
Abstract
In 2014 the United States Department of Energy and the China Institute of Atomic Energy collaborated in a study measuring the model ages of uranium certified reference materials. Lawrence Livermore National Laboratory (LLNL) and Los Alamos National Laboratory (LANL), and the Chinese Institute of Atomic Energy (CIAE) determined 230Th–234U model ages for uranium certified reference materials U010 and U850. The aim of this work was to collaborate with CIAE and compare methods for measuring the age of bulk uranium materials using the 230Th–234U radiochronometer. Accurate results for age-dating depend on the accurate calibration of the tracer materials (e.g., 229Th and 233U) used for the isotope dilution mass spectrometry (IDMS) analyses. To facilitate inter-comparison of results, samples of a 230Th standard reference material were distributed to each laboratory for 229Th tracer inter-calibration. The resulting 230Th–234U model ages from this collaboration for U010 ranged from March 1956 to January 1959, which agree with the known material production date of June 5th, 1958. The determined model ages for U850 were from December 1955 to October 1957, which agree with the material production date of December 31, 1957. All three laboratories used independent methods to determine model ages for uranium standards that agree with known production ages and with previously reported results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Following any potential illicit nuclear material seizure, a nuclear forensic investigation may include determining an “age” for the material found out of regulatory control. The age of an unknown material may provide information about when the material was produced that can support investigations. The most widely used method to determine the age of uranium materials is the application of the 230Th–234U radiochronometer. This chronometer utilizes the decay of the parent isotope, 234U to daughter isotope, 230Th, and has been applied to geologic materials, as well as nuclear materials, for decades (see [1]). Calculating an age with the 230Th–234U radiochronometer requires model assumptions that (1) there was complete separation of daughter isotope, 230Th, from parent, 234U, at that time, and (2) the sample has remained a closed-system (no loss or gain or either parent or daughter, except by radioactive decay). If these assumptions are valid, the model age determined with the 230Th–234U radiochronometer will agree with the date of nuclear material preparation.
Using the 230Th–234U radiochronometer, the model age or model production date can be determined, which for a nuclear forensics investigation, may provide important information about the material’s provenance. Many laboratories have developed analytical methods to date nuclear materials using this chronometer, however, the reproducibility of 230Th–234U age measurements are rarely tested. To examine the inter-laboratory reproducibility and accuracy of determining 230Th and 234U concentrations in uranium, three laboratories, Lawrence Livermore National Laboratory (LLNL), Los Alamos National Laboratory (LANL), and the Chinese Institute of Atomic Energy (CIAE) participated in a round-robin exercise measuring certified reference materials with known production dates. All participating laboratories applied the 230Th–234U radiochronometer to commercially available uranium certified reference materials produced by New Brunswick National Laboratory in the United States, U010 and U850. A record of the production of these certified reference materials is available providing known purification dates for each material [2]. These certified reference materials with known production dates were chosen for age determination quality control and validation [3, 4].
The following equation, Eq. (1) is used to determine the age of the material:
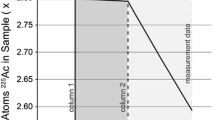
Using the above equation (Eq. 1), the model age, t, is calculated. The term, \(N_{{{}^{230}Th}} /N_{{{}^{234}U}}\), is the measured atom ratio for each sample. The half-lives, λ, used for the calculations here are from [5]. The 230Th half-life = 75,584 ± 55 years and the 234U half-life is 245,620 ± 150 years [5]. Model dates presented are calculated relative to a reference date, or date of Th–U separation in the laboratory. The model date of complete purification will agree with the date of the sample preparation when the model assumptions are met. If the daughter isotope is not completely purified from the parent isotope, then an older age than the production date will be calculated due to excess 230Th contaminant in the sample (Fig. 1).

Diagram showing the relationship between reference date, model age and model date
By using certified reference materials for this exercise, the date of sample preparation (the sample age) in known and the primary radiochronometry assumptions of complete daughter isotope purification and closed system behavior can be evaluated for validity.
Determining the age of uranium materials collaboratively establishes confidence in the analytical capabilities of participating countries and laboratories, thus providing assurance that each participating laboratory has the capability to provide accurate information for forensic investigations. The certified reference materials chosen for this study (U010 and U850) are considered to exhibit closed-system behavior based on the studies that show measured model ages closely match the production history of NBL U materials [3, 4, 6,7,8]. Therefore, any differences in model ages from the date of production may be due to initial 230Th, laboratory contamination, analytical uncertainty, or uncertainty in decay constants used [4]. This study compares LLNL, LANL, and CIAE laboratory methods for uranium sample preparation, purification, tracer calibration, and measurement technique to validate whether separate laboratories are capable of producing consistent and accurate ages (Fig. 2).

