
Abstract
In South Africa, the green seaweed Ulva lacinulata is grown in land-based integrated multi-trophic aquaculture (IMTA) farms with the abalone Haliotis midae. The Ulva serves as a biofilter and the co-produced Ulva is often used as feed for the abalone. To better understand the potential benefits and risks associated with this practice, this study characterised the bacterial microbiome associated with the seawater and Ulva raceways receiving abalone effluent (IMTA system) and compared this to Ulva tanks supplied with fertilised seawater (non-IMTA; control). Ulva samples were collected from each Ulva system, and water samples were collected at the inlet and outlet of each system. Bacterial communities were assessed using a culture-based approach and next-generation sequencing (NGS) of the V3-V4 16S rDNA region. It was observed that Ulva has the potential to reduce the bacterial load of abalone effluent, with the total number of potential culturable Vibrio species declining from 150×103 cells mL-1 in the inlet to 37×103 cells mL-1 in the outlet of the Ulva system. The NGS dataset supported these findings, with a reduction observed in Vibrio and Pseudoalteromonas from the inlet to outlet samples. A lower number of genera (p < 0.05) were observed on Ulva when compared with water samples, indicating that Ulva has a beneficial, modulatory effect on bacteria. These findings contribute towards the growing body of evidence for the benefits of seaweeds in IMTA and addresses the biosecurity concerns of abalone farmers wishing to improve the circularity of their farming activities by incorporating seaweeds.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The high stocking densities associated with most commercial aquaculture farms, including abalone farms, produces effluent water that often contains high concentrations of nitrogen and other dissolved and solid waste products. In partial and fully recirculating aquaculture systems (RAS), these waste products have been shown to impair growth, promote stress and/or disease, and can result in mortalities in more extreme cases (Primavera 2006). Additionally, opportunistic pathogen proliferation can be promoted by a nutrient-rich environment (Defoirdt 2016). Integrated aquaculture systems offer an environmentally friendly means to bioremediate nutrient-rich effluent water generated from monoculture (cultivation of single crop) facilities (Chopin et al. 2001). By using seaweeds to create an ecosystem-based/nature-based management approach to aquaculture, integrated multi-trophic aquaculture (IMTA) addresses some of the negative environmental impacts associated with high density monoculture (Soto et al. 2008), while allowing for the production of alternative/additional crops. For example, the green seaweed Ulva was identified as an ideal species for bioremediation in land-based aquaculture in the 1900s in Australia (Cotton 1910). In South Africa, Ulva spp. have been grown successfully in integrated systems since 2002; where Ulva is grown in abalone effluent and sometimes fed back to the abalone as a feed, thereby recycling nutrients sourced from feed and excreted by the animals (Bolton et al. 2009; 2016).
Extractive marine organisms from lower trophic levels (such as algae and bivalves), that are capable of utilising dissolved particulates and converting the inorganic nutrients into useful biomass can contribute towards improved sustainability of the aquaculture industry (Bolton et al. 2009). This biomass can be used for various applications, including their use as feeds or secondary crops, medicines (Mao et al. 2006; Hong et al. 2007), cosmetics (Farasat et al. 2014), fertilisers (Selvam and Sivakumar 2013) and other biorefined goods (Buchholz et al. 2012). The use of macroalgae as biofilters shows great promise as it reduces the impact of nutrient release into the environment (Vandermeulen and Gordin 1990; Chung et al. 2002; Nielsen et al. 2012; Lawton et al. 2013; Aníbal et al. 2014), resulting in improved nearby ecosystem health, social perception, and economic stability through crop diversification (Chopin et al. 2001; Soto et al. 2008). Furthermore, the use of Ulva as a biofilter in land-based aquaculture facilities has another advantage in that it can allow for partial wastewater recirculation in IMTA systems, substantially reducing pumping and energy costs on farms (Robertson-Andersson et al. 2008; De Prisco 2020) and contributing to the removal of common micropollutants (Hardegen et al. 2023).
Numerous facets of the land-based abalone-Ulva aquaculture systems in South Africa have been assessed, including nutrient recycling within the system, changes in water chemistry (De Prisco 2020; Geldart 2022), growth of farmed animals (abalone and urchins) fed diets supplemented with or without effluent grown macroalgae (Naidoo et al. 2006; Mulvaney et al. 2013; Cyrus et al. 2014, 2015a, 2015b; Kemp et al. 2015; Bansemer et al. 2016; Brink-Hull et al. 2022), and socio-economic evaluations (Troell et al. 2006). Nonetheless, very little attention has been devoted to assessing potential biosecurity risks associated with the recirculation of wastewater and use of effluent grown Ulva as a feed or feed additive. There has also been limited research on the potential health benefits of growing animals in land-based integrated systems and the effects of this seaweed integration on the microbiome. Managing aquatic environments is complex due to the intimate relationship(s) that exists between microorganisms and their hosts, especially in open flow-through or partially recirculating land-based (pump-ashore) aquaculture systems (Cripps and Bergheim 2000; Olafsen 2001). For example, water quality and disease control are directly impacted by the bacterial communities entering and/or residing within the system and these complex interactions are also affected by changes in biotic and abiotic factors (Moriarty 1997; Thompson et al. 2002). Due to the close association between bacteria and their microenvironment, it is vital to understand how communities assemble and function within integrated systems.
While a diverse array of microorganisms is capable of colonising marine macroalgal surfaces (Bolinches et al. 1988; Snoeijs 1994; Bengtsson et al. 2010; Godinho et al. 2013), bacteria are considered the primary colonisers of surfaces in marine environments and have the ability to impact subsequent colonisation by other organisms (Crisp and Ryland 1960; Bryers and Characklis 1982; Henschel and Cook 1990; Garrett et al. 2008). Bacterial colonisation of healthy hosts, as observed for algae (Ulva) and abalone on commercial IMTA farms in South Africa, suggests that a positive relationship exists between these hosts and the bacteria associated with them (collectively termed the “holobiont”). Interactions between macroalgae and bacteria have been widely documented and include both beneficial and pathogenic associations (Armstrong et al. 2001; Dale and Moran 2006; Goecke et al. 2010; Wichard 2015; Califano et al. 2020; Li et al. 2023). There are also possible communal relationships where host-associated bacteria do not impact host functionality (Macpherson and Harris 2004) or, in the case of aquaculture species, host productivity. However, Ulva performance/productivity (morphological development, growth and biochemical composition) has been previously linked to the seaweed beneficial microorganisms (SBMs) associated with them (Alsufyani et al. 2020; Polikovsky et al. 2020; Wichard 2015; 2023; van der Loos 2021; 2022; 2024; Li et al. 2023), releasing algal growth and morphogenesis promoting factors (AGMFs) (Ghaderiardakani et al. 2019). Microbial communities are thought to assemble and associate with hosts in one of two ways and these mechanisms are termed “the hologenome theory” and the “competitive lottery model”. The hologenome theory proposes that a stable core microbial community is present and, collectively with its host, acts as a selective force for evolution (Zilber-Rosenberg and Rosenberg 2008). In contrast, the competitive lottery model suggests that a microbial community forms as a result of/in response to its surrounding environment, where individual taxa simply co-exist in a variable environment (Chesson and Warner 1981). Burke et al. (2011) suggested that the assemblages of U. australis are determined by a lottery model as opposed to symbiotic interactions. However, several specific bacteria that are essential for settlement, growth and morphogenesis have been consistently found in associated with Ulva spp. (Spoerner, et al. 2012; Wichard 2015; Ghaderiardakani et al. 2019; Califano et al. 2020; van der Loos et al. 2021, 2022, 2024), suggesting that a holobiont exists but may be influenced by the environment (e.g., land-based vs open water aquaculture systems) and environmental parameters (e.g., water quality, seasons) to some extent.
