions combined in known quantities. For Pa, measurements were performed by peak-jumping on the center IC with integration of 2 s for 100 cycles. Mass bias correction was quantified by U010 and U050. Chemistry blanks were run in duplicate alongside samples and QC. They were quantified by IDMS. All chemistry blanks were < 0.01% of sample amounts and therefore considered negligible. Uncertainties have been propagated to provide a combined standard uncertainty in compliance with the recommendations made in the guide to the expression of uncertainty in measurement [34]. The relevant nuclear data and the Bateman equations were used for model age calculations [35,36,37,38,39,40,41,42].
LANL
The samples were received at LANL as fragments of cut metal per sample. Two fragments of each sample were used for analysis and in the case where three fragments were available, the third fragment was archived. Each metal sample fragment was individually pickled (acid leached) using 8 M HNO3 to remove surface oxidation in order to determine accurate metal sample masses. The pure DU metals were digested in pre-cleaned 60 mL Savillex jars on a hotplate at 130 °C using 8 M HNO3. Two fragments of each sample were digested together. Sample solutions were transferred to pre-cleaned Teflon bottles and were diluted to create 60 mL primary sample solutions in 4 M HNO3 + 0.005 M HF. The DU-Nb metal alloys were digested using a similar process; however, a larger acid volume and HF were included in the digestion in order to dissolve Nb precipitates from the DU-Nb matrix that are not soluble in small volumes of 8 M HNO3. Alloy sample solutions were transferred to pre-cleaned Teflon bottles and were diluted to create 125 mL primary sample solutions in 4 M HNO3 + 0.1 M HF. Process blanks were introduced during metal pickling and were treated identically to the samples. Secondary, tertiary, and quaternary serial dilutions of each subsample and associated process blanks were made for U isotope composition and assay determination using 4 M HNO3 + 0.005 M HF for metal samples and 4 M HNO3 + 0.1 M HF for alloy samples to keep Nb in solution.
The following spikes were used for IDMS measurements: for U, a LANL internal U-233 spike (0.5 ng 233U/g) calibrated through reverse IDMS using a gravimetric preparation of U metal standard NBS SRM 960 (CRM 112-A) - the calibration of the 233U spike concentration cross-checked against NBL CRM-145; for Th, the NFRM Th-1 229Th spike [11]; and for Pa, a 233Pa spike was prepared by separating 233Pa from a LANL legacy 237Np stock solution and calibrated using the NFRM Pa-1 231Pa standard [12]. Three mixtures between the 233Pa spike and NFRM Pa-1 were made for reverse IDMS calibration when the spike was initially prepared. Later in the spike’s life, another three mixtures of the 233Pa and 231Pa were made and a second round of reverse IDMS calibration was conducted. The average of the two spike calibration campaigns (decay corrected to a common date) was used as the final calibration of the 233Pa spike.
For quality control of LANL radiochronometry measurements, National Bureau of Standards (NBS) Standard Reference Material (SRM) 960 metal was used as a U assay standard, New Brunswick Laboratory (NBL) CRM 125-A was used as a 230Th/234U radiochronometry standard, and CRM U005-A was used as a U isotope composition QC material.
For U isotope composition analysis by MC-ICP-MS, duplicate volumetric aliquots from quaternary dilutions of the subsamples providing approximately 30 ng of U were transferred into pre-cleaned Savillex vials. At the time of aliquoting, aliquots of SRM 960, CRM U005-A, CRM 125-A, and process blanks were also taken for quality control. U assay was determined by IDMS using separate duplicate aliquots of each quaternary dilution taken gravimetrically to provide approximately 10 ng of U for purification and analysis. The U assay aliquots were spiked with 1 ng 233U spike. Both traced and untraced sample aliquots were dried and dissolved in 3 M HNO3 for chemical purification. U was purified using a 1 mL Eichrom UTEVA resin bed cleaned with 0.1 M HCl and conditioned with 3 M HNO3. Samples were loaded onto the column in 3 M HNO3 and sample impurities were then rinsed from the resin with 3 M HNO3 washes followed by 9 M HCl and 5 M HCl washes. U was eluted from the column using 0.1 M HCl. The samples were dried on a hotplate at 90 °C, dried with concentrated HNO3 to treat organics and convert the matrix to a HNO3 form, and then re-dissolved in 2% HNO3 for analysis by MC-ICP-MS.
For Th and Pa ID-MS, duplicate aliquots of each primary dilution were taken gravimetrically with the goal to provide approximately 10 to 100 pg of Th for purification and analysis. These aliquots provided between 1 and 2 pg of 231Pa for analysis. Aliquots were spiked with the 229Th NFRM Th-1 and 231Pa NFRM Pa-1. At the time of aliquoting, a process blank aliquot and an aliquot of certified reference material CRM 125-A were also taken and spiked with 229Th NFRM Th-1 and 231 Pa NFRM Pa-1. After spiking, samples were equilibrated on a hotplate, dried at 150 °C, and then dissolved in 9 M HCl + trace HNO3 and H3BO3 for chemical purification from bulk U. Th was initially separated from U and Pa using a 2 mL BioRad® AG1-X8 anion exchange column where Th has no sorption on the resin, and the resin binds U and Pa. Th fractions were evaporated to dryness on a hotplate and re-dissolved in 8 M HNO3. The Th was then further separated from bulk U using a second 2 mL BioRad® AG1-X8 anion exchange column where the sample was loaded in 8 M HNO3. The column was washed with 8 M HNO3, and Th was eluted with 9 M HCl. This separation was then repeated with a smaller 1 mL resin volume. The final Th purification was done using a 1 mL BioRad® AG1-X8 anion exchange column where the Th aliquot was loaded and eluted in 9 M HCl + 0.01 M HF. Purified Th fractions were evaporated to dryness on a hotplate and dissolved in 2% HNO3 + 0.005 M HF for analysis by MC-ICP-MS.