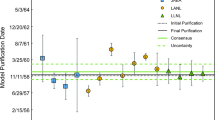
Model dates calculated for CRM U010 and CRM U850 compared to the paper age (purification date, dashed line) of U010 June 5, 1958; U850 December 31, 1957. Error bars are expanded uncertainties (k = 2)
Experimental
Uranium oxide certified reference materials (CRMs) U010 and U850 were independently acquired and dissolved at LLNL, LANL, and CIAE respectively. Each laboratory made assay analyses of 230Th and 234U by isotope dilution mass spectrometry (IDMS) using 229Th, 232Th (CIAE only), and 233U isotopic tracers.
To make high precision 230Th and 234U IDMS measurements, a tracer material, i.e., 233U, 229Th and 232Th, with a well characterized isotopic composition and concentration is necessary. Calibration of 233U tracers was done independently using U-oxide or U-metal materials chosen by each laboratory (e.g., CRM 145A, CRM 112A). Because each lab was using a different tracer to determine 230Th concentration, an inter-laboratory validation of each 229Th tracer calibration was made by distributing known amounts of SRM 4342A 230Th radioactivity standard prepared at LLNL to LANL and CIAE for 230Th assay. Aliquots of the SRM 4342A 230Th standard solution were individually weighed into 7 mL vials containing 0.5 mL 4 M HNO3, and were heated on a low-temperature hotplate until just dry. Four aliquots of the dried SRM 4342A 230Th standard were shipped to LANL and CIAE for analysis and comparison. The reproducibility of the concentration measurements of the 230Th radioactivity standard using separate tracer and calibration methods at LLNL, LANL, and CIAE was < 1%. USDOE laboratories (LLNL and LANL) used a 229Th tracer for all 230Th assay measurements in this study. CIAE used a combination of a 229Th tracer and a 232Th tracer (SPEX Certiprep, PLTH2-2Y) for IDMS measurement of 230Th. The 229Th tracer available to CIAE has 0.4% 230Th relative to 229Th; therefore, significant error is introduced to the 230Th measurement from the impurity of the 229Th tracer. The 232Th CIAE tracer has a higher purity making 232Th a more suitable tracer for Th measurements at CIAE, see [9] for a more detailed discussion of CIAE Th tracer calibration methods).
LLNL methods
Two primary solutions of U010 and were prepared by dissolving CRM U010 U-oxide (U010-1) and (U010-2) in 8 M HNO3 (all acids are from Seastar Chemicals, Inc.). Two primary solutions of U850 U-oxide (U850-1) and (U850-2) were prepared similarly. Following dissolution, the final primary solutions were diluted to approximately 2 M HNO3 and hydrofluoric acid was added to make the solutions 0.05 M HF [4]. The primary solution U010-1 was approximately 1000 µg-U/g solution and U010-2 was approximately 700 µg-U/g solution. The primary solution U850-1 was approximately 60 µg-U/g solution and U850-2 was approximately 16 µg-U/g solution.
Uranium concentration measurements were made for each primary solution by IDMS. First, quantitative serial dilutions were made of each primary solution, and the U IDMS aliquots which contained approximately 2–160 ng-U were spiked with approximately 2 ng 233U tracer (LLNL legacy material). The sample to tracer (238U/233U) ratios measured were between 1 and 60. The 233U tracer was calibrated using the NBL CRM 145 a standard prepared from CRM 112A (NBS SRM 960 U-metal), with additional measurements of an in-house CRM 112-A U-metal standard, as confirmation. Uranium IDMS aliquots for tracer calibration were not purified prior to mass spectrometry by multi-collector inductively coupled plasma mass spectrometry (MC-ICP-MS).
230Th concentrations were determined for each primary solution for U010 and U850. Aliquots containing 2.4–3.8 mg U (U010) and 37–64 µg U (U850) were spiked with a 229Th tracer. Sample to tracer (230Th/229Th) ratios of 0.2–0.6 were measured after spiking with approximately 100 pg of 229Th. The 229Th tracer was calibrated using the NIST 4342 230Th radioactivity standard (see discussion above).
