
Abstract
The computation of distribution coefficients between polar and apolar phases requires both an accurate characterization of transfer free energies between phases and proper accounting of ionization and protomerization. We present a protocol for accurately predicting partition coefficients between two immiscible phases, and then apply it to 53 drug-like molecules in the SAMPL5 blind prediction challenge. Our results combine implicit solvent QM calculations with classical MD simulations using the non-Boltzmann Bennett free energy estimator. The OLYP/DZP/SMD method yields predictions that have a small deviation from experiment (RMSD = 2.3 \(\log\) D units), relative to other participants in the challenge. Our free energy corrections based on QM protomer and \({\text{p}}K_{\text{a}}\) calculations increase the correlation between predicted and experimental distribution coefficients, for all methods used. Unfortunately, these corrections are overly hydrophilic, and fail to account for additional effects such as aggregation, water dragging and the presence of polar impurities in the apolar phase. We show that, although expensive, QM-NBB free energy calculations offer an accurate and robust method that is superior to standard MM and QM techniques alone.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The relative balance between hydrophilic and hydrophobic non-bonded molecular interactions is of central importance to the fields of chemistry [15], biophysics [70] and pharmacology [47]. Within the field of pharmacology, accurate characterization of these physiochemical properties is critical, as they affect all aspects of the drug design process, such as: availability [47], potency [75] and toxicity [48]. Tuning the hydrophobicity of a ligand affects its ability to diffuse across cellular membranes, alters its ability to bind to targets and impacts its clearance properties.
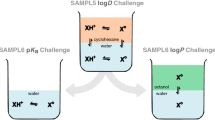
One way of rigorously quantifying a ligand’s hydrophilicity is the free energy required to transfer a molecule from a bulk apolar environment (k), e.g. octanol or hexane, to a bulk aqueous environment, \(\Delta G^{k \rightarrow {\text{aq}}}\), in the limit of infinite dilution. In the pharmaceutical sciences, this transfer free energy is often cast as a partition coefficient, \(P_{k}\), the ratio of concentrations of the solute in the two immiscible phases, which is trivially related to the transfer free energy, where \(k_{\text{B}}\) is the Boltzmann constant and T is the absolute temperature. Because of this, it is frequently expressed as a common logarithm (\(\log P_{k}\)).
Since many drug molecules contain ionizable groups that may exist in multiple protomeric states under physiological \({\text{pH}}\) conditions, the reality is often more complicated than the two-state model implied by partition coefficients. Solute aggregation, and the water dragging effect [18, 19] can also lead to non-negligible populations of solute molecules in “other” states. To account for these deviations from ideal behavior, we can combine the definition of a partition coefficient with a looser definition of relevant solute states. By including protonation, tautomerization, multimerization, etc., we obtain the definition of a distribution coefficient, \(D_{k}\) (Eq. 3).
The summation is over all states i and j in the apolar and aqueous environments, respectively. Equation 3 introduced the activity coefficient, \(\gamma\), to account for further, subtler deviations from ideal behavior, however in this work we will ignore this effect, and assume \(\gamma \equiv 1\).
While distribution coefficients are reliably and quickly characterized experimentally, [13, 46, 61] analogous physics-based computational predictions are expensive, and typically limited to neutral solutes [8, 32]. The accurate characterization of ionizable solutes, and thus \({\text{p}}K_{\text{a}}\) values remains a challenge. Most successful computational approaches to \({\text{p}}K_{\text{a}}\) prediction involve significant fitting to experimental data, [45, 77] or relative \({\text{p}}K_{\text{a}}\) calculations [11]. Accurately modeling the precise experimental conditions poses further challenges to the computational prediction of distribution coefficients. For example, the partitioning between aqueous and organic phases is exquisitely sensitive to the water content of the organic phase [1]. In experiments, both polar [29] and apolar [43] solutes are known to aggregate in the interfacial region, and thus deplete in the bulk phase.
Even without the complexities associated with predicting distribution coefficients, accurate prediction of partition coefficients still requires properly accounting for the change of solute–solvent interactions between apolar and polar phases. Previous SAMPL small molecule challenges have emphasized the calculation of hydration free energies for small molecules, [22, 55, 65] a not-dissimilar task from our current charge. The lessons learned in this regard from our previous work in SAMPL4, [40, 58] in which we showed the effectiveness of using quantum mechanical [39] based potential energy calculations in combination with the non-Boltzmann Bennett (NBB) free energy method [41], should be directly applicable in this current challenge [2].
In this work we predict the partitioning between aqueous and cyclohexane phases for 53 small drug-like molecules in the SAMPL5 blind prediction challenge (Figs. 1, 2). We use various computational techniques ranging from molecular dynamics (MD) simulations to quantum mechanical potential energy evaluations (QM), combining the best aspects of these approaches via the NBB free energy estimator. These QM-NBB calculations with implicit solvent yield predictions with a root mean squared deviation from experiment (RMSD) that ranks second among the various entries. We also attempt to account for deviations from non-ideal behavior using QM based \({\text{p}}K_{\text{a}}\) and protomeric calculations. When these corrections are applied to partition predictions, the resulting distribution predictions are found to correlate more strongly with experimental results, than those predictions made without the corrections. The results of the underlying molecular mechanics (MM) free energy simulations, as well as QM/MM multi-scale free energy results are discussed in a companion paper in the same issue [38]. The vast majority of the data in this work were generated in a blind fashion, before the conclusion of the SAMPL5 challenge, the exceptions being the inclusion of additional protomeric states of molecule 83 and additional dimerization states of molecule 50. These additional results are discussed in the body of this text, and do not appear in any tabulated results.