Pa was separated from the bulk DU matrices using a three-column ion-exchange procedure. The first column consisted of a 2 mL BioRad AG1-X8 resin bed. Samples were loaded in 9 M HCl + trace HNO3 + trace H3BO3 and sample impurities were rinsed from the resin with additional 9 M HCl washes. Pa was eluted from the columns using 9 M HCl + 0.05 M HF. The samples were dried again on a hotplate at 125 °C and re-dissolved in 2% HNO3 + trace H3BO3 prior to the second purification column. This column consisted of a 2 mL silica gel resin bed conditioned with 2% HNO3. The sample was loaded in 2% HNO3 + trace H3BO3 and sample impurities were rinsed from the resin with additional 2% HNO3 washes. Pa was then eluted using 2% HNO3 + 0.05 M HF. The samples were dried again on a hotplate at 125 °C and re-dissolved in 2% HNO3 + trace H3BO3. The silica gel column method was repeated for a final purification on the day of analysis. Samples were immediately transferred to mass spectrometry for same-day analysis to prevent the ingrowth of isobaric, 233U, from the decay of the 233Pa tracer.
Additional radiochemical purification was used to separate Pa from Nb for the DU-Nb alloy samples. Project research and development resulted in modifications to the procedure above, these modifications included: (1) an increase in anion resin column volume from 1 mL to 3 mL; (2) a decrease in the HF concentration of the Pa elution reagent of column 1 from 9 M HCl + 0.05 M HF to 9 M HCl + 0.02 M; and (3) the addition of concentrated HCl washes to column 2 to further remove Nb. The Pa recovery observed for the DU-Nb samples was approximately 50% lower relative to Pa recoveries observed for the pure DU metal samples.
Analytical Measurements were completed on a ThermoScientific Neptune Plus MC-ICP-MS. All purified U fractions were dissolved in 2% HNO3 and introduced into the mass spectrometry using a CETAC Aridus3 desolvating nebulizer system. Acid blank solutions with an identical matrix (2% HNO3) were analyzed before each sample. For U isotope composition measurements, NBL CRM U010 was used for mass bias and gain corrections. Reference standards IRMM 183 and IRMM 186 were measured throughout the analytical session as an unknown quality control (QC) measurement. For U assay, CRM IRMM 074/1 was used to calculate instrumental mass bias corrections; IRMM 074/2 and CRM U010 were measured throughout the analytical session for quality control. In addition, reference materials NBL CRM U005-A, CRM 125-A and NBS SRM 960 processed through the chemical separation procedure alongside the samples were analyzed for further quality control. Retarding Potential Quadrupole energy filters (RPQs) were utilized to decrease the contribution of tailing on 234U, 236U by 235U and 238U. Tail corrections were calculated by measuring four off-peak masses (−0.5, −0.35, + 0.35, and + 0.5 amu away from the peak center), fitting the points to an exponential curve, and subtracting the calculated tail contributions from the measured signal.
For Th, purified sample fractions were introduced into the mass spectrometer as 2% HNO3 + 0.005 M HF solutions and acid blank solutions with an identical matrix (2% HNO3 + 0.005 M HF) were analyzed prior to each sample. Purified samples contained variable Th concentrations which required two analytical sessions using a combination of ion counter and Faraday detectors and a third analytical session that used only Faraday detectors. For process blanks and subsamples of Metal-2, Alloy-4, and Alloy-5, isotope compositions were measured over two analytical sessions using a static multi-collection routine with 229Th and 230Th measured on ion counters and 232Th measured on a Faraday detector. Prior to the start of each analytical session, Faraday gain calibrations were performed using an internally supplied voltage. Certified reference material U010 was used to calculate instrumental mass bias corrections, and an internal Th isotope composition standard was used to determine ion counter gain corrections. For quality control, an additional internal Th isotope composition standard was analyzed throughout each analytical session along with fractions of quality control material CRM 125-A that were purified through chemistry with the samples.
For Metal-1 and Metal-3, all Th isotopes (229Th, 230Th, and 232Th) were measured on Faraday detectors in a single analytical session. Faraday gain calibrations were performed just prior to the analysis using an internally supplied voltage. Certified reference material IRMM 074/1 was used to calculate instrumental mass bias corrections. For quality control, an internal Th isotopic standard was measured throughout the analytical session, along with a fraction of quality control material CRM 125-A that was purified through chemistry with the samples.
For Pa, purified samples were introduced as 2% HNO3 + 0.05 M HF solutions. Pa measurements were made using a static technique with both 231 and 233Pa measured by ion counting. Blanks were measured before each sample using a 2% HNO3 + 0.05 M HF acid blank solution. CRM U010 was measured statically and used for mass bias and gain corrections, and CRM U005-A was measured for quality control of mass bias and gain corrections. SRM 960 was used for hydride corrections on 236U, and an internal Th standard was used for hydride correction on 233Pa. Hydride corrections at mass 233 resulting from the natural 232Th chemistry blank were < 5 counts per second. Aliquots of CRM 125-A that were processed through chemistry were measured for quality control. Nuclear data was used in line with previous publications [34].
