
Abstract
Radiochronometry analyses of three hydrolyzed UF6 gas samples were performed using the 230Th-234U and 231Pa-235U chronometers. All 230Th-234U model ages are younger than, and two of the three 231Pa-235U model ages overlap with, the known UF6 gas transfer dates. The 230Th-234U discordance is caused by 8–10% 230Th loss relative to the measurement reference date, likely during alkali hydrolysis. These results confirm that UF6 gas transfers effectively purify uranium from daughter progeny. Daughter progeny fractionation can occur if solutions are not kept within optimal conditions post-UF6 gas transfer and hydrolysis, and discordance between the 230Th-234U and 231Pa-235U chronometers may result from the laboratory procedures used to prepare samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
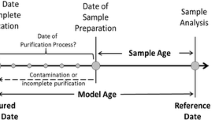
Radiochronometric dating is an important nuclear forensic tool that can be used to identify the history of an unknown nuclear material and potentially provide genetic links between different nuclear materials. In radiometric dating by mass spectrometry, the concentrations of radioactive parent, daughter, and in some cases, granddaughter, nuclides are measured to define a “model age,” or time that has elapsed since the latest chemical purification of the parent isotope from progeny nuclides. In most cases, this model age is interpreted as the date when the material was produced, although in practice this may not be the case if a material was not completely chemically purified of progeny isotopes upon production, or if it has not existed as a closed system (i.e., no gain or loss of nuclides from the bulk material) in the ensuing time. Although it is impossible to know whether these assumptions have been fully met, the measurement of multiple chronometers in the same sample provides a strong independent avenue to assess the validity of model age assumptions.
The degree to which these assumptions are met in diverse materials produced during different stages in the nuclear fuel cycle is an area of active research; different materials that are both consistent and inconsistent with model age assumptions have been recognized [1,2,3,4]. Furthermore, simply because a material is discovered to have discordant model ages does not preclude gaining useful information from the radiochronometric investigation. For example, the different chemical behavior of actinide elements along with the differing potential for these materials to be contaminated from the ambient environment can allow for indirect knowledge to be gained about a sample’s history from the discordant results. Here, we investigate the behavior of two daughter-parent radiochronometers in a common fuel cycle material to glean understanding of the meaning and significance of measured model ages.
Since World War II, gaseous diffusion was the primary method used to produce enriched 235U in the U.S. In this process, uranium is fluorinated to form a hexafluoride gas and propelled through a complex pressurized system of pipes and barriers to separate the uranium isotopes by mass, repeating the process hundreds if not thousands of times in an enrichment cascade to produce the desired 235U enriched product. Other enrichment approaches utilize the gas centrifuge process, which also utilizes UF6 gas as a feed material. UF6 gas and related phases from both enrichment processes may be stored in pressurized cylinders at a variety of stages throughout the conversion and enrichment process. However, U-series daughter and granddaughter progeny, including Th, Pa, Ac and Ra, theoretically do not form volatile fluorides at the pressure–temperature conditions under which UF6 is in the gas phase [5], so any conversion of a uranium hexafluoride gas to a different phase is predicted to reset the model age clock to the time the gas was converted into a form that was capable of retaining progeny nuclides, usually when converted to a solid fluoride or oxide form. Previous analysis of solid uranium hydrolysis products (“heels”) extracted from emptied UF6 gas cylinders found that these materials contain the bulk of daughter and granddaughter progeny produced from radioactive decay of the uranium, so that the model ages of the heels are well in excess of the known times of cylinder filling [3]. However, it is unknown via direct measurements of real-world nuclear fuel cycle materials whether the UF6 gas in equilibrium with the heels will exhibit complete radiochronometric resetting to the date when it was extracted from the cylinder and converted to a liquid or solid form. Here we investigate this process through radiochronometric analysis of a set of solid hydrolyzed uranium samples produced from highly enriched UF6 gas extracted from cylinders at a known time. The model ages of the cylinder heels from one of the same cylinders were determined in a previous study [3]. The full complimentary nature of all of these analyses allow for a comprehensive understanding of the behavior of parent and progeny isotopes in UF6 gas cylinders and provide the foundational knowledge vital towards interpretation of these signatures in similar, yet unknown, materials.