Thorium IDMS aliquots were purified using a three-step separation and purification procedure. All three column chemistry steps were done using columns for the resin beds from environmental express. Past experience showed that poor recoveries were obtained using plastic columns from other sources due to sorption of thorium on commercial column frits. The first Th separation from bulk-U is done using a 1 mL anion exchange resin bed (Bio-Rad AG 1X8 100–200 mesh). Uranium sorbs on the resin bed, while Th elutes in 9 M HCl + 0.05 M HF. This bulk separation is followed by a Th purification step using 1 mL TEVA (Eichrom) resin bed. Thorium is dissolved and loaded in 4 M HNO3, and eluted in 0.1 M HCl + 0.005 HF. The final step removes residual U separation from Th using a 1 mL anion exchange resin bed (AG 1X8) where Th is dissolved, loaded, and rinsed from the column using 9 M HCl + 0.01 M HF.
Both U and Th isotope dilution aliquots were analyzed using a Nu plasma high resolution (HR) multi-collector inductively coupled mass spectrometer (MC-ICP-MS). Uranium samples were dissolved in 2% HNO3 and Th samples were dissolved in 2% HNO3 + 0.005 HF. Uranium IDMS samples are measured using a static multi-collection routine where signals from 233U, 235U, and 238U are measured on Faraday collectors and 234U is measured on a secondary electron multiplier (ion counter). Instrumental mass bias corrections and ion counter gain calibrations for both U and Th measurements were made using CRM U010. Thorium IDMS samples were analyzed using a pulse-counting routine, where all three Th isotopes, 229Th, 230Th, and 232Th were measured on ion counters.
The analyses for the 230Th intercalibration exercises were performed by adding known amounts of 229Th tracer to the vials prepared by LLNL containing NIST 4342A 230Th standard, capping and heating to equilibrate, and then drying. The 229Th–230Th mixtures were not purified, and were analyzed by MC-ICP-MS.
LANL methods
Primary dissolutions of U010 and U850 were made by dissolving approximately 100 mg of U CRM material in 8 M HNO3 (Optima). Final primary solutions were stored in 8 M HNO3 + 0.01 M HF. Serial dilutions of the primary solution are prepared to measure the 234U concentration for each U standard. The first dilution, ‘A dilution’, is made by quantitatively transferring 0.20–0.25 g of the primary solution to a 30 mL Teflon bottle, adding 20–25 mL 8 M HNO3 and weighing the total solution mass. Similarly, a ‘B dilution’ is made from quantitatively transferring approximately 0.2–0.25 g of the ‘A dilution’ to a tared 30 mL Teflon bottle and obtaining a final mass. This is a dilution factor of 1:12,500. The ‘B dilutions’ have concentrations of approximately 100 ng-U/g.
For 234U IDMS measurements, aliquots of the ‘B dilution’ are spiked with a 233U tracer at appropriate sample to tracer ratios depending on the amount of U material dissolved. Uranium IDMS aliquots are purified using UTEVA resin, where the samples are loaded in 3 M HNO3 and U is eluted with 1 M HCl. Uranium isotopic compositions are measured on purified, untraced aliquots of ‘B dilution.’ Uranium isotopic composition samples are purified using the same UTEVA purification chemistry as the U IDMS samples. The 233U tracer, Δ17 (LANL legacy material), was calibrated using CRM-145 and NBS U960.
Thorium (Th) aliquots for radiochemistry are taken from the ‘A dilution,’ with an approximate 230Th concentration of 30 pg/g, spiked with a 229Th tracer, and purified using a three-step process, all using BioRad anion exchange resin, AG1-X8 (100–200 mesh). The first step is an initial bulk U–Th separation, where Th samples are loaded in 8 M HNO3 and eluted in 0.1 M HCl and then 9 M HCl. The second step repeats this U–Th purification column. The final Th purification step removes residual U. Thorium is loaded and eluted immediately in 9 M HCl.
Thorium and U IDMS samples were measured on a ThermoScientific Neptune Plus MC-ICP-MS. Uranium standards are used for detector gain calibration and mass bias correction for both U and Th analyses. The 234U/235U ratio of NBL CRM U500 is used for the ion counter gain calibration for Th IDMS samples. The 233U/235U ratio of IRMM 74/5 is used for Faraday-ion counter cross calibration and mass bias correction for 230Th/232Th atom ratio measurement. The standards, IRMM 74/1 and IRMM 74/2 are used for U mass bias corrections, measuring 233U, 235U, and 238U on Faraday collectors. Quality control standards used for these analysis routines included U010, U005A, U500, IRMM 74/1, IRMM 74/2, and IRMM 74/5.