LLNL
The following spikes were used for IDMS measurements: for U, an in-house 233U spike calibrated by a gravimetric natural U metal standard solution of CRM 112-A (NBS 960); for Th, the NFRM Th-1 229Th spike (Essex et al., 2017); and for Pa, a 233Pa spike produced internally from 237Np, calibrated using the NFRM Pa-1 231Pa standard (Essex et al., 2019), following procedures by Treinen et al. (2018) [43]. The 233Pa spike was calibrated using the combined results of two calibration analyses bracketing the beginning and end of the spike’s two-month lifespan. For quality control assessment, aliquots of a CRM 125-A solution were processed through separation and purification procedures alongside samples. Blanks were also prepared throughout chemistry procedures (“process blanks”) to trace potential cross contamination and complement information from dissolution blanks.
For U isotope ratio measurements, we processed sample aliquots of approximately 10–25 micrograms of U. For U assay measurements, aliquots were gravimetrically diluted and spiked with the in-house 233U spike. Both the pure DU and DU-Nb samples were purified for U assay measurement using Eichrom UTEVA resin in a BioRad Poly-Prep column. The column was conditioned with 4 M HNO3, and followed by the sample load dissolved in 4 M HNO3. Elemental impurities were then rinsed from the resin with 4 M HNO3 followed by 9 M HCl and 5 M HCl washes. U was eluted from the column using 0.1 M HCl. The samples were dried on a hotplate at 85°C and treated with concentrated HNO3 to remove any organics, and then re-dissolved in 2% HNO3 for analysis by MC-ICP-MS.
To determine the 231Pa and 230Th assay of the pure DU metal samples, we applied the procedure described in Treinen et al. (2018), wherein a single aliquot is processed for both Pa and Th assay. The procedure is briefly summarized as follows: an aliquot of 20–60 mg of U from the primary sample solution was spiked, equilibrated, dried and dissolved in 9 M HCl solution for the first purified of bulk uranium using AG1-X8 anion exchange resin in an Environmental Express column. Whereas U sorbs to this resin in 9 M HCl, Th does not, thus Th was eluted from the column with rinses of 9 M HCl. Subsequently, Pa is collected from the column with rinses of 9 M HCl + 0.05 M HF. With the Th and Pa fractions now separated, these solutions undergo two additional column purification steps to further remove U and any other matrix elements. The Th fraction is purified with Eichrom TEVA resin and a final AG1-X8 anion exchange resin, and the Pa fraction is purified with AG1-X8 resin and a final silica gel resin. The Pa fractions are eluted from the final Si-gel column in 2% HNO3 + 0.05 M HF and are analyzed by MC-ICP-MS in this solution. The Th fractions are then re-dissolved in 2% HNO3 + 0.005 M HF for analysis by MC-ICP-MS.
The determination of the Pa and Th assay of the DU-Nb samples posed a greater analytical challenge due to the presence of undissolved Nb precipitates in pure HCl solution. Therefore, the standard operating procedure for pure uranium metals was modified to include steps specifically for removing Nb. The experiments behind this method development are described in greater detail in Chen et al. (2022) [43]. In short, two additional and identical column purification steps were added to the beginning of the procedure summarized previously: an aliquot of 20–60 mg of U from the primary sample solution was dissolved in a 1 M HF solution instead of 9 M HCl. This solution was then loaded onto a column with 3 mL of AG1-X8 resin. Chen et al. (2022) observed that, for the most part, all elements of interest-U, Th, Pa, and Nb-sorb to the AG1-X8 resin in 1 M HF solution. Pa and Th are then eluted from the resin together with rinses of 9 M HCl. This column step effectively purified the sample of U and Nb by several orders of magnitude. To further increase this purification, this column procedure was repeated a second time. By removing Nb, these additional column steps allowed for the subsequent application of the procedure by Treinen et al. (2018) to separate and purify Th and Pa into different fractions.
Analytical measurements for U, Th, and Pa were performed on either a Nu Plasma 3 multi-collector ICP-MS or a Nu Plasma HR multi-collector ICP-MS, both outfitted with 1011 Ohm resistors on all Faraday collectors. Sample solutions were introduced to both instruments with a CETAC Aridus II desolvating nebulizer system. All measurements were corrected for spike contribution to non-spike masses, Faraday cup and ion counter baselines, instrument background, and instrumental mass bias and ion counter/Faraday gain with CRM U010 with 233U. Quality control standards included in mass spectrometry analytical sessions to verify corrections were CRM U050, CRM 129-A, and CRM 112-A.
For U isotopic and IDMS analyses, the 238U tailing correction on 236U was applied as part of the instrument baseline correction. For U isotopic composition analyses, static multi-collection was configured with 238U and 235U on Faradays and 233U, 234U, and 236U on ion counters. For U assay analyses spiked with 233U, static multi-collection was configured with 238U, 235U, and 233U on Faraday collectors. For Th analyses, no tailing correction was applied, and static multi-collection was configured with 232Th, 230Th, and 229Th on ion counters. For Pa analyses, static multi-collection was configured with 233Pa and 231Pa on ion counters. All analyses consisted of integration times of 10 s per cycle for 30 cycles, except for Pa, for which 40 cycles were measured. All chemistry blanks were <0.001% of sample amounts and considered negligible, and therefore no corrections for blanks were applied to samples.