Sample description & history
The samples analyzed in this study are highly enriched (HEU) hydrolyzed uranium hexafluoride gas extracted from three 2S cylinders that originated from the Portsmouth Gaseous Diffusion Plant in Pike County, Ohio. Photographs and descriptions of the sample history are presented in Fig. 1.

Photos of hydrolyzed uranium sample preparation, used with permission from Materials and Chemistry Laboratory, Inc (Oak Ridge, TN). a the gas extraction setup showing the 2S cylinder attached to a transfer system that contains a 50 mL stainless steel gas cylinder transfer vessel and two fluoropolymer hoke P10 tubes (photo from W. Bostick). b hydrolyzed UF6 solutions (photo from W. Bostick). c solid hydrolyzed uranium samples under ultraviolet light exhibiting strong luminescence (photo from W. Bostick). d hydrolyzed UF6 samples once converted to solids (photo from W. Bostick); samples were shipped to LLNL in these sample containers. e aliquoted samples before digestion at LLNL
The UF6 gas that produced 396HYD was stored in a 2S cylinder made of Monel while the UF6 gas sources of 255HYD and 146HYD were stored in nickel cylinders. These cylinders were filled directly from the enrichment cascade at the Portsmouth Gaseous Diffusion Plant (PORTS) and were originally used as reference standards in the plant analytical laboratory. PORTS operated between 1956 and 2001, and after shuttering of the plant some of these gas cylinders containing HEU materials were shipped to Nuclear Fuel Services (NFS) in Erwin, Tennessee. Five cylinders containing various levels of 235U enrichment were reserved for further sampling and analysis because of their well-known history.
Once the cylinders were at NFS, the extractable UF6 gas was removed via heated gas transfer and the cylinders were shipped to Materials and Chemistry Laboratories, Inc. (MCL, Inc.) in Oak Ridge, Tennessee for sampling and archival of the remaining residual uranium fluoride products (‘heels’). The dates of the gas transfers are reported in Table 1. After samples were received and weighed at MCL, Inc., the cylinders were attached to a gas manifold system that was new for each canister to prevent cross contamination (Fig. 1a). Non-condensable gasses hydrofluoric, nitrogen, and fluorine were removed by evacuation of the canister while under ice, followed by gentle heating in a warm water bath to sublimate UF6 gas to two P10 tubes after a brief collection and weighing period in the transfer volume chamber. If there was excess UF6 gas after filling the two P10 tubes, this material was stored in the transfer vessel for a short time before being transferred into a cleaned perfluoroalkoxy (PFA) tube. The UF6 was converted to a liquid via hydrolysis by adding de-ionized water to the PFA tube, producing uranyl fluoride (UO2F2) and hydrofluoric acid (Fig. 1b). After storage for several months, the HF was removed from solutions by the addition of ammonium carbonate and dried to form bright yellow solids that are highly luminescent under UV light (Fig. 1c). These solid uranyl compounds produced by drying down neutralized hydrolyzed uranium hexafluoride gas were shipped to Lawrence Livermore National Laboratory (LLNL) and analyzed as a part of this study (Fig. 1d and e). A schematic representation of the sample history and preparation of samples investigated in this report is shown in Fig. 2 (W. Bostick, personal communication).