Chemical structures of the molecules did not deviate significantly from their reference states (\({<}0.01\) kcal mol\(^{-1}\))

Chemical structures of the molecules that were determined to have multiple protomeric states
Methods
Free energy methods
We will first predict the partition coefficients, \(\log P_{\text{chex}}\), by calculating the transfer free energy from cyclohexane to water, \({\Delta } G^{{\text{chex}} \rightarrow {\text{aq}}} = {\Delta } G^{*}_{\text{aq}} - {\Delta } G^{*}_{\text{chex}}\), for the reference states of the molecules in the challenge, as they were provided by D3R. Here, the “\(*\)” denotes the standard state of 1 mol L\(^{-1}\), and will be implied for the rest of this work. The most straightforward approach for estimating the free energy difference between a sampled state i and an unsampled state j is by application of Zwanzig’s equation [80]. This approach is used to obtain the free energy difference by the following
where \(\beta ^{-1} = k_{\text{B}} T\) is the thermodynamic temperature, and \(U_i\) is the potential energy of a configuration evaluated using the indicated Hamiltonian, and the angular brackets indicate an ensemble average over state i. In principle this approach can be used to obtain a free energy value from an expensive QM based potential energy surface, using an ensemble generated using a cheaper MM based force field. This strategy is preferable to obtaining a free energy directly from ab initio MD, which would be prohibitively expensive. The accuracy of this approach is strictly limited by the similarity between the QM and the MM potential energy surfaces, as well as by the system size. Because of the presence of numeric instabilities in this method, alternative approaches are often preferable [5, 12, 17, 20, 23, 26, 30, 31, 33, 42, 54, 56, 60, 62, 63].
By drawing configurations from both states i and j, one can obtain the minimum variance estimate between these states by applying Bennett’s Acceptance Ratio (BAR) [6].
where f is the Fermi function
and C is a constant. An iterative solution is obtained, such that the ratio in Eq. 5 converges to unity. BAR is very commonly applied to studying free energy changes in chemical processes. More recently, a multistate variant has been derived [64], and it should be adopted when simultaneously considering the free energy differences between more than two states, such as in a chain of states during an alchemical transformation process. One strict disadvantage of using BAR is that it requires configurations to be drawn from both states i and j. This can make direct application of BAR to QM based calculation too computationally demanding.
Similar to the Zwanzig equation, we can use the non-Boltzmann Bennett method to estimate the free energy of an unsampled state i by using configurations drawn from a sampled state \(i^{\prime }\). This is accomplished by biasing the sampled states \(i^{\prime }\) and \(j^{\prime }\) using the potential energy difference between i and \(i^{\prime }\) by the following function.
The correct ensemble averages in the unsampled states i and j are then recovered from the biased states by applying Torrie and Valleau’s relationship [71] to calculate the unbiased ensemble average, \(\langle X \rangle _i\), from configurations taken from a biased state \(i^{\prime }\).
By combining Eqs. 5 and 8 one obtains the NBB equation, allowing us to estimate the free energy difference between two unsampled states i and j, that are typically too expensive to explicitly sample.
MD simulation
All MD simulations were carried out using the PERT module [10] of the CHARMM simulation package [9, 10] and the CHARMM General Force Field (CGenFF) for organic molecules. [73] The aqueous phase was modeled with 1906 TIP3P water molecules [34] and six pairs of sodium and chlorine ions, to approximately reproduce the ionic strength of the reported experimental conditions (\({\text{pH}}\) 7.4, 136 mM NaCl, 2.6 mM KCl, 7 mM \({\text{Na}}_3{\text{PO}}_4\), 1.46 mM \({\text{KH}}_2{\text{PO}}_4\), 0.27 M DMSO and 0.18 M acetonitrile). The cubic simulation boxes were pre-equilibrated with 0.5 ns of constant pressure dynamics, resulting in unit cells with edges varying between 38.55 and 38.75 Å in length. The apolar phase was modeled with 337 cyclohexane molecules and cubic box sizes with edges varying from 39.93 to 40.18 Å in length. Long range electrostatics were represented using smooth particle mesh Ewald summation [14], while Lennard–Jones interactions used a switching window at 10 Å, before being truncated at 12 Å. A Nosé-Hoover thermostat [28] maintained the canonical ensemble during the 0.5 ns equilibration runs, and during the 5 ns production runs. All simulations used a 1 fs timestep and SHAKE constraints on all hydrogen valence terms. Geometric configurations were saved every 1000 steps for later analysis and post-processing.
Transfer free energies were calculated by turning off all non-bonded solute interactions, both in the cyclohexane and the aqueous phases. This alchemical mutation was carried out in five steps. In step 1, the charges on the cyclohexane phase solute were decremented to zero over six states (\(\lambda = 0.00, 0.25, 0.50, 0.75, 0.90\) and 1.00). We refer to this process as “uncharging”. In step 2, we decremented the Lennard-Jones interactions in the gas phase over 24 equidistant states (\(\lambda = 0, {1}/{23}, \ldots , {22}/{23}, 1\)). We refer to this process as “vanishing”. For molecules 65, 83 and 92 an additional state at \(\lambda = 0.022\) was used to achieve convergence as these are the largest and most flexible molecules. In step 3, we transfer the non-interacting ligand, \({\text{A}}^{({\text{n}},\varnothing )}\), from the cyclohexane to the aqueous phase. The free energy of this process is equivalent to zero. Step 4 and step 5 negate the vanishing process and uncharging processes, respectively, in the aqueous phase, using the same alchemical scheme employed in the cyclohexane phase. The alchemical scheme is summarized in Eq. 10,
where “+” denotes the fully-charged states, and “n” denotes uncharged states.