Results and discussion
Sample selection, sub-sampling and determination of sample production histories
Determination of sample production histories involved consulting archival records of production as well as supporting casting operations in 2018-19 with samples archived to allow for in-growth of decay progeny. A set of five samples were chosen for this initial study of cast DU metal and DU alloy (Table 1). These were cast under known conditions and provided sampling location opportunities on the multi-gram scale. The first two metal samples are a paired feed and cast product from a VIM cast. Figure 4 is a summary of the relevant fuel cycle processes involved.

Summary flow diagram for relevant history of paired feed and product cast samples Metal-1 and Metal-2
Metal-1 is the feed to a VIM cast and has a sample history that is representative of common fuel cycle processes. The sample was cast into a metal plate produced from UF4 salt from a common feedstock. This metal plate was then cast by VIM as a billet on September-3-1987. The billet sample was then VIM cast to a plate on March-14-1995. Because the feedstock material is comprised of a single source, we can minimize possible confounding effects of feedstock recycling in our investigation of radiochronometric signatures of VIM casting processes in chronometric data. The material was assayed for specific reactive elements of interest for casting and material purity. The material was assayed using validated analytical methods as 99.9 ± 0.1 wt% U, (Davies-Gray potentiometric titration), with 60 ± 3 ppm of C (combustion IR), 32 ± 6 ppm of Fe (ICP-MS), 7 ± 2 ppm of Al (ICP-MS),7 ± 0.5 ppm Si (spectrophotometry) and 1.3 +/− 0.1 ppm hydrogen (combustion IR). The sample was cut by Electrical Discharge Machining (EDM) from the edge of the plate and sampled by Knipex end modified snips to provide similar location bulk samples to the laboratories for future studies (Fig. 5).

Example of DU feed Metal-1 cut from plate for intercomparison study
Metal-2 derives from a cast part from a bulk (i.e., multi-hundred kg) quantity of Metal-1 cast on June-05-2019 by VIM. The cast part was sub-sampled to produce samples from multiple locations on the multi-gram scale. This process provided a set of samples for interlaboratory comparison from near identical locations. For this intercomparison, Metal-2 was taken from one of the locations, at the top of the part (below the header) and sampled by hand tools to provide gram-scale samples to the laboratories for analysis. The trace progeny in this material can then be assayed and treated as the initial concentration in the feedstock prior to casting. Pieces of Metal-2 from all locations were analyzed by validated chemistry methods for a variety of relevant analytes. In the case of this sampling location, these were C (80 ± 8 ppm), Fe (31 ± 3 ppm), Al (10 ± 1 ppm), Si (14 ± 0.4 ppm) and U assay (99.9 ± 0.1 wt%) at k = 1 (Fig. 6).

Example VIM casting of Metal-2 through VIM furnace sight glass (left) and example cut Metal-2 for radio-chronometric studies (right)
To complement the study of paired samples Metal-1 and Metal-2, another U metal sample, Metal-3, cast by VIM, was procured from archive materials. Metal-3 is a DU metal sample cast by VIM on March-22-1983. This material was produced from six different feedstocks. Most of these feedstock materials were prepared by wet chemical processing. The material was cast in the same VIM furnace as Metal-2. Metal-3 material had previously been observed to have a discordant Pa age to the known processing history from AWE internal laboratory measurements. This material was cut into small pieces in batches from a location for analysis by EDM and hand tools to provide gram scale samples to the laboratories. Figure 7 summarises this process flow:

Example summary flowsheet for relevant history of DU metal sample with a mixed feedstock for inter-comparison
This sample is a useful comparator to Metal-1 and -2 as all three samples were cast using the same VIM furnace, with the main difference being the blending of feedstocks in Metal-3 and the re-use of the header material from a previous casting in Metal-2 (Fig. 8).

Example size reduced batch of Metal-3 used in this study
Alloy-1 was sampled from a known position on a casting of a DU-Nb alloy. The alloy was produced by the VIM-VAR-VAR method on June-05-1983 [24]. This alloy was then subsequently cast to a part by VIM on March-21-1994 and archived. This sample has undergone four casting processes prior to our analysis of the material. A summary of this material history is shown in Fig. 9.

Example flowsheet of casting history of Alloy-1
Alloy-2, another DU-Nb alloy, was sampled from a casting with a known processing history, produced by VIM from scrap material on Feburary-28-2018 from feedstocks produced in 1982 by VIM-VAR-VAR and offers a comparator to Alloy-1 with an additional casting step. This process is described in Figs. 10, 11:

Flow chart of casting history of recycled Alloy-2

Example DU Alloy-1 (left) and Alloy-2 (right) provided to laboratories for radiochronometry analysis
Analytical measurements of the intercomparison samples
The aim of this work was to determine 230Th/234U and 231 Pa/235U model ages of the five-cast U metal and alloy samples as part of a laboratory inter-comparison. This study was conducted to obtain information on hypothesized chemical fractionation of decay progeny during VIM and VAR casting methods. Additionally, this laboratory intercomparison allowed the three participating laboratories to demonstrate current radiochronometry analytical capabilities.