In South Africa, five large (each producing 200 – 600 t of abalone per annum) commercial abalone farms collectively cultivate more than 2 000 t of Ulva in large D-shaped paddle-raceways (ca. 30 m long, 8 m wide, 0.5 – 1 m deep), mostly in abalone effluent (Bachoo et al. 2023). Despite this, no published information exists on the microbial communities associated with the seawater and Ulva in these intensive land-based pump-ashore mariculture systems. This information is particularly important given that some of these farms utilise the Ulva as feed for the abalone, which has been shown to have consumption, growth and health benefits for the abalone (Naidoo et al. 2006; Mulvaney et al. 2013; Kemp et al. 2015; Bansemer et al. 2016). However, the biosecurity concerns associated with feeding effluent-grown Ulva is preventing further IMTA uptake in South Africa and abroad (Bolton et al. 2009).
To better understand potential benefits and risks associated with the use of effluent-grown Ulva as a feed, as well as the role of Ulva in these IMTA systems, the bacterial microbiome associated with seawater and Ulva grown in non-recirculating (flow-through) raceways receiving either abalone effluent (IMTA) or fertilised seawater (non-IMTA; control) was assessed in this study. By improving the understanding of integrating Ulva into aquatic systems and its impact on the microbiome, this study could contribute to the improvement of existing recirculating aquaculture technologies and increase the uptake of the integration of Ulva in aquaculture practices, thereby improving the sustainability, productivity and profitability of the aquaculture sector.
Materials and methods
Study site and sampling
Water and Ulva lacinulata samples were collected from Ulva paddle-raceway systems at Irvine & Johnson (I&J) Cape Abalone Farm at Danger Point, Gansbaai (34°34’60 S; 19°21’0 E) in August 2017. Samples were collected from two separate Ulva production systems on the farm (Fig. 1). One system consisted of tanks that received seawater supplemented with inorganic fertiliser (non-IMTA/Seawater), whereas the other system was comprised of raceways receiving abalone effluent water (IMTA/Effluent). Triplicate Ulva samples were collected from within each system, whereas the water samples (in triplicate) were collected at the inlet and outlet of each system. In total, two seawater tanks (there were only two on the farm at the time of this study) and five effluent raceway systems were sampled (Fig. 1), resulting in n = 15 samples (five inlet, outlet and Ulva) in the IMTA systems and n = 6 samples in the non-IMTA systems (two inlet, outlet and Ulva), respectively.

Schematic of the abalone effluent wastewater Ulva raceways (UR) at I&J abalone farm showing the Ulva raceways receiving effluent water from the abalone tanks (UR_Effluent_In) and water going out of these tanks (UR_Effluent_Out) (scale bar = 5 m) (a), as well as the location of these Ulva raceways (b). Seawater systems consisted of two tanks containing Ulva (U_Seawater) that received non-effluent seawater (UR_Seawater_In) and releasing water to the sea (UR_Seawater_Out) (c), as opposed to the Ulva raceways bio-remediating abalone effluent water (d)
The non-IMTA systems serve as algal “seeding tanks” for the larger Ulva raceways and are based on the system previously described by Robertson-Andersson (2003). These systems are comprised of two semi-circular, PVC lined containers with an approximate surface area of 3 m × 1 m and a depth of 0.5 m supported on a wooden frame. Fresh seawater is pumped directly from the adjacent surf zone into the tanks via a sub-surface filter system and exits the tanks passively. Tanks are actively aerated and pulse-fertilised with ammonium phosphate twice weekly to promote algal growth.
The effluent (IMTA) Ulva raceways systems receive nutrient rich effluent water from the abalone raceways. Water gravity fed from the abalone culture tanks enters through a central canal, after which it passively flows through each of the 13 parallel raceways (L × W × D; 27.7 m × 8.23 m × 0.45 m) via openings along the channel (Fig. 1). No mixing of water occurs between the raceways. A paddlewheel is used to circulate and agitate the Ulva within each raceway, as well as to oxygenate the water. The effluent raceways are pulse-fertilised with nitrogen and phosphorus every second week to reduce nutrient stress on the Ulva, as the nutrients provided by the abalone effluent wastewater alone is insufficient. Each effluent Ulva raceway consistently produces 1.5 to 1.75 t wet biomass per month (Shuuluka 2011; Bolton et al. 2016) and is utilised as a feed for abalone on the farm.
Healthy vegetative Ulva thalli (approximately 15 g fresh weight), with no necrotic white tissue or noticeable epiphytes, were collected from the seawater and effluent systems and immediately transferred into separate pre-labelled sterile 50 mL conical centrifuge tubes that had been placed in a cooler box with ice. Thereafter, samples were transferred to the Department of Forestry, Fisheries and the Environment (DFFE) Marine Research Aquarium (MRA) in Cape Town, South Africa, within three hours for further processing. At the research facility, the freshly collected Ulva was rinsed with 10 mL of 0.22 μm filtered autoclaved seawater via inversion for 20 s, to remove any loosely attached organisms and debris. Directly after rinsing, a portion of the Ulva (approximately 20 mg) was used to quantify the culturable bacteria. The remaining Ulva was ground to a fine powder using a mortar and pestle with liquid nitrogen and the ground samples were stored at -80°C prior to next-generation sequencing (NGS).
Inlet and outlet water samples (500 mL) were collected in triplicate from the seawater (non-IMTA) and effluent (IMTA) systems in sterile 500 mL Schott bottles. Samples were stored on ice and transferred to the MRA in Cape Town, where they were processed for enumeration of culturable bacteria and NGS as described below.