Representation of the history of gas cylinders sampled in this study. At some time in the past (t = 0), cylinders were filled with UF6 from the Portsmouth Gaseous Diffusion Plant and set aside for several years, during which time small amounts of UF6 gas may have been extracted for use as a reference material. At t = 1, here in late 2011- early 2012, the filled gas canisters were shipped to Nuclear Fuel Services (Erwin, TN) where the bulk of the UF6 gas was extracted (t = 2). The canisters were then shipped to Materials and Chemistry Laboratory, Inc (Oak Ridge, TN), during which time some of the solid UF5 sublimated to produce a small amount of UF6 gas (t = 3), which was then extracted in several evacuation steps at MCL, Inc. The gas extracted at t = 4 was reacted with water to create hydrolyzed uranium solutions. These hydrolyzed uranium solutions were stored for several months until they were reacted with ammonium carbonate to neutralize the high fluorine concentrations (t = 5). These solutions were dried down to form solid uranyl compounds and transferred to long-term storage containers for shipment to LLNL (t = 6). After all the UF6 gas was extracted from the canisters, they were sawed open and the solid “loose heel” material was extracted and analyzed in a parallel study [3]
After extraction of the UF6 gas, the cylinders were cut open to extract solid hydrolysis uranium materials (i.e., the “loose heel” material) that could be poured out of the cut cylinders, and wall deposit material that was scraped from the sides of the cylinders. These materials were investigated in Rolison and Williams [3].
Experimental
Radiochronometry analyses at LLNL use ultra-high purity acids and water purified to > 18.2 MΩ for labware cleaning and analytical dilutions. The procedure used in this study follows the method outlined in detail in Treinen et al. [6].
Single aliquots of each hydrolyzed uranium sample were digested by weighing approximately 200 mg of solid material into a cleaned quartz tube and dissolving with 8 M HNO3 for several hours while heating. After this time there were no insoluble residues observed. Samples were then transferred into cleaned PFA vials and diluted into a final solution of 4 M HNO3 + 0.05 M HF; this acid matrix has been demonstrated to stabilize uranium and progeny isotopes in solution for long periods of time [1]. The concentration of U in these primary solutions was 9–10 mg U/g solution. Separate aliquots of the digested sample solutions were taken for isotope dilution uranium assay and uranium isotopic composition analysis. Uranium concentrations were determined by isotope dilution mass spectrometry after a series of dilutions were made to achieve optimal sample-spike ratios of 235U with a very high purity 233U spike (> 99.9877% n (233U)/n(U)) calibrated in-house using NBL CRM 112A (dilutions for uranium isotope dilution measurements had approximately 8 ng U/g solution in the analytical aliquot). The isotope dilution aliquots were spiked and equilibrated and then both the spiked and the unspiked aliquots were purified by extraction chromatography using UTEVA resin (Eichrom Technologies Inc.).
Th and Pa concentrations were measured on the same aliquot via isotope dilution mass spectrometry. Weighed sample aliquots were spiked with a high-purity nuclear forensic reference material (NFRM) 229Th spike [7] and a 233Pa spike that is produced by separation of 233Pa from a 20–60 mg 237Np parent solution; the resulting 233Pa spike is calibrated with a 231Pa high purity standard [8] before use (see [6] for a more detailed discussion of 233Pa spike production and calibration). Th and Pa are then separated using a series of anion, TEVA, and silica gel purifications to produce analytical aliquots free of uranium (key for protactinium analyses, where the continual production of 233U from decay of the 233Pa spike must be minimized for accurate results).
All isotopic analyses were completed using a Nu Plasma HR MC-ICP-MS using CRM U010 to correct for instrumental mass bias and Faraday/ion counter gain factor; corrections were applied using the sample-standard bracketing method. CRMs U630, U850, and U930 were analyzed with samples for uranium isotopic quality control materials; all analyses were within error of certificate values. Uranium isotope dilution samples were analyzed with 238U, 236U, 235U, 234U and 233U masses measured concurrently on Faraday detectors; uranium isotopic composition samples were analyzed with 236U, 234U, and 233U measured on ion counter detectors and 238U and 235U measured on Faraday detectors. Thorium isotopes were measured both using a peak jumping program where the 229Th and 230Th beams were jumped onto the same ion counter to eliminate error introduced by relative ion counter gain factor correction on the 229Th/230Th measurement and with 229Th and 230Th concurrently analyzed on separate ion counters. A second analytical aliquot was prepared and analyzed by Th isotope dilution mass spectrometry about 8 months after the original analysis to confirm the analytical results; replicate Th analyses, corrected for radioactive decay of 234U between the times of the two analytical sessions, yield 230Th concentrations that are identical within analytical uncertainty. All protactinium isotopes are analyzed concurrently on separate ion counters as soon as possible after final purification. Uncertainties on all isotope analyses are calculated following the guidelines laid out by GUM [9].