Radiochronometry measurements of the DU-Nb alloy samples required analytical method development as all laboratories had variable prior experience with DU-Nb alloy digestion and separation. Thus, this exercise served as a realistic test of an unknown forensic sample, where additional method development, validation and verification may be required to produce the required analysis for a customer. Niobium is a homologue of the 231Pa daughter nuclide with similar chemical behavior. The Nb is present in weight% abundance, while 231Pa concentrations were on the order of femtograms. This allowed for the inter-comparison to be used for research and development for the highly selective separation of Pa from Nb.
As shown in Table 2; Figs. 12, 13, 14 and 15, the measured 230Th/234U and 231 Pa/235U model ages are generally reproducible among the three laboratories. The samples analyzed for the intercomparison had no true declared value within experimental design, with only QC samples and reference to the known date of casting to answer the relevant research hypotheses and to assess measurement quality.

Comparison of model ages produced on Metal-1 and Metal-2 by the three laboratories. (k = 2). Orange highlighted area demonstrates that the 231Pa-235U bomb reduction model age is generally preserved through recasting within the approximate timescale (e.g., 1980-81). In contrast, the 230Th/234U model age appears to be partially reset by casting where a header is reused

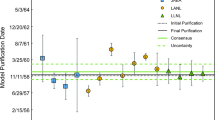
Comparison of model ages produced on Metal-3 by the three laboratories. Uncertainties at k = 2. The last date of processing of the wet chemically processed feedstocks was in 1980, aligning with the measured Pa ages

Comparison of model ages produced on Alloy-1 by the three laboratories. Uncertainties at k = 2

Comparison of model ages produced on Alloy-2 by the three laboratories. Uncertainties at k = 2
In the case of Metal-1, all laboratories determined model ages that are consistent to within 1 to 2 years but not within measurement uncertainty (k = 2). There is a five year discordance (i.e. 13% relative bias) in the 230Th/234U model age relative to the known date of VIM casting in March 1995. The Pa model ages also show a discordant 231Pa/235U age of up to 13 years relative to to the known date of casting. The older Pa age may reflect the feedstock age prior to casting i.e. the date of bomb reduction in 1980–1981. The material was last processed prior to the VIM in March-14-1995 on September-03-1987 according to the records provided. The only prior stage to this was the date of bomb reduction from UF4 into metal in 1980-81 [24]. Further archive work is underway to confirm this date precisely but currently this is consistent with the material production history from U feedstocks. Measurement by three independent laboratories allows for the Th and Pa concentrations in this feedstock material to be well-characterised prior to VIM casting of Metal-1 to generate Metal-2.
Metal-2 is a sub sample of the part that was cast using Metal-1 as the feedstock. The results from three laboratories for both chronometers are again consistent within a 1–2-year time window. Analytical biases could result from different spike calibrations or the use of different certified reference materials for instrument bias and gain corrections during mass spectrometry. LANL and LLNL processed larger aliquots of U than AWE to separate the progeny isotopes but otherwise followed similar methodologies. The metal samples were taken from the cast part as close together spatially as possible. This difference can be investigated in a future inter-comparison by providing each laboratory an aliquot of a single dissolved sample solution for radiochronometry analysis. Measurements of CRMs by all laboratories were reported within control with the certified value for the analysis batches. Spatial variance can be investigated in future studies by analyzing samples from another location on this cast U product. The approach chosen here highlights the next set of logical sampling needs for inter-comparisons. If we do assume sample heterogeneity at sub-gram scale from the above results for a trace impurity, this informs future sampling for materials on the multi-gram scale to remove this variable.
As observed for Metal-1, the model ages for Metal-2 reported by the three laboratories do not reflect the VIM cast date of June-05-2019. There is an offset to relatively older 230Th/234U model ages versus the known casting date. This could be due to incomplete separation of 230Th in the casting process or could result from additional 230Th from the header used in these casts being present in the product material when re-melted.
This data suggests that specific processing, such as the re-use of header material from a previous cast can bias Th/U model ages significantly relative to the age of the sample. This is up to five years in this case, which relative to a three old sample is a major source of bias that reduces as a relative bias the sample ages to percentage levels. Future studies aim to access the feed, product, and header from a cast to further investigate these contributions from casting. The 231Pa/235U model ages for Metal-1 and Metal-2 agree within a 1–2 year window between the laboratories. This finding of similar 231Pa-235U model ages for both a feed and cast product is the first documented observation of the preservation of a radiochronometric signature after subsequent metal casting. This key observation suggests that 231 Pa is not removed during metal casting and has many favorable implications for the use of 231Pa-235U radiochronometry as a fingerprint for metal production activities preceding the last applied casting process. These data suggest that 231Pa is not removed during metal casting. Therefore, 231Pa concentrations in a VIM cast U metal may preserve the model age of the material prior to the casting production. In the case of the paired Metal-1 and Metal-2 materials in this study, we observe potential preservation of a historic 1980–1981 bomb reduction signature. This key observation may support future interpretation efforts for 231Pa-235U radiochronometry when applied to cast samples.