Enumeration of bacteria in seawater and on Ulva
Three types of microbiological media were used to enumerate live culturable bacteria in seawater and on Ulva: Tryptic Soy Agar (TSA; Difco), Thiosulfate Citrate Bile Sucrose Agar (TCBS; Sigma Aldrich), and Ulvan Agar (UA; Jaulneau et al. 2010). TSA is a general-purpose growth medium frequently used for the isolation of a wide variety of marine microorganisms (Lemos et al. 1985; Romanenko et al. 2008; Rodrigues and de Carvalho 2022). TCBS Agar is used for the isolation of potential culturable Vibrio spp. (Pfeffer and Oliver 2003), whereas UA is a minimal medium supplemented with Ulvan, the primary carbohydrate of Ulva (prepared according to Jaulneau et al. 2010). Though it should be noted that a limited number of bacteria are culturable using standard agar plate techniques and laboratorial conditions (Rodrigues and de Carvalho 2022), this approach allowed for the evaluation of differences in bacterial colony forming units across different system types (IMTA vs non-IMTA) and between samples collected from different system compartments (i.e. inlet vs outlet vs Ulva). The culture-based approach also allowed for the isolation, purification and identification of bacteria that appeared in high abundance for comparison with the next-generation sequencing (NGS) approach.
Preparation of media
The TSA and TCBS agar plates were prepared as per the manufacturer’s instructions and supplemented with 2.0% and 1.5% NaCl (w/v), respectively. The UA plates consisted of a basic minimal media containing 0.1% (w/v) yeast extract, 0.5% (w/v) ulvan extract (described below), and 15 g L-1 of bacteriological agar in 1000 mL sterile autoclaved seawater. Ulvan was extracted from dry Ulva by homogenising 50 g of dried Ulva into a coarse powder (< 1 cm) in a food processor before diluting the Ulva powder with 450 mL Millipore water and autoclaving at 120ºC for 15 min. After allowing partial cooling, the algal suspension was filtered through a sieve (0.25 mm) and centrifuged at 5000×g for 5 min. The pellet was discarded and 2.5 volumes (1 125 mL) of 70% ethanol was used to precipitate the ulvan from the supernatant. After one hour, the white dough-like ulvan precipitate was removed and washed with 96.9% ethanol. The precipitate was centrifuged at 5000×g for 5 min, washed with 96.9% ethanol and vacuum dried in a centrifuge (1 200 rpm) at 30ºC for 4 h. The dry ulvan was ground into a fine powder using a mortar and pestle and aliquots were stored in Eppendorf tubes in a -20ºC freezer until needed. On average, 50 g of dried Ulva yielded 4.612 g of ulvan (9.2% dw).
Enumeration of bacteria in seawater and on Ulva
For the isolation and enumeration of culturable bacteria from seawater, the 500 mL water samples collected from the seawater (n = 2 per sample type) and effluent water systems (n = 5 per sample type) were aseptically filtered through 0.22 μm-pore-size filter membranes (47 mm diameter, Millipore Corp., USA). The filter membranes were gently scraped with a sterile scalpel blade to loosen the bacterial cells before transferring each filter and its accompanying cells into separate sterile hawk tubes containing 10 mL sterile autoclaved seawater (ASW). Tubes were vortexed for 5 min to remove any remaining bacteria attached to the membrane. Thereafter, the membranes were discarded, and 0.1 mL aliquots of the re-suspended cells were serially diluted (10-2 – 10-5) and plated in triplicate (100 µL aliquots) on TSA, TCBS and UA plates. The remaining 9.9 mL of seawater containing bacteria from each sample was centrifuged for 10 min at 10 816×g to concentrate the bacterial cells. Following centrifugation, the supernatants were carefully decanted and the tubes containing the pellet were stored at -80°C until required for DNA extractions and NGS.
Bacteria were isolated from fresh vegetative thalli of Ulva using a modified version of the method described by Nakanishi et al. (1996). Algal thalli were gently washed in autoclaved seawater (ASW) to detach any loosely associated debris, after which approximately 0.5 g of thallus was vortexed vigorously in 10 mL ASW for 10 min to detach the associated epibacteria. The thallus was then removed, and the remaining supernatant was serially diluted (100 – 10-3) and 100 µL aliquots were plated in triplicate onto the three selective media. Immediately following plating, petri dishes were sealed with Parafilm, and the TSA and TCBS plates were incubated at room temperature for two days, whereas UA plates were incubated at room temperature for up to 7 days (accounting for slow growth) and monitored daily for growth. Bacterial colonies were counted using a dissecting microscope and recorded for each plate following the respective incubation periods. The number of culturable bacteria per mL seawater or g of Ulva for each dilution series was calculated and averaged. Thereafter, assay replicates for each experimental replicate (n = 5 for IMTA, and n = 2 for non-IMTA) were averaged and data expressed as the mean ± SE for each media type.
Isolation, identification and storage of culturable bacteria
Following 2 – 7 days of incubation, bacterial colonies of interest were selected from each media type and transferred to fresh plates of the corresponding media type to isolate pure colonies. Bacterial colonies that appeared unique, based on colony form (shape, colour, size and texture), elevation (flat, raised, umbonate, convex or pulvinate) and margin (entire, undulated, lobate or filiform), or occurred in high abundance on the different media types were selected for further analysis. Colonies were sub-cultured until pure/monocultures were obtained and used for long-term storage. For long-term storage, glycerol socks (20% sterile glycerol in pre-autoclaved Tryptic Soy Broth solution (TSB); 0.625 g NaCl, 0.6 g TSB powder in 20 mL Millipore water) were prepared for each isolate and stored in a -80ºC ultra-freezer.
Genomic DNA was extracted from the chosen bacterial isolates and subjected to 16S rDNA amplification and Sanger sequencing to identify the isolated bacteria (Supplementary Table 1). Genomic DNA was isolated by picking cells from a single colony and suspending them in 300 μL of sterile Millipore water in a 1.5 mL micro-centrifuge tube. Each tube was vortexed vigorously and centrifuged at 16 000×g for 5 min. The supernatants were carefully removed, and the remaining pellets re-suspended in 300 μL sterile water. Bacterial pellets were homogenised for 1 min using a hand-held pellet pestle and transferred to new sterile 1.5 mL microcentrifuge tubes containing 0.04±0.05 g Chelex-100 beads. Chelex-100 is comprised of negatively charged microscopic beads that chelate metal ions which are required as catalysts or cofactors in enzymatic reactions. The samples containing Chelex-100 beads were briefly vortexed and incubated for 20 min at 56ºC on a heating block. Post incubation, samples were briefly vortexed and incubated for a further 30 min at 95ºC to lyse bacterial cells. The lysed samples were placed on ice to cool for 5 min, vortexed briefly and centrifuged at 16 000×g for 5 min.