Total procedural blanks were below the detection limit for 230Th, 0.05 fg/g for 231Pa, and 8.5 pg/g for U, respectively, and were measured in spiked blank aliquots processed alongside samples. Because these amounts of blank contribution are negligible compared to analyte in samples, no blank subtraction correction was applied to the results. Model ages are calculated by solving the Bateman equations for generalized radioactive decay assuming there were no daughter nuclides present at t = 0 [10] (Eq. 1).
The decay half-lives used in model age calculations are 230Th = 75,690 ± 115 years, 234U = 245,250 ± 490 years, 231Pa = 32,760 ± 110 years, and 235U = 7.0381 × 108 ± 4.8 × 105 [11,12,13].
Results
All hydrolyzed uranium samples are highly enriched uranium (HEU) that contain the anthropogenic isotopes 236U and 233U, indicating that reprocessed uranium was used in the feed material of the enrichment cascade (Table 2). The samples have 91.21, 93.25, and 88.27 atom percent 235U for samples 146HYD, 255HYD, and 396HYD, respectively.
Thorium assay and isotopic compositions are reported in Table 3. As is apparent from the low overall concentrations of 232Th (6–10 ppb), the sample preparation of the hydrolyzed UF6 gas at both MCL, Inc and LLNL were very clean for Th, and it is therefore unlikely that samples are contaminated with appreciable amounts of 230Th introduced during processing since the UF6 gas was extracted.
231 Pa-235U and 230Th-234U radiochronometry results
Radiochronometry analyses were performed using 230Th-234U and 231Pa-235U isotopic systems. The model age results measured in the dried uranyl compounds are presented in Table 4 and Fig. 3. These results are not corrected for initial intrinsic 229Th that existed in the samples at the time of ThID measurement from the decay of 233U, as the amount of 233U in the samples was so small that this correction results in a change in the Th-U model age of a few hours, negligible compared to the uncertainty of the radiochronometric measurement itself.

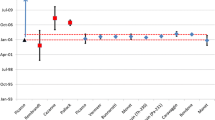
Radiochronometry results of hydrolyzed UF6 gas; diamonds are the 231Pa-235U age and circles are the 230Th-234U age. Error bars indicate the combined and expanded uncertainty (k = 2). The NFS bulk UF6 gas removal for the cylinders is labeled, and the other dashed lines indicate the first, second, third (if applicable) gas transfer dates at MCL, Inc, and the date the gas was hydrolyzed, if known. Also shown for reference is the date that the ammonium carbonate was added to samples, followed by immediate dry down
All 230Th-234U model ages are younger than the gas transfer dates, indicating 230Th that originated from decay of UF6 gas since the time the cylinders were originally filled at the PORTS plant was quantitatively left behind by the extraction of UF6 gas from the fuel cylinders (Fig. 3). For samples 396HYD and 146HYD, the 231Pa-235U age overlaps with the date of gas transfer at MCL, Inc, while the 231Pa-235U model age of sample 255HYD plots between the gas extraction dates at NFS and MCL, Inc. If the 255HYD 231Pa-235U model age is considered to be erroneously older than the gas transfer date at MCL, Inc, the amount of 231Pa that is in excess is on the order of 4 × 107 atoms, or < 0.0004% of the amount measured in the PaID measurement (and less than the analytical blank); it is probable this amount of 231 Pa was inadvertently added during sample processing. There is discordance between the Th-U and Pa-U model ages, as the model ages of 230Th-234U ages are younger by several months than the Pa ages.