To enhance sampling, \(\lambda\)-Hamiltonian Replica Exchange [67, 68] was used to attempt exchanges between neighboring \(\lambda\)-states every 1000 steps. Because these \(\lambda\)-states are already required for the underlying BAR free energy calculation, multiplexing the alchemical states together via replica exchange provides accelerated convergence, for marginal cost. Soft-core potentials were used to avoid the endpoint problem [7, 76].
QM calculations
All QM calculations in this work were performed using Gaussian 09 [21]. Transfer free energies were calculated by using a standard QM optimization approach. To calculate QM based partition coefficients, We used an “adiabatic” protocol at the M06-2X/6-31+G(d) level of theory [78, 79] with the SMD implicit solvent [50, 51, 59]. In this scheme, geometry optimizations are carried out in both the cyclohexane and aqueous phases. Next, the Hessian matrices are computed for both phases, and are used to compute the thermal corrections (to 298.15 K) for each molecule in the harmonic limit. Finally, a single point calculation (SPC) was computed on the static geometries using a larger (6-311++G(d,p)) basis set, in both phases, to attempt to further improve the computed transfer free energies, and to explore the efficacy of the 6-311++G(d,p) basis set. All QM optimizations were performed with “Tight” wave function and geometry convergence criteria and by using“UltraFine” numerical quadrature as required by M06-2X.
Due to the large size of molecule 83, QM optimizations on this ligand instead used the cheaper BLYP/6-31G(d) [4, 44, 53] method in conjunction with the SMD implicit solvent We estimated the transfer free energy as the difference of vertical solvation free energies from the gas phase into the appropriate bulk phase. Specifically, this was calculated as the hydration free energy less the solvation free energy in cyclohexane. The default options for wavefunction and geometric convergence, as well as default numerical quadrature were also used to speed up the calculations. Harmonic entropy contributions were ignored, as the frequency calculations were too expensive. Some of our previous work [37] has indicated the effectiveness of the BLYP functional for HFE predictions, despite its simplicity (and significantly reduced cost) with respect to M06-2X.
QM-NBB calculations
We also estimated the transfer free energies using NBB combined with two different QM methods: M06-2X/6-31+G(d) and OLYP/DZPFootnote 1 [16, 25, 27, 44, 53]. In this approach, configurations are drawn from the explicit solvent MD calculations, the explicit solvent is removed and energies are computed using single point QM calculations with the SMD implicit solvent. Because the solvent degrees of freedom are treated implicitly, there now exists sufficient overlap, with NBB biasing, to connect the cyclohexane state to the aqueous state directly. In this case 4N QM calculations are required, where N is the number of configurations drawn from the two chemical states, and the NBB equation simplifies to the following.
While this approach requires a large number of single point QM calculations, \(4 \times 5000\) per molecule in this study, these costs can be mitigated by the use of looser wave function convergence criteria and coarser numerical quadrature than was was used for the analogous QM optimization calculations. This increased performance ca. fivefold and incurred a loss of \({<}0.005\) kcal mol\(^{-1}\) in precision. These calculations also have the advantage of being “embarrassingly” parallel, allowing us to efficiently use any and all available computer resources, especially older marginal hardware with poor networking capabilities.
Protomer and \({\text{p}}K_{\text{a}}\) corrections
Because the goal of the SAMPL5 challenge is to predict the distribution coefficients between cyclohexane and water, rather than the partition coefficients, we must incorporate contributions from states that significantly deviate from the neutral reference structures. Using QM based \({\text{p}}K_{\text{a}}\) calculations [11, 49], we will account for populations of the acidic and basic ligands in their conjugate forms (\({\Delta } G_{{\text{p}}K_{\text{a}}}\)). Our corrections will also address the presence of protomers (\({\Delta } G_{\text{taut}}\)). While our submissions did not include corrections for the effects of dimerization (\({\Delta } G_{\text{dimer}}\)) or water dragging (\({\Delta } G_{\mu {\text{solv}}}\)) [18, 19], we will demonstrate that ignoring these phenomena may diminish the accuracy of distribution predictions as well.
Our \({\text{p}}K_{\text{a}}\) calculations used both an “absolute” and a “relative” protocol [11, 49]. In the absolute protocol we use the usual thermocycle (Fig. 3) to obtain an expression for the free energy of deprotonating \({\text{AH}}^{+}\), in the aqueous phase. Values for \(G({\text{AH}}^{+}_{\text{aq}})\) and \(G({\text{A}}_{\text{aq}})\) are obtained directly from the QM calculations. The value of \(G({\text{H}}^{+}_{\text{gas}})\) is analytic [52], while \({\Delta } G_{\text{solv}}({\text{H}}^{+})\) is experimentally determined [69]. A final factor of \(R T \ln (24.46)\) is also included to account for change of standard state from 1 atm L\(^{-1}\), denoted “\(\circ\)”, in the gas phase to 1 mol L\(^{-1}\) in the aqueous phase. Physically, this term corresponds to the loss of entropy when compressing an ideal gas from 1 to 24.46 atm (1 M), and is 1.89 kcal mol\(^{-1}\) at 298.15 K. Errors from the QM calculation of hydrating the charged ligand and uncertainties associated with the experimental value of hydrating a free proton (\({\Delta } G_{\text{solv}}({\text{H}}^{+}) = -265.9\) kcal mol\(^{-1}\)) [69], are thought to limit the accuracy of the absolute scheme [11]. Once the quantity \({\Delta } G_{\text{aq}}\) has been obtained, it can be readily converted into a \({\text{p}}K_{\text{a}}\) value using Eq. 13, where \(R = k_{\text{B}}/ N_{\text{A}}\) is the usual gas constant.