The universal broad-spectrum bacterial primers 16S F-fD1 and 16SR-Rp2 (Supplementary Table 1; Lane 1991) were used to amplify an approximately 1.5 kb fragment of the 16S rRNA gene region of each isolate. PCR reaction mixtures (25 μL; performed in triplicate) consisted of 1 μL genomic DNA (ca. 25 ng), 12.5 μL KapaTaq ReadyMix (Kapa Biosystems; Cat#KK1006), 10.5 μL Millipore water, and 0.5 μL of each primer (400 nM). Amplification was conducted using a Labnet Multigene Thermal Cycler (Labnet International Inc.) and consisted of an initial denaturation at 96ºC for 3 min, followed by 40 cycles of 45 s at 96ºC, 30 s at 57ºC, and 1 min at 72ºC, with a final extension of 72ºC for 5 min. Known amounts of bacterial genomic DNA, previously analysed by PCR and subjected to sequencing, were included as positive controls during the PCR and non-template controls (PCR-grade H2O) were included as negative controls. The amplified PCR products, as well as the positive and negative controls were visualised on a 0.8% agarose gel electrophoresis to assess reaction specificity and fragment size (ca. 1.5 kb fragment expected). The PCR product obtained from each bacterial colony was purified and sequenced as described above at the Stellenbosch University Central Analytical Facilities (CAF). The universal 16S forward primer F-fD1 was used for sequencing (Supplementary Table 1). The sequences were edited using CLC Main Workbench and homology searches were carried out using the BLASTN algorithm (Altschul et al. 1990) provided by the National Centre for Biotechnology Information (NCBI).
Statistical analyses for colony forming unit (CFU) data
SigmaPlot 12.0 software was used to perform all CFU data statistical analysis. To determine whether the number of culturable bacteria differed between the inlet (incoming) and outlet (outgoing) water within and between each system (n = 2 per sample type across 2 independent non-IMTA systems; n = 5 per sample type across 5 independent IMTA systems), including the abalone effluent water and seawater system (CFU mL-1), as well as that on Ulva from each system (CFU.g-1 of Ulva), a one-way analysis of variance (ANOVA) was performed (statistically significance at p < 0.05), with post-hoc Holm-Sidak tests for comparisons between samples. Data for the different media types (TCBS, TSA and UA) were treated separately. All data were tested for normality (Kolmogorov-Smirnov test) and equal variance. Data were square root transformed prior to statistical analysis. Data values on all figures refer to mean±standard error of the mean (SE).
Bacterial community profiling by 16S rDNA sequencing
DNA extraction, library preparation and sequencing
Microbial genomic DNA was isolated from frozen ground Ulva samples (to target both epi- and endophytic bacterial communities) and bacterial pellets, obtained from the seawater samples, using the QIAamp DNA Micro Kit (Qiagen, Cat. No. 56304) following the manufacturer’s instructions. The entire bacterial pellet obtained from each seawater sample was used for genomic DNA isolation, whereas approximately 5 mg of each frozen ground Ulva sample collected from the seawater and effluent water systems, respectively, was used for DNA extraction. A total of 14 frozen bacterial pellets and 7 ground Ulva samples were processed. DNA concentration, integrity and fragment size were determined via spectrophotometry and 0.8% agarose gel electrophoresis.
The 16S_341F and 16S_805R primer pair (Klindworth et al. 2013), with added Illumina adapter overhang nucleotide sequences (Supplementary Table 1), were used to amplify the 16S V3-V4 hypervariable 16S gene region of each sample. Each PCR reaction (25 μL; performed in triplicate) consisted of 1 – 5 μL of genomic DNA (ca. 25 ng total), 12.5 μL 2×KAPA HiFi HotStart ReadyMix for hot-start PCR (Kapa Biosystems; Catalog #KM2605), 5.5 – 10.5 μL ddH2O, and 0.5 μL of each primer (200 nM). Touchdown PCR amplifications were carried out in a Bio-Rad CFX96 Real-Time PCR Detection System Instrument (Bio-Rad Laboratories) using a modified version of the cycling conditions outlined by the Illumina 16S metagenomics sequencing library preparation manual. The PCR conditions were as follows; an initial denaturation at 95ºC for 5 min, 10 cycles of touchdown PCR (30 s at 95ºC, 30 s at 65ºC, with a 1ºC decrement per cycle, and 30 s at 72 ºC), followed by an additional 25 cycles of PCR (30 s at 95ºC, 30 s at 55ºC, and 30 s at 72ºC), and a final extension step for 10 min at 72ºC. Negative controls, containing all components other than DNA templates, were run in parallel. Amplified PCR products and negative controls were electrophoresed as above to confirm successful amplification, reaction specificity and fragment size (ca. 460 bp fragment expected).
Post validation, PCR purification, indexing and library preparation were performed at the Next Generation Sequencing Facility (NGSF) at the University of the Western Cape (UWC), where the PCR products from each sample were used to create libraries with unique barcodes (n = 21 samples/libraries). Briefly, PCR products were purified to remove free primers and primer dimer species using AMPure XP beads (Beckman Coulter), as per manufacturer’s instructions. Thereafter, dual indices and Illumina sequencing adaptors were attached to the purified PCR products using the Nextera XT Index Kit, followed by a second PCR purification step using AMPure XP beads, whereafter final library quantification and normalisation were performed. Normalised libraries were pooled prior to paired-end sequencing (2×250 bp) on the Miseq Illumina sequencer, as per manufacturer protocols.
16S raw data processing
Analysis of the 16S V3-V4 rDNA sequences generated using Illumina’s MiSeq platform were performed using the open-source Quantitative Insights into Microbial Ecology 2 (QIIME2 version 2020.11) software (Caporaso et al. 2010). The DADA2 pipeline (Callahan et al. 2016) was used to process a total of 6 016 889 demultiplexed sequences across 21 samples (average of 286 519 reads per sample), where forward and reverse reads were trimmed to lengths of 227 and 203 bp, respectively, to remove low quality reads (Q20 score < 20). Subsequently, DADA2 was used to filter, denoise and merge reads, which resulted in reads with a final average read length of 419±11 bp (Supplementary Table 1). An average of 7.63% of the total reads were detected as chimeric and were removed from analyses. After filtering, denoising, merging and removing chimeric reads, a total of 2 138 906 dereplicated sequences were included in subsequent analyses, with an average of 101 853 reads per sample. Taxonomy was assigned using a naive Bayes classifier trained on the SILVA database (release 132) (Quast et al. 2013; Yilmaz et al. 2014; Glöckner et al. 2017) for each amplified sequence variant (ASV), representing each unique read of the sequenced gene region (Callahan et al. 2017) up to genus-level.