Discussion
The uranium isotopic compositions of the hydrolyzed UF6 gas and the solid cylinder heel material are indistinguishable within error and agree with the isotopic compositions reported for this material in original documents from Portsmouth, indicating that these materials were not contaminated by extraneous U, and by extension, significant amounts of Th or Pa, during processing or handling. The discordance in the ages between the 230Th-234U and 231Pa-235U chronometers was unexpected, as the controlled UF6 gas extraction was assumed to have completely purified the parent uranium atoms from daughter progeny. Uranium hexafluoride will be sublimated starting at approximately 56.5°C at room pressure. Protactinium will not form a fluoride gas at all, while thorium requires temperatures in excess of 800–850°C to form a ThF4 gas [14, 15]. Previous work has shown that Th and Pa are elevated above the expected concentrations for samples of their age in uranium cylinder heel material because Th and Pa do not form fluoride gasses under the conditions that UF6 gas is extracted from cylinders, and are therefore quantitatively retained in the solid cylinder heel material while UF6 gas is evacuated [3].
The discordance of the 230Th-234U chronometer with the known gas transfer dates represents an age bias of < 10% at the time of measurement and indicates that there may have been some additional loss of 230Th after the time the UF6 was hydrolyzed; this could have occurred during reaction of the solutions with ammonium carbonate, storage in solution form, or processing at LLNL. It is unlikely that the uranium isotopes extracted from the UF6 cylinder would be fractionated by mass, as the fractionation factor of UF5 sublimating at < 100°C are so small that no measurable fractionation would occur. Furthermore, the uranium isotopic composition between the hydrolyzed uranium samples studied here and the cylinder heels analyzed elsewhere agrees to less than 1% relative standard deviation on all isotopes, suggesting isotopic fractionation is negligible. Therefore, the likely scenario is loss of a small amount of 230Th during preparation of these samples.
The deficit in 230Th in the hydrolyzed uranium samples is equivalent to 6–8 months, or approximately 31–40 ng of 230Th per gram of U (Table 5). It is apparent that 230Th is very sensitive to environmental conditions and laboratory processing post UF6 gas extraction, effects that are exacerbated by the fact that most of the investigated materials were dated relatively soon after extractions of the UF6 gas (i.e., young 230Th-234U ages). For the hydrolyzed uranium samples, it is likely that the sample processing method post UF6 hydrolyzation resulted in the loss of < 10% 230Th from the solutions. This percentage is calculated based on the theoretical amount of 230Th that should be present in samples based on radioactive decay equations and the time elapsed between the known gas extraction age and the reference date when samples were measured. We explore the possible mechanisms for this below.
When the cylinders were received at MCL, Inc, they were weighed, photographed, and drained of all remaining UF6 gas through a gentle heated gaseous transfer on a manifold extraction system. Samples collected were stored in PFA tubes until they were hydrolyzed soon after by the addition of water to the frozen UF6 gas. This step produces a vigorous reaction resulting in aqueous uranyl fluoride and hydrofluoric acid. The resulting solutions had a pH ≈ 2 and a high free fluoride ion concentration, optimal for keeping trace actinides in solution. Uranium hydrolysis proceeds by the generalized reaction [16]:
Samples were stored in these low-pH and high fluorine concentration solutions for approximately 1.6–1.8 years. Then, in order to mitigate the hazardous hydrofluoric acid concentrations and low pH of these solutions, ammonium carbonate was added to samples in amounts assumed to fully react with the quantity of HF present in the hydrolyzed uranium solutions (here approximately 8 mol ammonium carbonate for every one mole of uranium). This reaction results in a basic solution (pH ≈ 9) of dissolved ammonium fluoride salt, water, and eventual formation and release of gaseous carbon dioxide after the formation of an intermediate carbonic acid. The generalized reaction is:
After this step, no precipitate was observed in these solutions, which were transferred to glass containers for drying down on a hot plate. The PFA tube was rinsed once with deionized water and added to the solutions drying down. Excess ammonium carbonate will break down upon heating and be fumed from the solution as a mixture of carbon dioxide, ammonia, and water; similarly, excess HF will be fumed off as a gas. Once fully dry, the solid materials were transferred to a TFE container for shipment to LLNL.