The thermodynamic cycle used for absolute \({\text{p}}K_{\text{a}}\) calculations in this work
Alternatively, relative \({\text{p}}K_{\text{a}}\) corrections may be preferable (Eq. 14), as the two main sources of error stated above are explicitly removed. The correctness of relative \({\text{p}}K_{\text{a}}\) calculations instead depends upon the choice of an appropriate analog ligand, L, and the availability of reliable experimental data, \({\text{p}}K^{\text{exp}}_{\text{a}}\), obtained under conditions (temperature, concentration and ionic strength) mirroring those for the system of interest. If any of these conditions are not sufficiently met, the relative \({\text{p}}K_{\text{a}}\) calculations can vastly underperform their absolute counterparts. For more information about the specific analogs used in this work, please see Table 3 and Figure S1.
Both \({\text{p}}K_{\text{a}}\) schemes can be combined with either adiabatic or vertical hydration free energy (HFE) calculations from QM. The adiabatic scheme is as described above. In the the vertical solvation scheme, gas phase optimized geometries optimized at the M06-2X/6-31+G(d) level of theory are used for a single point energy calculation in the aqueous phase at the same level of theory in the SMD implicit solvent. This approach neglects solvent relaxation effects during solvation process and may not be appropriate for some of the larger more flexible molecules in the SAMPL5 data set. A simple combination of these various approaches yields the four total \({\text{p}}K_{\text{a}}\) correction schemes we used in our submissions. Once we calculated the \({\text{p}}K_{\text{a}}\) values from our various approaches, we obtained relative populations of conjugate pairs using the Henderson–Hasselbalch equation at \({\text{pH}} =7.4\). These populations are then converted into free energy corrections (\({\Delta } G_{{\text{p}}K_{\text{a}} }\)) from the neutral reference state.
Other corrections, such as \({\Delta } G_{\text{taut}}\), can be obtained by appropriately combining Eqs. 1 and 3. We then cast the difference between QM calculated \(\log P_k\) and \(\log D_k\) values as a free energy correction (Eq. 15) from the reference transfer free energy, to a transfer free energy that has additional states included to model the correction of interest. This correction, originally derived from QM calculations, may then be applied to a transfer free energy obtained from any method of choice (Eq. 16).
Results and discussion
In this section, individual and collective descriptors, such as RMSD, of partition and distribution coefficients will be given in logarithmic units, which are dimensionless, and thus will not be explicitly listed. These results can be expressed as free energies using the conversion \(1 \log = 1.36\) kcal mol\(^{-1}\), at \(25~^{\circ }{\text{C}}\). When comparing predictions with an experiment, a “−” sign indicates that the prediction is more hydrophilic than experiment, while a “+” indicates that our prediction is too hydrophobic.
Being one of the most popular and effective quantum chemistry methods in use today, the M06-2X/6-31+G(d)/SMD level of theory yielded \(\log P_{\text{chex}}\) predictions that served as a good reference point by which we could evaluate the accuracy and efficiency of the rest of our submissions to the SAMPL5 challenge. When combined with the vertical solvation protocol (the adiabatic protocol performs similarly, submission 28), these predictions agreed relatively well with experiment, sixth overall (submission 27, RMSD = 2.58), but correlated poorly with experiment (Kendall’s \(\tau = 0.46\)). While we chose to include both frequency and single point corrections with a triple-\(\zeta\) basis set, with our adiabatic protocol, neither of these corrections changed the collective behavior of our predictions significantly (Figure S2). The most significant outlying result, by far, is for 83. We did not identify the correct protomeric state for this molecule in either the cyclohexane or aqueous phases. Using the incorrect protomer as the basis for our predictions, our value for \(\log P_{\text{chex}}({\mathbf{83}})\) is too hydrophilic by 12.45. The results from these submissions are explicitly tabulated in Table 1.
After consulting with other participants at the D3R meeting, and then identifying more stable protomers in both phases, our predicted partition value is in much better agreement with experiment, but is still far too hydrophilic \({\Delta }_{\text{exp}} = -7.11\). The RMSD for this submission is also significantly reduced to 2.25 units by using the proper tautomers for 83, now ranking it amongst the best submissions by RMSD. The correlation with experiment is still very poor however, and is significantly worse than the result obtained by the top performing COSMO-RS submission (submission 16, \({\text{RMSD}} = 2.1 \pm 0.2\), \(\tau = 0.73 \pm 0.04\)) [36, 35]. The extreme sensitivity of these results to the inclusion of two additional protomers for a single molecule in the data set, dramatically underscores the difficult nature of these calculations.
While a detailed analysis of the results from the underlying MM free energy simulations are discussed in a companion paper to this work, [38] it is important to briefly introduce and discuss them. Running the simulations using reference states where all protonizable groups are neutral, and protomers are incorrectly assigned for at least three molecules (50, 56 and 83), yields extremely poor results. The CGenFF fixed charge force field, in combination with the BAR free energy estimator, provides partition predictions that significantly deviate from experiment (submission 38, \({\text{RMSD}} = 5.6 \pm 0.4\), \(\tau = 0.25 \pm 0.08\)). Applying our corrections based on absolute \({\text{p}}K_{\text{a}}\) calculations (Table 3) and adiabatic solvation free energy calculations, improves this result dramatically (Fig. 4), reducing the deviation from experiment and increasing the correlation (submission 10, \({\text{RMSD}} = 3.14\), \(\tau = 0.49\)).