Data filtering and within sample diversity assessments
The ASV dataset was filtered and normalised using MicrobiomeAnalyst (Dhariwal et al. 2017). Data was filtered for low abundance reads (minimum count of 2 with 20% prevalence in samples), whereby ASVs containing mostly zeros were removed to account for potential sequencing errors and low-level contaminations (Dhariwal et al. 2017). This filtered dataset was used to calculate alpha diversity (within sample) statistics (Chao1, Shannon and Simpson index).
Bacterial community alpha diversity was evaluated using the R phyloseq (McMurdie and Holmes 2013) and vegan (Dixon 2003) packages implemented in MicrobiomeAnalyst. The Chao1 (Chao 1984), Shannon (Shannon 1948) and Simpson (Simpson 1949) non-parametric diversity indexes were evaluated to assess species richness, evenness and uniqueness, respectively. Statistical significance between cohorts was assessed with an analysis of variance (ANOVA; p < 0.05) for each index. Lastly, rarefaction curves were constructed in MicrobiomeAnalyst (Dhariwal et al. 2017) by randomly sampling a fixed number of reads to assess whether there was sufficient coverage to capture the bacterial diversity in each sample, which was further assessed by calculating Good’s coverage indices across samples.
Normalisation and between sample comparisons
Low variance reads (10% based on standard deviation), unlikely to be associated with study conditions, were removed to reduce the effects of multiple testing on downstream comparative analyses. Filtered data was normalised through relative log expression (RLE) transformationto account for sparsity, under-sampling and uneven sequencing depth in downstream beta diversity analyses.
Beta diversity analyses were conducted in MicrobiomeAnalyst using the R phyloseq and vegan packages. A NMDS analysis, based on Bray-Curtis dissimilarity indices, was performed using a non-rarefied dataset. These analyses were conducted at the genus level to provide an overview of the microbial community structure in the seawater (non-IMTA) and abalone effluent water (IMTA) systems, as well as the microbiota on the Ulva within each system. Statistical significance (p < 0.05) was assessed using a permutational multivariate analysis of variance (PERMANOVA; Anderson 2001), which tests for homogeneity across data points. Additionally, to further assess the degree of dissimilarity across samples, a permutational analysis of multivariate dispersions (PERMDISP; Anderson et al. 2006) and an analysis of similarities (ANOSIM; Clarke 1993) was conducted. The online webtool, Jvenn (Bardou et al. 2014), was used to construct a Venn diagram by assessing common bacterial ASVs at genus level in the non-IMTA and IMTA system, respectively, without incorporating relative abundance information. Lastly, hierarchical relationships between cohorts were assessed at genus level through a sample-based clustering analysis, based on the Bray-Curtis distance matrix and Ward clustering algorithm.
MicrobiomeAnalyst was also used to assess taxonomic abundance across the respective groups, where abundance tables were constructed based on relative abundances (%). The 30 most represented ASVs were displayed and the remaining ASVs were combined and denoted as “Others”. Differential abundance across the water and Ulva samples was assessed using the DESeq2 (Love et al. 2014). The false discovery rate (FDR) was calculated to adjust p-values for multiple comparisons, mitigating the possibility of type I errors (Benjamini and Hochberg 1995).
Functional profiling across water and Ulva-associated microbiomes
The functional potential of the bacterial communities from the seawater system, effluent system and Ulva in each system was assessed by mapping RLE normalised ASVs to Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) (Kanehisa and Goto 2000) using the QIIME against SILVA database annotation pipeline of the R package Tax4Fun (Aßhauer et al. 2015) in the Marker Data Profiling (MDP) module of MicrobiomeAnalyst. Subsequently, the Shotgun Data Profiling (SDP) module was used to analyse the differences in KEGG metabolism, where putative enzymes are mapped onto known metabolic pathways, across water and Ulva samples. The statistical software JASP (JASP Team 2017) was used to assess data for normality (Shapiro-Wilk test) and homoscedasticity (Levene’s test). As data were not normally distributed, a Kruskal-Wallis test with a post-hoc Dunn’s test was performed to test for statistically significant (p < 0.05) differences in putative functional capabilities between cohorts.
Results
Enumeration of culturable bacteria in seawater and on Ulva
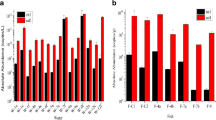
Culturable bacterial numbers were significantly (p < 0.05; one-way ANOVA) higher in the systems receiving effluent water from the abalone raceways, when compared with the Ulva tanks receiving seawater that was fertilised (Fig. 2A). The bacterial abundance associated with Ulva cultured in the IMTA systems was also significantly higher than on Ulva cultured in the non-IMTA systems on all three selective media types (Fig. 2B). In both systems, the macroalga Ulva appeared to have a strong modulatory effect on bacteria, with culturable numbers on all media types decreasing from the inlets to the outlets of the Ulva raceways. This modulatory effect was more pronounced in the IMTA systems, particularly for the total number of potential Vibrio (indicated by growth on TCBS), which decreased significantly (p = 0.032; one-way ANOVA) from the inlet (150×103 cells mL-1) to the outlet (37×103 cells mL-1) of the IMTA systems (Fig. 2A). In contrast, there was no significant decline in the total number of general marine bacteria between the inlets and outlets of the IMTA systems (p = 0.096), as indicated by growth on TSA (Fig. 2A), though this result could have been influenced by low sample numbers. There was a high abundance of ulvan specific bacteria associated with the Ulva collected from both the IMTA and non-IMTA systems, with the abundance of these bacteria shown to be significantly (p = 0.005) higher in the IMTA than in the non-IMTA systems (Fig. 2B).

Number of culturable bacteria per mL seawater (a) collected from the inlets (abalone effluent water entering tanks) and outlets (bioremediated water exiting tanks) of the fertilised (non-IMTA; n = 2 per sample type in triplicate) and effluent (IMTA; n = 5 per sample type in triplicate) Ulva raceway systems. (b) Culturable bacteria per gram of Ulva collected from the non-IMTA and IMTA systems. Bacteria were cultivated on three selective media: Tryptone Soy Agar (TSA), Thiosulfate Citrate Bile Sucrose agar (TCBS) and Ulvan Agar. All data represents the means (±SEM) of triplicate readings across sample types from the respective systems. An asterisk represents a significant difference (p < 0.05; t-test) between the inlet and outlet or non-IMTA verses IMTA Ulva on a specific selective media
Identification of culturable bacteria
The 11 bacterial isolates evaluated in this study included three isolates from ulvan (U2, U10 and U14), five from TSA (T3, T5, T6, T8, and T10) and three from TCBS (V1, V2, and V3). These isolates were evaluated based either on their unique morphology or their elevated abundance on the three media types. The nucleotide sequences obtained following PCR amplification and sequencing of the forward read using primer (F-fD1) of the 16S rRNA gene region of each of the isolated bacteria of interest resulted in sequences of approximately 460 bp. A BLAST search on GENBANK revealed that most of the sequences showed high similarity to organisms that are frequently isolated and identified from marine environments, including several Vibrio and Pseudoalteromonas species (Table 1).