For the 230Th standard measurements, the 229Th tracer was added directly to the standard vials provided by LLNL. The 230Th–229Th mixture was dried to equilibrate and was not purified prior to analysis by MC-ICP-MS [10].
CIAE methods
CIAE conducted two dissolution campaigns for 230Th–234U radiochronometry of U010 and U850. The first campaign resulted in one model age for U010 and two model ages for U850. The second campaign involved a second dissolution of U010 and resulted in two additional model ages, and one additional model age for the original dissolution of U850. The first dissolution campaign is described below, followed by the second dissolution campaign.
For the preparation of a U850 primary solution, 1.5 mg U-oxide was dissolved in ultra-pure 6 M HNO3 (Beijing institute of chemical reagents, BV-III). Serial dilutions were made to measure 234U by IDMS. The U850 primary solution was diluted by a factor of 1:1000 and three aliquots of approximately 5 µg-U of this dilution were spiked with 25, 37.5, and 50 ng 233U tracer, respectively. Uranium IDMS aliquots were not purified prior to analysis by MC-ICP-MS. Separate isotopic analyses were made on unspiked aliquots.
The first primary solution for U010 was made by dissolving 3.4 mg U-oxide ultra-pure 6 M HNO3. This primary solution was diluted by a factor of 0.01. This solution was used for the 234U IDMS measurement aliquots. Three separate aliquots of approximately 10 µg-U were spiked with 25 ng 233U tracer. For the second dissolution, a freshly prepared primary solution was made by dissolving 2.8 mg U010 in ultra-pure 6 M HNO3. Three aliquots were taken and spiked with 125 ng 233U tracer. Uranium IDMS aliquots were not purified prior to analysis by MC-ICP-MS. CIAE calibrated their 233U tracer using CRM 112A and CRM 145.
The Th IDMS aliquots for U850 and U010 were taken from the primary solutions. The U850 Th IDMS aliquots were spiked with approximately 200 pg 229Th tracer (229Th separated from 233U at CIAE) and analyzed in December. Aliquots for U010 Th IDMS were spiked with 80 ng 232Th tracer and analyzed in July 2014. Subsequent Th IDMS aliquots were taken from a new dissolution of U010 resulting in two Th IDMS samples spiked with 229Th tracer. Another Th IDMS aliquot of the original dissolution of U850 was spiked with 232Th. All Th IDMS aliquots were purified using Eichrom TEVA resin. The first column is a bulk U-Th separation, where samples are dissolved and loaded in 3 M HNO3 and Th is eluted in 8 M HCl and 4 M HCl. The Th is further purified using the same column. Samples were taken to dryness and dissolved in 2% HNO3 for mass spectrometry (Table 1).
Both U and Th IDMS measurements were made on a Micromass-VG Instruments Isoprobe MC-ICP-MS. For the first measurement campaign, U850 Th IDMS samples were analyzed by a peak-jumping multi-collection routine, with 229Th and 230Th measured on the Daly detector, and 232Th measured on Faraday detectors. Corrections for instrumental mass bias and relative detector gain factors were made by measuring IRMM-199, CRM U030 and UTB900 (CIAE in-house U material calibrated using the CRM series of standards). For U010 samples spiked with 232Th, a static multi-collection routine was used, where 230Th was measured on the Daly detector and 232Th on a Faraday collector. CRM U030 and CRM U200 were used for gain calibration correction and IRMM-199 was used for instrumental mass bias correction. For the second measurement campaign, U010 was spiked with 229Th and all three Th isotopes, 229Th, 230Th, and 232Th were measured on ion counters. CRM U030 and IRMM-199 were used to correct for instrumental mass bias and the detector cross-calibration factors.
Results and Discussion
The measured 230Th/234U and uncertainties (k = 1), calculated model ages and model dates, and expanded uncertainties (k = 2) are given in Table 2. LLNL, LANL, and CIAE calculated model dates for U010 range from March 1956 to January 1959. The known purification date of U010 is June 5, 1958 [2]. These model dates agree within uncertainty with previously published work and the known purification date of the material [4, 8].
For U850, the calculated model dates from all laboratories are between December 1955 and October 1957. The known purification date of U850 is December 31, 1957. The LLNL, LANL, and CIAE calculated dates agree with previously published work and the known purification date [3, 4]. Only one age, CIAE result U010-2 (model date of January 1959) is younger than the production age.