Our partition estimations from MM BAR (submission 38) plotted against experiment. We have applied our QM based free energy corrections (adiabatic/absolute scheme, submission 10), shifting the predicted values towards more hydrophilic values. These corrections account for multiple protomeric states and for ligand ionization due to the presence of protonizable groups. These corrections substantially reduce the RMSD and increase the correlation of these predictions with respect to experimentally determined values
The predicted partition coefficients (Table 1) using the QM-NBB free energy estimator combined with the OLYP/DZP level of theory had a relatively low deviation from experiment (submission 02, \({\text{RMSD}} = 2.3 \pm 0.3\), \(\tau = 0.48 \pm 0.07\)), ranking second by RMSD, but a relatively mediocre correlation (Fig. 5). After applying our free energy corrections based on absolute \({\text{p}}K_{\text{a}}\) calculations and adiabatic solvation free energy, the resulting distribution coefficients deviate further from experiment, however the correlation with experiment increases (submission 54, \({\text{RMSD}} = 2.68\), \(\tau = 0.53\)). While we did not address dimerization in our SAMPL5 submissions, our subsequent analysis indicated that these effects can be substantial. For example, molecule 50 will likely dimerize in the apolar phase, significantly decreasing its lipophobicity. Similarly, for molecule 74, the water dragging effect may diminish its lipophobicity as well, as its many alcohol groups can strongly coordinate a water molecule. Similarly the effect of polar impurities in the apolar phase was not investigated either. Our QM-NBB calculations using M06-2X did not perform significantly differently from the analogous OLYP calculations. This is an advantageous result from an efficiency perspective, as OLYP is a pure functional, and does not have a kinetic energy density term, nor a Hartree–Fock exchange, making it significantly cheaper than M06-2X. However, this result is also disappointing, because it closes an obvious path for trivially improving the quality of partition predictions by improving the quality of our QM functional.

Partition coefficient estimations from our QM-NBB OLYP/DZP free energy calculations (left, submission 02). After application of our free energy corrections (adiabatic/absolute scheme, the resulting distribution coefficients correlate more strongly with experiment, but are significantly too hydrophilic, and systematically overestimate the hydrophilicity (right, submission 54)
The quality of \({\text{p}}K^{\text{rel}}_{\text{a}}\) calculations (Table 2) is exquisitely dependent upon the choice of analog molecule (Table 3). In many cases, an obvious choice will present itself, and the resulting \({\text{p}}K^{\text{rel}}_{\text{a}}\) calculation is likely to be more accurate than its absolute analog. In other cases, choosing an appropriate chemical analog will be difficult or impossible. One example is the acidic phenolic hydrogen in 17. Phenol is a poor choice of analog for this system, because this proton is stabilized by an intramolecular hydrogen bond with the neighboring basic heterocyclic nitrogen. By directly comparing the \({\text{p}}K^{\text{rel}}_{\text{a}}\) and \({\text{p}}K^{\text{abs}}_{\text{a}}\) predictions (Fig. 6), we may be able to blindly assess the quality of our free energy corrections without any a priori knowledge of the distribution coefficients.

Differences in free energy corrections calculated using the two different \({\text{p}}K_{\text{a}}\) calculation schemes, both of the free energy corrections in this figure used the adiabatic solvation protocol. When the two sets of corrections strongly deviate, it may be a sign of poor analog selection in the \({\text{p}}K^{\text{rel}}_{\text{a}}\) calculations
Conclusions
The OLYP/DZP QM method with SMD implicit solvation model performed very strongly relative to other submissions when combined with the NBB free energy estimator (submission 02). Overall, this submission ranked second by RMSD, but had only a mediocre correlation as estimated by Kendall’s \(\tau\). While this particular combination of density functional and basis set is unusual, this protocol [58] was designed using HFE data from the SAMPL4 challenge [55] as a target. The cost of the QM-NBB approach is relatively high relative to simple QM optimization, due to the large number of configurations that must be evaluated (\({\approx }4 \times 5000\)) for each molecule. This cost is mitigated somewhat by the embarrassingly parallel nature of these energy evaluations.
The M06-2X/6-31+G(d) QM optimization calculations with SMD implicit solvent also performed well, ranking sixth overall by RMSD (submission 27). This submission was made because the required QM calculations were a strict subset of the calculations required for our \({\text{p}}K_{\text{a}}\) predictions. The M06-2X and SMD approaches are ubiquitous in the literature, [50, 59] and serves as a good “control” to help us understand how our more complicated and more expensive free energy methods compare against other popular approaches. These predictions also had mediocre correlation as estimated by Kendall’s \(\tau\).
By including our \({\text{p}}K_{\text{a}}\) and protomeric corrections with our partition predictions (specifically our corrections based on adiabatic solvation free energies and an absolute \({\text{p}}K_{\text{a}}\) scheme), our resulting distribution predictions enjoyed increased correlation for all tested methods. Unfortunately, in many of our best performing methods, such as QM-NBB with OLYP/DZP, our corrections increased our RMSD values. This occurred because our \(\log P_{\text{chex}}\) predictions were already too hydrophilic relative to experiment. Our corrections, as submitted to the SAMPL5 challenge, exacerbated this problem, further increasing the hydrophilicity of our predictions, because our corrections summed over additional aqueous phase states, further tipping the balance of our predictions towards the hydrophilic.
Our \({\text{p}}K_{\text{a}}\) corrections indicated that some of our reference states, under which our MD simulations were performed, were very far from equilibrium. Molecule 83 for example, has a protomer in the apolar phase that is ca. 10 kcal mol\(^{-1}\) from the state we modeled with MD. Differences this large, cannot likely be corrected for using QM optimization calculations on one configuration.
Our \({\text{p}}K_{\text{a}}\) corrections were performed using the QM optimization protocol, which, while successful overall, suffers from over representing the global minimum structure, as conformational entropy of neighboring low-lying configurations is neglected. This effect should be particularly troublesome for larger molecules that were very common in this challenge, as well as for the many ionic conjugates that were ubiquitous in this data set. The accuracy of our \({\text{p}}K_{\text{a}}\) corrections could likely be improved by using a NBB scheme here as well. This approach will be the subject of follow up work.