Bacterial community profiling by 16S rDNA sequencing
Raw data processing, filtering and normalisation
Microbial diversity in the seawater (non-IMTA) system, abalone effluent water (IMTA) system and on the Ulva grown in both systems was assessed using next-generation sequencing of the V3-V4 region of the 16S rDNA. After quality control steps (trimming and filtering), a total of 2 138 906 sequence reads were retained across the 21 samples included in this study, with an average of 101 853±17 703 reads per sample (Supplementary Table 2) and all samples reaching a plateau in the rarefaction analysis (Supplementary Fig. 1). Raw sequence data is publicly available under project accession number PRJNA1075670 on the NCBI sequence read archive (SRA) database.
Library sizes ranged from 25 765 to 312 621 reads prior to data normalisation (Supplementary Table 2). All rarefaction curves approached the saturation plateau and reached Good’s coverage indices ranging from 99.993 – 100%, indicating that there was sufficient sequence reads within each of the 21 samples (Supplementary Fig. 1). Overall, 481 genus-level ASVs were identified after mapping reads to the SILVA reference database. A total of 197 low abundance features were removed based on prevalence prior to conducting alpha diversity analyses and for the construction of relative abundance plots, and a total of 53 low variance features were removed based on standard deviation, resulting in the total removal of 250 features prior to differential abundance assessments using DESeq2. A total of 231 genus-level ASVs were included in downstream analyses.
Within sample diversity
Overall, water samples collected from the seawater tanks had the highest ASV abundance, when compared with Ulva samples (Fig. 3). Moreover, bacterial diversity in terms of evenness in the distribution of bacterial genera and the uniqueness of these communities across cohorts did not vary significantly across the samples collected from effluent water, fertilised seawater and Ulva, as no significant differences were observed for both the Shannon and the Simpson diversity indexes, with ANOVA F-statistics of 1.13 (p = 0.39) and 1.52 (p = 0.24), respectively (Fig. 3). In contrast, the richness estimator Chao1 was significantly higher for the water samples (both IMTA and non-IMTA) than for the Ulva samples from both systems (F = 7.11, p = 0.001), indicating a high degree of unique ASVs in the water column (Fig. 3).

Average within sample diversity measures (Chao1, Shannon and Simpson) for samples collected from seawater (non-IMTA) and effluent (IMTA) systems at genus level, where the median and standard error for each is indicated. The ANOVA F-values and corresponding p-values are also indicated for each diversity index. UR – Ulva raceway; U – Ulva
Bacterial community structure in a non-IMTA vs IMTA system
Clustering analysis, based on Bray-Curtis matrices, showed that the 21 samples are broadly arranged into two clades (Fig. 4A). The first clade is composed of water samples from both the IMTA and non-IMTA systems, whereas the second clade is primarily composed of Ulva samples from both systems. It appears as if several subclades exist within the dendrogram. For instance, the inlets and outlets of the effluent water appears to be further subdivided into subclades (Fig. 4A). The Ulva samples are also grouped as such and form their own subclades (i.e., Ulva growing in fertilised seawater and Ulva growing in effluent water). However, the seawater raceway inlet and outlet samples seem to be randomly distributed, which may partly be due to small sample size.

Between sample (beta) diversity analyses including (a) dendrogram analysis using the Ward linkage method and Bray-Curtis distance measure, where sample sub-clades are also indicated; (b) non-metric multi-dimensional scaling analysis (NMDS) based on Bray-Curtis distance matrices at genus level showing a distinction between water and Ulva samples with ellipses indicating clustering at a 95% confidence level; and (c) a Venn diagram of overlapping bacterial communities across water samples collected from inlets and outlets, as well as bacterial communities associated with Ulva in the (i) non-IMTA system (n = 6) and (ii) IMTA system (n = 15). UR – Ulva raceway; U – Ulva
Non-metric multidimensional scaling (NMDS) at genus level shows three clusters, belonging to the effluent system (UR_Effluent_In, UR_Effluent_Out, and U_Effluent), with some degree of overlap for samples collected from inlets and outlets of the Ulva raceway (UR) and distinct clustering of the Ulva (U) samples (Fig. 4B). It can also be observed that the UR_Seawater_In and UR_Seawater_Out samples cluster closely with the effluent system inlets and outlets. Similar clustering patterns were observed at all other hierarchical levels (data not shown). The NMDS stress of 0.07 implies a good fit, allowing confident inferences. The samples collected from the seawater system did not form clusters with 95% confidence intervals as a result of the small sample size (n = 2 samples per sample type). A shift in the overall structure of the microbiota from the inlets to the outlets of both raceway types, towards a community structure with greater similarity to the Ulva samples, can be observed, with some of the outlet samples grouping with the Ulva samples to some extent. The bacterial communities associated with Ulva from the effluent and seawater systems also cluster together, suggesting that Ulva has a distinct microbial profile that is similar across non-IMTA and IMTA environments. These clustering patterns are also supported by the analysis of similarities (ANOSIM) with an R-value of 0.79 (p < 0.001) and permutational analysis of multivariate dispersions (PERMDISP) with an F-value of 10.13 (p < 0.001) (Fig. 4B), all of which indicate some degree of dissimilarity in bacterial community profile. As observed in the dendrogram analysis, the 21 samples are broadly arranged into two clades, i.e. a water clade, and an Ulva clade (Fig. 4). A further separation could be observed for the inlet and outlet water samples collected from the IMTA (effluent) system.
Lastly, Venn diagrams indicated that 133 genera were shared by all samples collected from non-IMTA systems (Fig. 4C). A further 89 genera were shared by water collected from inlets and outlets of the non-IMTA Ulva raceway, whereas only 8 and 6 genera were common between the samples collected from Ulva and the inlets and outlets, respectively. Similarly, in the effluent Ulva raceways, 125 genera are shared by all cohorts, with 117 genera being shared between water samples from inlets and outlets but only 16 genera being shared between outlet and Ulva samples (Fig. 4C). No genera were shared between the inlet water and Ulva from the IMTA system. The pattern observed in the NMDS plot was also observed here, where the number of genera exclusively associated with the outlet samples (number of genera = 9 and 2, respectively) was lower than those exclusively associated with inlet water samples (number of genera = 12 and 5, respectively) for both the non-IMTA and IMTA systems.