Metal-3 is a DU metal produced in the same VIM furnace as Metal-2 from a variety of feedstocks. Records also show that this cast did not re-use the header material. Interlaboratory data show measured 230Th/234U model ages that are consistent with the known casting date of March-22-1983. These data suggest that the cause of the 3–5 year bias towards 230Th/234U model ages older than the date of VIM casting may result from instances where header material was added from a previous casting. The 231Pa/235U model ages are again discordant with the measured 230Th/234U model ages and are older than the casting date. However, the difference in model age between chronometer pairs is smaller on a relative basis than observed in Metal-1 and 2. The older 231 Pa/235U model ages may be an indication of an earlier wet chemical processing date of the feed material prior to VIM casting. This observation is in-line with the material’s provenance where feedstocks were last processed in 1980, using a mixed feedstock with some residual decay products which is preserved in the measured Pa model age by three laboratories in the 1978-80 timeframe.
Alloy-1 is a depleted U-Nb alloy sample, cast in 1994 by VIM using feed produced from multi-stage casting processes in 1983. The VIM-VAR-VAR process is reported elsewhere, but for completeness a U-Nb alloy is produced from prior VIM cast feedstocks [30]. The cast alloy product is subsequently processed by VAR at least twice to ensure the alloy is homogeneous. The important point here is that continued casting (five times) of the material retains a discordant Th and Pa radiochronometry signature in the model ages of the material relative to the last known casting date.
There is variability between laboratory results. More study will be needed to further probe the cause of this as stated above. The results are, however, within agreement within a 2-year window. The 230Th/234U model ages are up to a decade older than the known last date of casting. The larger relative discordance in the alloy samples could be an artefact from the multi-stage casting process and material recycling during alloy production processes relative to VIM casting. An alternative hypothesis could be that the alloy inhibits efficient segregation of Th from the bulk molten metal. The fact that Nb and Pa are homologs, and that Nb alloys with U, is consistent with the observation that Pa is not effectively segregated from the bulk metal.
The 231 Pa/235U model ages are again discordant relative to the known casting date in 1994 and the last date of the feedstock casting in 1983. This again may retain a prior processing stage signature in the material from the late 1970s. This is evidence to suggest that even through complex casting processes, a U-Nb alloy may retain a 231 Pa/235U model age representing earlier production processes. This provides additional rationale to measure repeatability in different materials cast under the same conditions, as well as measurement of U alloys to add to documented observations of discordance in a diversity of sample types.
Alloy-2 is the second depleted U-Nb alloy sample. This was again produced by more complex VIM and VAR processes, but with a recycled, unprocessed feedstock. The sample was cast from 1980s scrap alloy feed in 2018 by VIM. The material therefore has seen one additional casting stage in comparison to Alloy-1. The 230Th/234U model ages are consistent between the laboratories within measurement uncertainty and are again discordant by more than 14–15 years relative to the casting date in 2018. Model ages for alloyed metals that use a recycled feed show larger 230Th/234U discordance from the known casting date. The 231 Pa/235U model age is up to three decades discordant relative to the known casting date. The larger discordance in this system again implies a prior processing stage in the 1970s, relative to the known ages of the scrap material feed in 1980–1983. This adds further evidence to show the retention of earlier model ages in U materials relative to known casting operations. Overall, both alloy samples allowed for method development and a sample test for the laboratories on Nb/Pa separations. This is the first data on discordant model ages from complex alloy casting processes and gives a platform to plan future work.
Conclusions
A radiochronometry laboratory inter-comparison on five DU metal and alloy samples was completed within a set six-month timeline by three laboratories. The published literature indicates that there is limited understanding of how to interpret discordance between 230Th-234U and 231Pa-235U chronometers. This study of samples of known provenance and casting history provides some of the first data to assist interpretation of discordant model ages in U metals and alloys. This includes the analysis of the first paired feed and product to a VIM cast. This inter-comparison was aligned with research and development needs for the radiochronometry community and demonstrates the reproducibility of radiochronometry model ages for samples from the nuclear fuel cycle and current measurement techniques. This work is the first multi-lab study of its kind and starts to identify a clear research path and ideal samples to use for laboratory inter-comparisons to allow for an understanding of decay product fractionation in relevant nuclear fuel cycle processes. All laboratories generated reproducible 230Th/234U model ages for CRMs used and near identical ages within 1–2 years on cast samples on the sub-gram scale from real production processes. More variability was observed in the interlaboratory 231Pa/235U model age results; however, all 231Pa/235U model ages reported in this study agree within 3 years. This work provides information on the impact of processing on Th and Pa during VIM and VIM-VAR casting. For application to nuclear forensics, the 230Th/234U model ages of VIM cast metals studied here are at most 5 years older than the known casting date. Used in this way, the 230Th/234U chronometer could potentially discriminate samples with ages that differ by a similar relative bias. However, Th/U ages are less accurate for determining alloy metal casting production dates and may significantly older (i.e., larger relative percentage bias) than the true date of production. Other analytical methods then might need to be employed to constrain questions relating to the history of the material. This is evident from the discordance observed for the samples with repeated casting stages. More data from further studies will improve our ability to interpret radio-chronometric ages for U materials produced through complex casting processes. The outcomes will aid the planning of future inter-comparisons on cast samples and inform controlled castings targeting feed, product, and header samples in the future. Lessons learnt allow us to aim to look at spatial heterogeneity, analysis of master dissolutions and targeting specific locations and samples in future studies to identify the causes of radio-chronometric discordance. Overall this will aim to add confidence to our ability to apply these techniques to samples from within the nuclear fuel cycle to answer questions relevant to law enforcement.