Notes
This is the version of Dunning’s DZP basis set that appears in the Psi4 quantum chemistry package [72].
References
Abraham MH, Zissimos AM, Acree WE Jr (2001) Partition of solutes from the gas phase and from water to wet and dry di-n-butyl ether: a linear free energy relationship analysis. Phys Chem Chem Phys 3:3732–3736. doi:10.1039/B104682A
Bannan CC, Burley KH, Mobley DL (2016) Blind prediction of cyclohexane-water distribution coefficients from the SAMPL5 challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9954-8
Bausch M, Selmarten D, Gostowski R, Dobrowolski P (1991) Potentiometric and spectroscopic investigations of the aqueous phase acidbase chemistry of urazoles and substituted urazoles. J Phys Org Chem 4(1):67–69. doi:10.1002/poc.610040111
Becke A (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38(6):3098–3100. doi:10.1103/PhysRevA.38.3098
Beierlein FR, Michel J, Essex JW (2011) A simple QM/MM approach for capturing polarization effects in protein-ligand binding free energy calculations. J Phys Chem B 115(17):4911–4926. doi:10.1021/jp109054j
Bennett CH (1976) Efficient estimation of free energy differences from Monte Carlo data. J Comput Phys 22:245–268
Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF (1994) Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett 222:529–539
Bhatnagar N, Kamath G, Chelst I, Potoff JJ (2012) Direct calculation of 1-octanolwater partition coefficients from adaptive biasing force molecular dynamics simulations. J Chem Phys 137(1):014502. doi:10.1063/1.4730040
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J Comput Chem 4:187–217
Brooks B, Brooks C III, Mackerell A Jr, Nilsson L, Petrella R, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner A, Feig M, Fischer S, Gao J, Hodošček M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor R, Post C, Pu J, Schaefer M, Tidor B, Venable R, Woodcock H, Wu X, Yang W, York D, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30(10, Sp. Iss. SI):1545–1614. doi:10.1002/jcc.21287
Casasnovas R, Ortega-Castro J, Frau J, Donoso J, Muoz F (2014) Theoretical pKa calculations with continuum model solvents, alternative protocols to thermodynamic cycles. Int J Quantum Chem 114(20):1350–1363. doi:10.1002/qua.24699
Cave-Ayland C, Skylaris CK, Essex JW (2015) Direct validation of the single step classical to quantum free energy perturbation. J Phys Chem B 119(3, SI):1017–1025. doi:10.1021/jp506459v
Comer J, Tam K (2007) Lipophilicity profiles: theory and measurement. Verlag Helvetica Chimica Acta, pp 275–304. doi:10.1002/9783906390437.ch17
Darden T, York D, Pedersen L (1993) Particle mesh Ewald—an N Log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Du Q, Freysz E, Shen YR (1994) Surface vibrational spectroscopic studies of hydrogen bonding and hydrophobicity. Science 264(5160):826–828. doi:10.1126/science.264.5160.826
Dunning TH (1970) Gaussian basis functions for use in molecular calculations. I. Contraction of (9s5p) atomic basis sets for the firstrow atoms. J Chem Phys 53(7):2823–2833. doi:10.1063/1.1674408
Dybeck EC, König G, Brooks BR, Shirts MR (2016) A comparison of methods to reweight from classical molecular simulations to QM/MM potentials. J Chem Theory Comput. doi:10.1021/acs.jctc.5b01188
Fan W, Tayar NE, Testa B, Kier LB (1990) Water-dragging effect: a new experimental hydration parameter related to hydrogen-bond-donor acidity. J Phys Chem 94(12):4764–4766. doi:10.1021/j100375a003
Fan W, Tsai RS, Tayar NE, Carrupt PA, Testa B (1994) Soluble-water interactions in the organic phase of a biphasic system. 2. Effects of organic phase and temperature on the “water-dragging” effect. J Phys Chem 98(1):329–333. doi:10.1021/j100052a054
Fox SJ, Pittock C, Tautermann CS, Fox T, Christ C, Malcolm NOJ, Essex JW, Skylaris CK (2013) Free energies of binding from large-scale first-principles quantum mechanical calculations: application to ligand hydration energies. J Phys Chem B 117(32):9478–9485. doi:10.1021/jp404518r
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, revision B.01. Gaussian, Inc., Wallingford
Geballe MT, Guthrie JP (2012) The SAMPL3 blind prediction challenge: transfer energy overview. J Comput Aided Mol Des 26(5):489–496. doi:10.1007/s10822-012-9568-8
Genheden S, Ryde U, Söderhjelm P (2015) Binding affinities by alchemical perturbation using QM/MM with a large QM system and polarizable MM model. J Comput Chem 36(28):2114–2124. doi:10.1002/jcc.24048
Hall HK (1957) Correlation of the base strengths of amines. J Am Chem Soc 79(20):5441–5444. doi:10.1021/ja01577a030
Handy NC, Cohen AJ (2001) Left-right correlation energy. Mol Phys 99(5):403–412. doi:10.1080/00268970010018431
Heimdal J, Ryde U (2012) Convergence of QM/MM free-energy perturbations based on molecular-mechanics or semiempirical simulations. Phys Chem Chem Phys 14:12,59212,604. doi:10.1039/c2cp41005b
Hoe WM, Cohen AJ, Handy NC (2001) Assessment of a new local exchange functional OPTX. Chem Phys Lett 341(34):319–328. doi:10.1016/S0009-2614(01)00581-4
Hoover WG (1985) Canonical dynamics—equilibrium phase-space distributions. Phys Rev A 31:1695
Hu YF, Lv WJ, Shang YZ, Liu HL, Wang HL, Suh SH (2013) Dmso transport across water/hexane interface by molecular dynamics simulation. Ind Eng Chem Res 52(19):6550–6558. doi:10.1021/ie303006d