Taxonomic identification and abundance of bacteria associated with Ulva and water samples
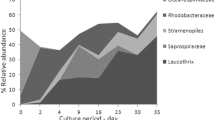
At phylum level, majority of the bacteria identified in this study belonged to Proteobacteria (60%), which was followed by Bacteroidota (25%) and Fusobacteriota (4%). At class level, the three most abundant classes identified were Gammaproteobacteria (50%), Bacteroidia (25%) and Alphaproteobacteria (10%). At family level, most of the bacteria belonged to the families Pseudoalteromonadaceae (16%), Saprospiraceae (12%), Thiotrichaceae (11%), Flavobacteriaceae (10%), Vibrionaceae (7%) and Rhodomonadaceae (5%).
The three most prevalent ASVs at genus level across samples included Pseudoalteromonas (15%), Leucothrix (9%) and Vibrio (7%) (Fig. 5; Supplementary Fig. 2), all of which vary in abundance across the sample types included in this study. For example, there is a notable reduction in abundance of the genera Pseudoalteromonas and Vibrio from inlet to outlet samples, across both non-IMTA and IMTA systems. Other high abundance features at genus level consisted of members of the Saprospiraceae family (5%), as well as the genera Psychromonas (4%), Polaribacter (4%) and Psychrilyobacter (4%). In total, 125 and 140 significant differentially abundant ASVs (FDR-corrected p < 0.05) were observed at genus level. Of these, 99 genus-level ASVs had a higher abundance in water samples (Fig. 6) when compared to the Ulva samples, in both the non-IMTA and IMTA systems, with the top 25 differentially abundant genera being displayed. Interestingly, the majority of the bacterial genera isolated and sequenced in the culture-based technique were in low abundance in the Illumina based assessment. For example, the Celeribacter (U2), Roseobacter (U10), Modestobacter (U14), Agarivorans (T3) and Halomonas (T8) were present in low abundance, whereas the Pseudoalteromonas (T5) and Vibrio (T6, T10, V1 and V2) were present in high abundance.

Relative ASV abundance (percentage abundance) across 21 samples at genus level from Ulva (U) samples and water samples collected flowing into and leaving the Ulva raceway (UR) in the seawater/non-IMTA and effluent water/IMTA systems, respectively. Less abundant ASVs (counts < 2500) were merged and denoted as “Others”

Differentially abundant ASVs at genus level that had a higher abundance in water samples (UR_Seawater_In; UR_Seawater_Out; UR_Effluent_In; UR_Effluent_Out) when compared with Ulva samples (U_Seawater; U_Effluent), where the p-value and false discovery rate (FDR) are indicted. Each boxplot corresponds to one of the top 25 differentially abundant ASVs across the cohorts
Various marine bacteria, including the genera Pseudoalteromonas, Vibrio, Shewanella, Psychromonas and Tenacibaculum, were identified as differentially abundant (Fig. 6; Fig. 7). Interestingly, similar levels of abundance of these genera were observed for water samples collected from the non-IMTA and IMTA systems (Fig. 6), with a slightly higher abundance in these genera in the samples collected from the non-IMTA systems in most instances. Some exceptions to this include Lutibacter, Psychrilyobacter and Marinifilum that have a higher abundance in effluent water samples (Fig. 6). Limited differences were observed between non-IMTA and IMTA systems, but in both systems, there is a general decline in abundance of taxa at genus-level when comparing water collected from inlets and outlets. An overall lower abundance of these bacteria in samples collected from Ulva was observed. For example, a very low abundance of genera Shewanella, Alteromonas, Desulfotalea, Halarcobacter, Formosa, Oleispira, Marinicella, Lutimonas, Wenyingzhuangia and members of the families Cyclobacteriaceae and Saccharospirillaceae were observed on Ulva relative to the water samples (Fig. 6).

Differentially abundant ASVs at genus level that had a higher abundance in Ulva samples (U_Seawater; U_Effluent) when compared to water samples (UR_Seawater_In; UR_Seawater_Out; UR_Effluent_In; UR_Effluent_Out), where the p-value and false discovery rate (FDR) are indicted. Each boxplot corresponds to one of the top 25 differentially abundant ASVs across the cohorts
In contrast, some bacterial communities had a higher abundance (p < 0.05) on Ulva when compared to water samples (Fig. 7). This was particularly evident for genera such as Portibacter, Rubidimonas, Octadecabacter, Hellea, Leucothrix, Chitinophagales, Granulosicoccus, Algibacter and Lewinella. Along with this, a higher abundance of these bacteria associated with Ulva was observed in outlet samples in both the seawater and effluent systems (Fig. 7), further supporting the modulating effect Ulva may have on the bacterial communities in IMTA systems.
Putative functional roles of bacteria in the IMTA system
The functional potential of bacterial communities associated with Ulva showed that the microbiome is capable of being involved in various metabolic processes, such as the metabolism of amino acids, carbohydrates, lipids and other secondary metabolites (Fig. 8). An upregulation potential for xenobiotic (chemical or substance that is not naturally produced by the organism) biodegradation and metabolism could be observed for the bacteria associated with Ulva samples (Fig. 8). However, these results should be treated with caution given that it is based on 16S data and requires further validation.

The Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) metabolic pathways based on total KO hits averaged across inlets (UR_Seawater_In; UR_Effluent_In), outlets (UR_Seawater_Out; UR_Effluent_Out) and associated with Ulva (U_Seawater; U_Effluent) in the IMTA and non-IMTA systems, respectively, where the standard error for each mean is indicated. Letters indicate statistically significant differences (p < 0.05; Kruskal-Wallis test with Dunn’s post-hoc test)
Discussion
Enumeration of culturable bacteria in seawater and on Ulva
Microbial communities are often host-specific (Taylor et al. 2004; 2005; Reis et al. 2009), and numerous studies have demonstrated host-specific differences between the microorganisms present on macroalgae and in the surrounding seawater (Staufenberger et al. 2008; Lachnit et al. 2009; Califano et al. 2020), between alga of different species and even on a single species from different locations (Bolinches et al. 1988; Tujula et al. 2010; Singh and Reddy 2014; Califano et al. 2020). In the current study, in both fertilised seawater tanks (non-IMTA systems) and abalone effluent raceways (IMTA systems), the macroalga Ulva had a strong inhibitory effect on Vibrio within the systems, as indicated by the significant decrease in bacteria growing on TCBS agar (a Vibrio selective media) between the inlets and the outlets of both systems (Fig. 2). The total number of potential culturable Vibrio from Ulva samples was higher in the IMTA systems than in the non-IMTA system, and reflect values typically found in seawater and on seaweeds in the natural environment (values range from 102 – 104 CFU g-1 seaweed; Mahmud et al. 2008). Moreover, the inhibitory effect appears to be nutrient dependent, with a substantial decrease in Vibrio numbers observed in the effluent water systems. These findings support results of Lu et al. (2008) who demonstrated a decrease in a strain of V. anguillarum in the presence of U. clathrata, which was enhanced following the addition of nitrogen and phosphorus. However, this inhibitory effect can be driven by the microbial communities or by Ulva itself (Lemos et al. 1985; Egan et al. 2000; Lu et al. 2008; Penesyan et al. 2009).