Uranium assay values of the final dried solid are low compared to what would be expected for a pure uranyl fluoride (UO2F2·H2O, 0.73 g U/g sample), and is instead much closer to the stoichiometric assay of ammonium uranyl carbonate (56 wt % U) and ammonium uranyl fluoride (62 wt % U; see Table 6). Ammonium uranyl carbonate and ammonium uranyl fluoride are highly fluorescent while UO2F2 is not; the hydrolyzed uranium samples analyzed here are highly fluorescent (see Fig. 2). The U assay values suggest that uranyl fluoride produced by the hydrolysis of UF6 gas was quantitatively converted to ammonium uranyl carbonate by the alkali hydrolysis.
Therefore, the entire sample chemical processing can be described by the generalized equation [17]:
There are several potential areas for loss of thorium during this preparation. Hydrolysis of uranium hexafluoride is sometimes accomplished by reaction of the UF6 with pure water as it was here, and other times it is reacted with low molarity nitric acid solutions. Thorium is predominately in the 4+ oxidation state when it is in solution, and is very stable in solutions of nitrate, chloride, sulphate, and perchloric acids [18]. When uranium hydrolysis is carried out with nitric acid solutions, the resulting thorium compound is the highly soluble Th(NO3)4.6H2O. However, in very strong solutions of hydrofluoric acid, thorium will form insoluble fluorides that are pure if dried down multiple times in the presence of NH4F, otherwise it is a white, gelatinous precipitate of ThF4 hydrate that is actually a mix of ThF4 + ThO2 + ThOF2 [19, 20]. This step may be a possible candidate for the fractionation of thorium from protactinium because protactinium is very stable in strong complexing acids such as hydrofluoric and sulfuric acids [21, 22]. However, if the formation of thorium fluoride was quantitative as the thorium grew from uranium decay and left behind as a gelatinous precipitate on the PFA walls when the solution was transferred, it would have resulted in a complete resetting of the 230Th-234U age to the age of the alkaline hydrolysis and dry down rather than only losing ~ 10% of the 230Th that should have decayed between the known gas transfer date and the measurement reference date. Therefore, it is likely most of the thorium fluoride precipitate was transferred with the solution and rinse to the final sample dry-down container.
The second candidate for loss of 230Th is the alkaline hydrolysis of the solutions before the final dry down. Typically, when the pH of a solution containing Th4+ is elevated to a pH above 3.5, the thorium will be hydrolyzed to form a Th(OH)4 gelatinous precipitate [20, 23, 24]. However, this reaction will not occur if there is excess carbonate in the solution to complex with the thorium as would have been the case with the addition of so much ammonium carbonate. In an excess of carbonate, thorium will be precipitate as (NH4)2Th(CO3)3·6H2O [20]. It is possible that at some point there was an incomplete reaction of thorium that left a small 10% amount of the total 230Th as a thorium fluoride hydrate stuck to the PFA tube walls, or as a small amount of thorium hydrolysis products that stuck to the PFA tube wall; targeted experiments could test which scenario is the most likely mechanism for thorium loss.