While the model ages determined by all laboratories are in good agreement, there is some variation in model age determinations between laboratories. In addition, the range of model ages differs somewhat from the known sample production date. For both CRMs, the composite results indicate slightly older model dates than the paper dates (ca. 7 months for U010, and 11 months for U850), and while this “old-bias” is not outside of uncertainty, it suggests that the purification of Th from U was not complete on the production date, or the materials were subsequently contaminated with Th. This is consistent with these production dates giving the maximum model-age, or a maximum age for the U material that was used to produce these CRMs. The model ages for U010 determined by all three laboratories agree within analytical uncertainty, but the CIAE results for U850 show a bias to older model ages relative to the other two laboratories. The variation in model age between laboratories in likely explained by the calibration materials and methods, as well as the instrumentation used for analyses. While heterogeneity of CRM U850 is possible, the CIAE deviation from LLNL and LANL results is more likely to be the result of environmentally sourced Th contamination of the CIAE material at some point in its history. Also, the CIAE results obtained using the 229Th tracer were less precise than results using the 232Th tracer. This may be due to two factors; the inherent 230Th present in the CIAE 229Th tracer, and the inherent issues with calibrating the 229Th tracer. Access to high purity and well calibrated tracers for 230Th and 234U determination by IDMS is paramount to attain accuracy and precision in radiochronometry.
Conclusion
The 230Th–234U model dates measured by all three laboratories for CRM U010 (between March 1956 and January 1959), and for U850 (between December 1955 and October 1957), are consistent between laboratories and with the known purification dates of these materials. Except for one result, all model ages calculated are slightly older than the purification dates suggesting incomplete 230Th purification from U at that time. The reproducibility of the analyses illustrates that the different sample preparation methods, unique Th tracers and tracer calibration techniques, and different mass spectrometric techniques used by these laboratories are equally valid for measurement of 230Th and 234U to age-date such uranium materials. Through international collaborations such as this, participants gain mutual confidence in the validity of these measurements that can be extended to future nuclear forensic investigations.
References
Ivanovich M, Harmon RS (1982) Uranium-series disequilibrium. Oxford University Press, Oxford
Petit GS (1960) Preparation of uranium isotopic standards for the National Bureau of Standards. Union Carbide Nuclear Company, Oak Ridge, Tennessee. Oak Ridge National Laboratory, Report #KL-8, Department of Energy/K25 Archives
Wallenius M, Morgenstern A, Apostolidis C, Klaus M (2002) Determination of the age of highly enriched uranium. Anal Bioanal Chem 374:379–384
Williams RW, Gaffney AM (2011) 230Th–234U model ages of some uranium standard reference materials. Radiochim Acta 1:31–35
Cheng H, Edwards RL, Shen C, Polyak VJ, Aserom Y, Woodhead J, Hellstron J, Wang Y, Kong X, Spotl C, Wang X, Alexander A Jr (2013) Improvements in 230Th dating, 230Th and 234U half-life values, and U–Th isotopic measurements by multi-collector inductively coupled plasma mass spectrometry. Earth Planet Sci Lett 371–372:82–91
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Varga Z, Nicholl A, Wallenius M, Mayer K (2012) Development and validation of a methodology for uranium radiochronometry reference material preparation. Anal Chim Acta 718:25–31
Varga Z, Wallenius M, Klaus M (2010) Age determination of uranium samples by inductively coupled plasma mass spectrometry using direct measurement and spectral deconvolution. J Anal Spectrom 25:1958–1962
Chen Yan, Zhao Yong-Gang, Li Li-Li, Chang Zhi-Yuan, Liu-Chao Zhu, Xiao Guo-Ping, Huang Sheng-Hui (2017) A comparison of two calibration methods of the 229Th spike for uranium age dating by 230Th/234U radiochronometer. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-017-5396-6
Gaffney AM, Hubert Am, Kinman WS, Magara M, Okubo A, Pointurier F, Schorzman KS, Steiner RE, Williams RW (2016) Round-robin 230Th–234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem 307:2055–2060
Acknowledgements
This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Releasable to external audiences, LLNL-JRNL-739136.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Treinen, K.C., Kinman, W.S., Chen, Y. et al. US-DOE and CIAE international cooperation in age-dating uranium standards. J Radioanal Nucl Chem 314, 2469–2474 (2017). https://doi.org/10.1007/s10967-017-5593-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5593-3