References
Nuclear Forensics in Support of Investigations, IAEA Nuclear Security Series No. 2-G (International Atomic Energy Agency, Rev (2015) 1), IAEA, Vienna, 2015
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Kristo M, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. Annu Rev Earth Planet Sci 44(6):555–579
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U-231 Pa age dating of U materials for nuclear forensic investigations. J Anal At Spectrom 28:666–674
Wallenius M, Morgenstern A, Apostolidis C, Mayer K (2002) Determination of the age of highly enriched uranium. Anal Bioanal Chem 374:379–384
Pointurier F, Hubert A, Roger G (2013) A method for dating small amounts of uranium. J Radioanal Nucl Chem 296:593–598
Nguyen CT, Zsigrai J (2006) Basic characterization of highly enriched uranium by gamma spectrometry. Nucl Instrum Methods B 246:417–424
Morgenstern A, Apostolidis C, Mayer K (2002) Age determination of highly enriched uranium: separation and analysis of 231Pa. Anal Chem 74:5513–5516
Keegan E, Kristo MJ, Colella M, Robel M, Williams R (2014) Nuclear forensic analysis of an unknown uranium ore concentrate sample seized in a criminal investigation in Australia. Forensic Sci Int 240:111–121
Kraiem M, Essex RM, Mathew KJ, Orlowicz GJ, Soriano MD (2013) Re-certification of the CRM 125-A UO2 fuel pellet standard for uranium isotopic composition. Int J Mass Spectrom 352:37–43
Essex RM, Williams RW, Rogers KT, Hexel CR, Parsons-Davis T, Treinen KC (2021) A new highly enriched 233U reference material for improved simultaneous determination of uranium amount and isotope amount ratios in trace level samples. Talanta. https://doi.org/10.1016/j.talanta.2020.121638
Essex RM, Williams RW, Treinen KC (2019) Preparation and calibration of a 231Pa reference material. J Radioanal Nucl Chem 322:1593–1604. https://doi.org/10.1007/s10967-019-06711-6
William RW, Gaffney AM (2011) 230Th-234U model ages of some uranium standard reference materials. Proc Radiochem. 1:31–35. https://doi.org/10.1524/rcpr.2011.0005
Wallenius M, Mayer K (2000) Age determination of plutonium material in nuclear forensics by thermal ionisation mass spectrometry. Fresenius J Anal Chem 366:234–238
Kayzar TM, Williams RW (2016) Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Nucl Chem 307:2061–2068
Taylor F, Higginson M, Marsden O, Schwantes J (2020) State of practice and emerging application of analytical techniques of nuclear forensic analysis: highlights from the 5th Collaborative Materials Exercise of the Nuclear Forensics International Technical Working Group (ITWG). J Radioanal Nucl Chem 323:415–430
Kristo MJ, Tumey SJ (2013) The state of nuclear forensics. Nucl Instrum Methods Phys Res Sect B 294:656–661
Higginson MA, Thompson P, Dawkins B, Taylor F, Kaye P (2020) Application of uranium radio-chronometry to interpret uranium samples of known provenance. Environ Radiochemical Anal. https://doi.org/10.1039/9781788017732-00106
Tandon L, Kuhn K, Martinez P, Banar J, Walker L (2009) Establishing reactor operations from uranium targets used for the production of plutonium. J Radioanal Nucl Chem 282:573–579
Sturm M, Richter S, Aregbe Y, Wellum R, Mialle S (2014) Evaluation of chronometers in plutonium age determination for nuclear forensics: what if the ‘Pu/U clocks’ do not match? J Radioanal Nucl Chem 302:399–411
Kristo M, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. Annu Rev Earth Planet Sci 44:555–79
Hanlen R (2011) Round Robin 3 Exercise After Action and Lessons Learned Report. PNNL-20079. Pacific Northwest National Laboratory, U.S. Department of Energy, Richland, WA, p 78
Martin FS, Miles GL, Series III (1956) Process chemistry. In: Bruce FR, Fletcher JM, Hyman HH, Katz JJ (eds) McGraw-Hill Book Col, New York, pp 407
Feder HM, Chellew N, Ader M, Series III (1956) Process chemistry. In: Bruce FR, Fletcher JM, Hyman HH, Katz JJ (eds) McGraw-Hill Book, New York, pp 407
Whitman CI, Compton V, Holden RB (1957) Zone melting of uranium. J Electrochem Soc 104:240–244
Antill JE, Barnes E, Gardner M (1959) Zone melting of uranium. In: Finniston HM, Howe JP (eds) Progress in nuclear energy. Series V, Metallurgy and Fuels. McGraw-Hill, New York, pp 9–18
Schwantes JM, Marsden O (2020) Twenty years of collaborative materials exercises by the nuclear forensics International Technical Working Group. United States, IAEA-TECDOC—1896, ISBN 978-92-0-100920-3
Kovarik L, Lach T, Reilly DD (2019) Characterization of slag and metal from uranium bomb reduction: morphology, speciation, and the search for Th. Mater Charact. https://doi.org/10.1016/j.matchar.2019.109948
Harrington CD, Ruehle AE (1959) Uranium production technology. D. Van Nostrand Co., Inc., New York, pp 262–269
Wyatt LM (1956) The fabrication of uranium and alloys, progress in nuclear energy, V, metallurgy and fuels. Pergamon Press, New York, pp 39–61