Conversely, in concentrated solutions of hydrofluoric acid, protactinium exists in the Pa5+ oxidation state as incredibly stable PaF72− anions [22]. There would therefore have been excellent stability of the protactinium in solution during UF6 hydrolyzation and subsequent storage as low pH, high F− molarity solutions. When solutions of protactinium in hydrofluoric acid are dried down, the result is readily soluble Pa2OF5 salts [25]. Similarly, alkali hydrolysis and carbonate precipitation will result in the precipitation of protactinium that will not undergo a reverse reaction to re-dissolve in the solution [21]. There is, consequently, conservation of all the protactinium in the system. It should be noted that even though protactinium is notoriously sticky to glassware, the strong complexation of the protactinium with fluorine, followed possibly by carbonate, resulted in the complete conservation of protactinium in this system [21, 25]. It is unknown whether the final solid product of protactinium was a fluoride or carbonate compound.
The coherence of the chemistry of these samples is supported by the fact that all three samples exhibited very similar losses of thorium; each hydrolyzed uranium sample lost between 7.8 and 9.9% of the amount of 230Th that is predicted based on the time elapsed between the date of UF6 gas extraction and the measurement reference date. This suggests that the process that resulted in the loss of 230Th is reproducible within 2%, which appears systematic rather than random. Overall, there is agreement that the extraction of UF6 gas from a fuel cylinder separates the parent isotope uranium from daughter progeny during low-temperature controlled releases of gas, even in materials in real-world use. Furthermore, if conservation of daughter progeny is desired in the resulting hydrolyzed uranium, the solutions should be dried down immediately to prevent fractionation in the 230Th-234U chronometer.
Conclusion
The results found in this study support the hypothesis that the removal of UF6 gas from the cylinder effectively purifies the uranium from daughter progeny quantitatively as only the uranium parent atoms form a hexafluoride gas that is extractable by low-temperature gaseous transfer. This scenario satisfies the underlying assumptions of radiochronometry model ages, and the 230Th-234U and 231Pa-235U model ages should record the date of UF6 gas transfer if care is taken to ensure progeny nuclides are conserved in the subsequent prepared UF6 hydrolyzed solution. However, the fractionation of daughter progeny can easily occur if solutions are not kept under optimal conditions, and discordance between the 230Th-234U and 231Pa-235U radiochronometers in hydrolyzed uranium samples may indicate such steps occurred during sample processing.
References
Williams RW, Gaffney AM (2011) 230Th-234U model ages of some uranium standard reference materials. Radiochim Acta 1:31–35
Gaffney AM, Hubert A, Kinman WS, Magara M, Okubo A, Pointurier F, Schorzman KC, Steiner RE, Williams RW (2016) Round-robin 230Th-234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem 307:2055–2060
Rolison JM, Williams RW (2018) Application of the 226Ra-230Th-234U and 227Ac-231Pa-235U radiochronometers to UF6 cylinders. J Radioanal Nucl Chem 317:897–905
Varga Z, Mayer K, Bonamici CE, Hubert A, Hutcheon I, Kinman W, Kristo M, Pointurier F, Spencer K, Stanley F, Steiner R, Tandon L, Williams RW (2015) Validation of reference materials for uranium radiochronometry in the frame of nuclear forensic investigations. Appl Radiat Isot 102:81–86
Shimada S, Okumura I, Higashi K (1973) Experimental study on fluoride volatility process for thorium fuels. J Nucl Sci Tech 10(11):689–695
Treinen KC, Gaffney AM, Rolison JM, Samperton KM, McHugh KC, Miller ML, Williams RW (2018) Improved protactinium spike calibration method applied to 231Pa-235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem 318:209–219