Hudson PS, White JK, Kearns FL, Hodošček M, Boresch S, Woodcock HL (2015) Efficiently computing pathway free energies: new approaches based on chain-of-replica and Non-Boltzmann Bennett reweighting schemes. Biochim Biophys Acta Gen Subj 1850(5, SI):944–953. doi:10.1016/j.bbagen.2014.09.016
Hudson PS, Woodcock HL, Boresch S (2015) Use of nonequilibrium work methods to compute free energy differences between molecular mechanical and quantum mechanical representations of molecular systems. J Phys Chem Lett 6(23):4850–4856. doi:10.1021/acs.jpclett.5b02164
Ingram T, Storm S, Kloss L, Mehling T, Jakobtorweihen S, Smirnova I (2013) Prediction of micelle/water and liposome/water partition coefficients based on molecular dynamics simulations, cosmo-rs, and cosmomic. Langmuir 29(11):3527–3537. doi:10.1021/la305035b
Jia X, Wang M, Shao Y, König G, Brooks BR, Zhang JZH, Mei Y (2016) Calculations of solvation free energy through energy reweighting from molecular mechanics to quantum mechanics. J Chem Theory Comput 12(2):499–511. doi:10.1021/acs.jctc.5b00920
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79(2):926–935. doi:10.1063/1.445869
Klamt A (2011) The COSMO and COSMO-RS solvation models. Wiley Interdiscip Rev Comput Mol Sci 1(5):699–709. doi:10.1002/wcms.56
Klamt A (2016) Placeholder: Cosmo-rs sampl5 results. J Comput Aided Mol Des. doi:10.1007/s10822-016-9927-y
König G, Mei Y, Pickard FC, Simmonett AC, Miller BT, Herbert JM, Woodcock HL, Bernard BR, Shao Y (2016) Computation of hydration free energies using the multiple environment single system quantum mechanical/molecular mechanical method. J Chem Theory Comput 12(1):332–344. doi:10.1021/acs.jctc.5b00874
König G, Pickard FC, Huang J, Simmonett C, Tofoleanu F, Lee J, Dral PO, Prasad S, Jones M, Shao Y, Thiel W, Brooks BR (2016) Calculating distribution coefficients based on multi-scale free energy simulations an evaluation of MM and QM/MM explicit solvent simulations of water-cyclohexane transfer in the SAMPL5 challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9936-x
König G, Hudson PS, Boresch S, Woodcock HL (2014) Multiscale free energy simulations: an efficient method for connecting classical MD simulations to QM or QM/MM free energies using Non-Boltzmann Bennett Reweighting schemes. J Chem Theory Comput 10(4):1406–1419. doi:10.1021/ct401118k
König G, Pickard FC, Mei Y, Brooks BR (2014) Predicting hydration free energies with a hybrid QM/MM approach: an evaluation of implicit and explicit solvation models in SAMPL4. J Comput Aided Mol Des 28(3):245–257. doi:10.1007/s10822-014-9708-4
König G, Boresch S (2011) Non-Boltzmann sampling and bennett’s acceptance ratio method: how to profit from bending the rules. J Comput Chem 32(6):1082–1090. doi:10.1002/jcc.21687
König G, Brooks BR (2015) Correcting for the free energy costs of bond or angle constraints in molecular dynamics simulations. Biochim Biophys Acta Gen Subj 1850(5):932–943. doi:10.1016/j.bbagen.2014.09.001
Kunieda M, Nakaoka K, Liang Y, Miranda CR, Ueda A, Takahashi S, Okabe H, Matsuoka T (2010) Self-accumulation of aromatics at the oilwater interface through weak hydrogen bonding. J Am Chem Soc 132(51):18281–18286. doi:10.1021/ja107519d
Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. doi:10.1103/PhysRevB.37.785
Lee AC, Yu Yu J, Crippen GM (2008) pKa prediction of monoprotic small molecules the smarts way. J Chem Inf Model 48(10):2042–2053. doi:10.1021/ci8001815
Lin B, Pease JH (2013) A novel method for high throughput lipophilicity determination by microscale shake flask and liquid chromatography tandem mass spectrometry. Comb Chem High Throughput Screen 16(10):817–825. doi:10.2174/1386207311301010007
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) In vitro models for selection of development candidates experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1):3–25. doi:10.1016/S0169-409X(96)00423-1
Lipnick RL (2008) Environmental hazard assessment using lipophilicity data. Wiley-VCH Verlag GmbH, pp 339–353. doi:10.1002/9783527614998.ch19
Liptak M, Shields G (2001) Accurate pKa calculations for carboxylic acids using complete basis set and Gaussian-n models combined with CPCM continuum solvation methods. J Am Chem Soc 123(30):7314–7319. doi:10.1021/ja010534f
Marenich AV, Cramer CJ, Truhlar DG (2009) Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J Phys Chem B 113(14):4538–4543
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396
McQuarrie DA (1976) Statistical mechanics. Harper and Row, New York
Miehlich B, Savin A, Stoll H, Preuss H (1989) Results obtained with the correlation energy density functionals of becke and lee, yang and parr. Chem Phys Lett 157(3):200–206. doi:10.1016/0009-2614(89)87234-3
Mikulskis P, Cioloboc D, Andrejić M, Khare S, Brorsson J, Genheden S, Mata RA, Söderhjelm P, Ryde U (2014) Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid host-guest binding energies. J Comput Aided Mol Des 28(4):375–400. doi:10.1007/s10822-014-9739-x
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) Blind prediction of solvation free energies from the SAMPL4 challenge. J Comput Aided Mol Des 28(3):135–150. doi:10.1007/s10822-014-9718-2