Baird JE, Carson NJ (1957) Melting and casting of uranium, zirconium, niobium alloys. Nucl Metall 4:31–38
Higginson M, Gilligan C, Taylor F et al (2018) Development of rapid methodologies for uranium age dating. J Radioanal Nucl Chem 318:157–164. https://doi.org/10.1007/s10967-018-6021-
Rolison JM, Treinen KC, McHugh KC, Gaffney AM, Williams RW (2017) Application of the 226Ra-230Th-234U and 227Ac-231 Pa-235U radiochronometers to uranium certified reference materials. J Radioanal Nucl Chem 314:2459
Taylor BN, Kuyatt CE (1994) Guidelines for evaluating and expressing the uncertainty of NIST measurement results. NIST Technical Note 1297
Kayzar-Boggs TM, Treinen KC, Okubo A (2020) An interlaboratory collaboration to determine consensus 231 Pa/235U model ages of a uranium certified reference material for nuclear forensics. J Radioanal Nucl Chem 323:1189–1195
Browne E, Tuli JK (2007) ENSDF insertion: 2007–04. Publ Nucl Data Sheets 108:681
Browne E, Tuli JK (2014) ENSDF insertion: 2014–11. Publ Nucl Data Sheets 122:205
Browne E, Tuli JK (2008) ENSDF insertion: 2008–10. Publ Nucl Data Sheets 109:2657
Browne E, Tuli JK (2012) ENSDF insertion: 2012–09. Publ Nucl Data Sheets 113:2113
Browne E, Tuli JK (2013) ENSDF insertion: 2013–06. Publ Nucl Data Sheets 114:751
Singh B, Tuli JK, Browne E (2020) ENSDF insertion: 2020–12. Publ Nucl Data Sheets 170:499
Bateman H (1910) The solution of a system of differential equations occurring in the theory of radioactive transformations. Proc Cambridge Philos Soc 15:423–427
Chen CY, Kayzar-Boggs TM, Higginson M, Denton JS, Dunne J, Edwards MA, Eng C, Engel J, Gaffney AM, Gilligan C, Morris MN, Rolison JM, Sanborn ME, Wende AM (2022) Refining the isolation and purification of protactinium from uranium-niobium alloys for 231Pa-235U radiochronometry for nuclear forensics. Int Conf Methods Appl Radioanal Chem 2:5548
Treinen KC, Gaffney AM, Rolison JM, Samperton KM, McHugh KC, Miller ML, Williams RW (2018) Improved protactinium spike calibration method applied to 231Pa–235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem 318(1):209–219
Acknowledgements
We thank AWE for funding this work at AWE. LANL and LLNL thank the NA-83 Office of Nuclear Forensics for funding the laboratory intercomparison. A portion of this work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344, LLNL document ID - LLNL-JRNL-834140. Work performed at Los Alamos National Laboratory (operated by Triad National Security, LLC) was performed under the auspices of the U.S. Department of Energy under contract 89233218CNA000001. LANL document ID - LA-UR-22-26640
Author information
Authors and Affiliations
Contributions
M.H., A.M.G., C.Y.C. and T.K.-B. were lead investigators for the intercomparison. J.D., S.C., C.G., J.D., M.A.E., M.S. and A.W. were involved in the formal analysis and investigation and review and editing of the manuscript. C.Y.C. performed radiochronometric analyses by LLNL and led the Pa-Nb separations method development work with assistance from M.N.M. and expertise by J.M.Rolison and A.G. C.Y.C. also performed microscopy on samples guided by expertise of C. Eng. Higginson: Funding Acquisition, Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Project Administration, Software, Supervision, Validation, Visualization, Writing– Review and Editing. Kayzar-Boggs: Conceptualization, Funding Acquisition, Data Curation, Formal Analysis, Investigation, Methodology, Project Administration, Software, Supervision, Validation, Visualization, Writing – Review and Editing. Chen: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Project Administration, Supervision, Validation, Visualization, Writing – Review and Editing. Cross: Formal Analysis, Investigation, Writing – Review and Editing. Denton: Formal Analysis, Investigation, Methodology, Writing – Review and Editing. Dunne: Formal Analysis, Investigation, Writing – Review and Editing. Edwards: Formal Analysis, Investigation, Writing – Review and Editing. Eng: Investigation. Gaffney: Conceptualization, Funding Acquisition, Project Administration, Resources, Supervision, Writing – Review and Editing. Gilligan: Formal Analysis, Investigation, Writing – Review and Editing. Morris: Investigation, Methodology. Rolison: Conceptualization, Investigation, Methodology, Supervision. Sanborn: Formal Analysis, Investigation, Writing – Review and Editing. Wende: Formal Analysis, Investigation, Writing – Review and Editing.
Corresponding author
Ethics declarations
Conflict of interest
All authors have declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Higginson, M.A., Kayzar-Boggs, T.M., Chen, C.Y. et al. Establishing discordance as a radiochronometric signature for nuclear forensic investigations: a multi-laboratory intercomparison exercise. J Radioanal Nucl Chem 331, 4799–4815 (2022). https://doi.org/10.1007/s10967-022-08428-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-022-08428-5