Essex RM, Mann JL, Williams RW, Kinman WS, Hubert A, Bennett ME, Gurgiotis A (2018) A new thorium-229 reference material. Appl Radiat Isot 134:23–31
Essex RM, Williams RW, Treinen KC, Colléfitzgearld R, Galea R, Keightley J, LaRosa J, Laureano-Pérez L, Nour S, Pibida L (2019) Preparation and calibration of a 231Pa reference material. J Radioanal Nucl Chem 322:1593–1604
BIPM, IEC, IFCC, ISO, IUPAC, IUPAP, OIML (1995) Guide to the Expression of Uncertainty in Measurement. International Organization for Standardization, Geneva. ISBN 92-67-10188-9, First Edition 1993, corrected and reprinted 1995. (BSI Equivalent: BSI PD 6461: 1995, Vocabulary of Metrology, Part 3. Guide to the Expression of Uncertainty in Measurement. British Standards Institution, London)
Bateman, H (1910) Solution of a system of differential equations occurring in the theory of radioactive transformations. Proc Camb Philos Soc 15(V): 423–427
Jaffey AH, Flynn KF, Glendenin LE, Bentley WC, Essling AM (1971) Precision measurement of half-lives and specific activities of 235U and 238U. Phys Rev C 4:1889–1906
Cheng H, Edwards RL, Hoff J, Gallup CD, Richards DA, Asmerom Y (2000) The half-lives of uranium-234 and thorium-230. Chem Geol 169:17–33
IAEA (2004) Handbook of nuclear data for safeguards: Database extensions, August 2008. International Nuclear Data Committee INDC (NDS)-0534
Lau KH, Brittain RD, Hildenbrand DL (1989) High temperature thermodynamic studies of some gaseous thorium fluorides. J Chem Phys 90:1158–1164
Andrews L, Thanthiriwatte KS, Wang X, Dixon DA (2013) Thorium fluorides ThF, ThF2, ThF3, ThF4, ThF3(F2), and ThF5- characterized by infrared spectra in solid argon and electronic structure and vibrational frequency calculations. Inorg Chem 52:8228–8233
Hu S-W, Lin H, Wang X-Y, Chu T-W (2014) Effect of H2O on the hydrolysis of UF6 in the gas phase. J Mol Struct 1062:29–34
Baran V, Škvor F, Voseček V (1984) Formation of the ammonium-uranyl-carbonate complexes of the type (NH4)4[UO2(CO3)3], prepared by precipitative re-extraction. Inorgananica Chim Acta 81:83–89
Shimada-Fujiwara A, Hoshi A, Kameo Y, Nakashima M (2009) Influence of hydrofluoric acid on extraction of thorium using a commercially available extraction chromatographic resin. J Chromatogr A 1216:4125–4127
Stoll W (2000) In: Ley C, Elvers B, and 19 others (ed) Ullmann’s encyclopedia of industrial chemistry. Wiley‐VCH Verlag GmbH & Co
Kirby HW (1959) The radiochemistry of protactinium. National Academy of Sciences National Research Council Nuclear Science Series NAS-NS 3016
De Sio SM, Wilson RE (2014) Structural and spectroscopic studies of fluoroprotactinates. Inorg Chem 53:1750–1755
Shinnosuke H (1959) Determination of the solubility of thorium hydroxide. Bull Instit Chem Res Kyoto Uni 37(3):200–206
Neck V, Kim JI (1999) Solubility and hydrolysis of tetravalent actinides. Forschungszentrum Karlsruhe Technik und Umwelt, Wissenschaftliche Berichte FZKA 6350
Brown D, Easey JF (1970) Protactinium(V) fluorides. J Chem Soc A. https://doi.org/10.1039/j19700003378
Sakanoue M, Abe M (1967) Adsorption method for the separation of protactinium isotopes. Radioisotopes 16(12):645–651
Acknowledgements
We would like to thank the Department of Homeland Security for postdoctoral support to LNH, MCL, Inc., for helpful discussion and information on the samples used in this study, and one anonomous reviewer for comments that improved this work. This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 and supported by funding from the Department of Homeland Security. Releasable to external audiences, LLNL-JRNL-818999.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Harrison, L.N., Gaffney, A.M. 230Th-234U and 231Pa-235U radiochronometry of hydrolyzed uranium hexafluoride gas. J Radioanal Nucl Chem 329, 1513–1521 (2021). https://doi.org/10.1007/s10967-021-07903-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-021-07903-9