Ollson MA, Söderhjelm P, Ryde U (2016) Converging ligand-binding free energies obtained with free-energy perturbations at the quantum mechanical level. J Comput Chem 37(17):1589–1600. doi:10.1002/jcc.24375
Perrin DD, Dempsey B, Serjeant EP (1981) pKa prediction for organic acids and bases. Chapman and Hall, London
Pickard IV FC, König G, Simmonett AC, Shao Y, Brooks BR (2016) An efficient protocol for obtaining accurate hydration free energies using quantum chemistry and reweighting from molecular dynamics simulations. Bioorg Med Chem. doi:10.1016/j.bmc.2016.08.031
Ribeiro RF, Marenich AV, Cramer CJ, Truhlar DG (2010) Prediction of SAMPL2 aqueous solvation free energies and tautomeric ratios using the SM8, SM8AD, and SMD solvation models. J Comput Aided Mol Des 24(4):317–333. doi:10.1007/s10822-010-9333-9
Rodinger T, Pomès R (2005) Enhancing the accuracy, the efficiency and the scope of free energy simulations. Curr Opin Struct Biol 15:164–170
Rustenburg AS, Dancer J, Lin B, Feng JA, Ortwine DF, Mobley DL, Chodera JD (2016) Measuring experimental cyclohexane-water distribution coefficients for the SAMPL5 challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9971-7
Ryde U, Söderhjelm P (2016) Ligand-binding affinity estimates supported by quantum-mechanical methods. Chem Rev 116(9):5520–5566. doi:10.1021/acs.chemrev.5b00630
Sampson C, Fox T, Tautermann CS, Woods C, Skylaris CK (2015) A “Stepping Stone” approach for obtaining quantum free energies of hydration. J Phys Chem B 119(23):7030–7040. doi:10.1021/acs.jpcb.5b01625
Shirts MR, Chodera JD (2008) Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 129(12):124105. doi:10.1063/1.2978177
Skillman AG, Geballe MT, Nicholls A (2010) SAMPL2 challenge: prediction of solvation energies and tautomer ratios. J Comput Aided Mol Des 24(4):257–258. doi:10.1007/s10822-010-9358-0
Speight JG (2005) Lange’s handbook of chemistry, 16th edn. McGraw-Hill Education, New York
Sugita Y, Kitao A, Okamoto Y (2000) Multidimensional replica-exchange method for free-energy calculations. J Chem Phys 113:6042–6050
Sugita Y, Okamoto Y (1999) Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 314:141–151
Tissandier MD, Cowen KA, Feng WY, Gundlach E, Cohen MH, Earhart AD, Coe JV, Thomas R, Tuttle J (1998) The proton’s absolute aqueous enthalpy and gibbs free energy of solvation from cluster-ion solvation data. J Phys Chem A 102(40):7787–7794. doi:10.1021/jp982638r
Tofoleanu F, Brooks BR, Buchete NV (2015) Modulation of Alzheimers a protofilament-membrane interactions by lipid headgroups. ACS Chem Neurosci 6(3):446–455. doi:10.1021/cn500277f
Torrie GM, Valleau JP (1977) Nonphysical sampling distributions in monte carlo free-energy estimation: umbrella sampling. J Comput Phys 23:187
Turney JM, Simmonett AC, Parrish RM, Hohenstein EG, Evangelista FA, Fermann JT, Mintz BJ, Burns LA, Wilke JJ, Abrams ML, Russ NJ, Leininger ML, Janssen CL, Seidl ET, Allen WD, Schaefer HF, King RA, Valeev EF, Sherrill CD, Crawford TD (2012) Psi4: an open-source ab initio electronic structure program. Wiley Interdiscip Rev Comput Mol Sci 2(4):556–565. doi:10.1002/wcms.93
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD Jr (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31(4):671–690. doi:10.1002/jcc.21367
Verdolino V, Cammi R, Munk BH, Schlegel HB (2008) Calculation of pka values of nucleobases and the guanine oxidation products guanidinohydantoin and spiroiminodihydantoin using density functional theory and a polarizable continuum model. J Phys Chem B 112(51):16860–16873. doi:10.1021/jp8068877
Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL, Masse C, Knight JL, Steinbrecher T, Beuming T, Damm W, Harder E, Sherman W, Brewer M, Wester R, Murcko M, Frye L, Farid R, Lin T, Mobley DL, Jorgensen WL, Berne BJ, Friesner RA, Abel R (2015) Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc 137(7):2695–2703. doi:10.1021/ja512751q
Zacharias M, Straatsma TP, McCammon JA (1994) Separation-shifted scaling, a new scaling method for Lennard-Jones interactions in thermodynamic integration. J Chem Phys 100:9025–9031
Zhang S, Baker J, Pulay P (2010) A reliable and efficient first principles-based method for predicting pKa values. 1. Methodology. J Phys Chem A 114(1):425–431. doi:10.1021/jp9067069
Zhao Y, Truhlar DG (2007) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor Chem Acc 120:215–241
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41:157–167
Zwanzig RW (1954) High-temperature equation of state by a perturbation method. 1. Nonpolar gases. J Chem Phys 22:1420–1426
Acknowledgments
This work was supported by the intramural research program of the National Heart, Lung and Blood Institute of the National Institutes of Health and utilized the high-performance computational capabilities of the LoBoS and Biowulf Linux clusters at the National Institutes of Health. (http://www.lobos.nih.gov and http://biowulf.nih.gov)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pickard, F.C., König, G., Tofoleanu, F. et al. Blind prediction of distribution in the SAMPL5 challenge with QM based protomer and pK a corrections. J Comput Aided Mol Des 30, 1087–1100 (2016). https://doi.org/10.1007/s10822-016-9955-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-016-9955-7