The high number of bacteria that grew on the minimal media containing ulvan, the main carbohydrate component of Ulva (Ray and Lahaye 1995), suggests that there are numerous bacteria capable of growing on Ulva within the water column. Several bacteria colonising algal surfaces are known to benefit from the organic components of algae, such as polysaccharides including ulvan, which the bacteria utilise for growth and biofilm formation (Erasmus et al. 1997; Steinberg et al. 2002; Lachnit et al. 2009). The high abundance of ulvan-specific bacteria in the effluent inlet water may be explained by the fact that abalone naturally feed on various wild seaweeds (Simpson and Cook 1998; Naidoo et al. 2006), and abalone in the present study were fed Ulva as well as kelp (Ecklonia maxima). Therefore, a high abundance of bacteria capable of degrading ulvan is expected in this aquaculture environment. Additionally, the incoming water is pumped from the adjacent coastline where there are kelp beds that could harbour bacterial communities capable of degrading algae.
Identification of culturable bacteria
Most of the marine bacteria identified from the culture plates in this study are frequently isolated from the marine environment, including Pseudomonas, Alteromonas and Vibrio (Rengpipat et al. 2003; Romanenko et al. 2004; Infante-Villamil et al. 2021). A BLAST search of the bacteria isolated on the ulvan substrate, isolates U2 and U10, showed high sequence similarity to Celeribacter and Roseobacter, respectively (Table 1). Both Celeribacter and Roseobacter are members of the family Rhodobacteraceae, which is a major chemo-organotrophic phylogenetic assemblage found within marine surface waters (Giovannoni 2000; Baek et al. 2014) and can cause host-specific adaptations that may be commensal or parasitic (Buchan et al. 2005; Sison-Mangus et al. 2014).
Bacterial isolate T3, isolated on TSA from the effluent system inlet water, showed high sequence similarity to the genus Agarivorans, which displays agarolytic activity. Agarolytic bacteria are common inhabitants of marine environments, particularly where there is a high abundance of macroalgae rich in this polysaccharide, such as the globally significant kelp forest ecosystem in South Africa where the farm is located and extracts its seawater from for the abalone raceways (Erasmus et al. 1997; Bolton et al. 2016). Kelp is also used as a feed for the abalone at I&J Cape Abalone Farm. The genus was first isolated from a healthy marine mollusc (Omphalius pfeifferi) (Kurahashi and Yokota 2004), and has since been isolated from several marine environments including the marine water column (Park et al. 2014), tidal flats (Kim et al. 2016a; 2016b) and macroalgae (Du et al. 2011). Isolate T8, also obtained from the effluent system inlet water, showed high sequence similarity to the genus Halomonas (Arahal et al. 2002). Certain species of Halomonas are known to display pathogenic potential in molluscs (Rojas et al. 2009), whereas others have been used as probiotics in aquaculture species (Gao et al. 2019) or to improve water quality of intensive recirculating aquaculture production systems (Sangnoi et al. 2017; Hastuti et al. 2020).
Isolate T5 shared a high level of sequence similarity with the genus Pseudoalteromonas, which belongs to the family Pseudomonadaceae. Pseudoalteromonas associated with Ulva australis has been shown to inhibit the attachment of fouling organisms and act as a Vibrio control agent (Rao et al. 2007; Aranda et al. 2012). This genus was also recently found in relatively high abundance on U. fenestrata and U. linza in a flow-through land-based laboratory in Sweden (van der Loos et al. 2024). Moreover, several members including P. auranitia (Gauthier and Breittmayer 1979), P. luteoviolacea (Gauthier and Flatau 1976) and P. rubra (Gauthier 1976) release high molecular weight antibiotics. Unlike the Modestobacter and Vibrio genera, which can be isolated from both terrestrial and marine habitats, the genus Pseudoalteromonas is restricted to marine waters and has been isolated from various marine environments, including from sea urchins and macroalgae (Holmström and Kjelleberg 1999; Rao et al. 2007; Brink et al. 2019). Nonetheless, some members of this genus are known to induce disease in macroalgae, including the kelp Saccharina japonica (as Laminaria japonica, Sawabe et al. 1998) and the red macroalga Gracilaria gracilis (Schroeder et al. 2003), but this is mostly when environmental conditions are not optimal and the macroalgae are under stress.
The bacterial isolates T6, T10, V1 and V2 shared high sequence similarity with members of the genus Vibrio. The genus Vibrio falls within the family Vibrionaceae which contains several marine pathogens that are known to infect a wide range of marine organisms, including abalone (Nicolas et al. 2002), marine bivalves (Paillard et al. 2004), corals (Ben-Haim and Rosenberg 2002), macroalgae (Largo et al. 1995), finfish (Kraxberger-Beatty et al. 1990), prawns (Jiravanichpaisal et al. 1994) and captive bred seahorses (Balcázar et al. 2010). For example, mass mortalities have been previously attributed to V. harveyi, which has induced white foot lesions on abalone in Japan (Nishimori et al. 1998) and France (Nicolas et al. 2002). Mass mortality in larvae of the abalone H. rufescens has also been observed as a result of V. alginolyticus at concentrations above 103 cells mL-1 (Anguiano-Beltrán et al. 1998). Nonetheless, the isolated Vibrio spp. in this study may be pathogenic, probiotic, opportunistic, or represent normal bacterial presence, as low concentrations of diverse Vibrio species have been isolated from both healthy and diseased marine organisms (Nakanishi et al. 1996; Macey and Coyne 2006; Marshall et al. 2006; Brink et al. 2019). For instance, V. gallaecicus, V. xuii, V. ichthyoenteri and V. parahaemolyticus have been isolated from marine aquaculture environments, of which only the latter two are often associated with disease (Zorrilla et al. 2003; Gauger et al. 2006; Martins et al. 2013). In the current study the isolated bacteria identified as belonging to the genus Vibrio were all isolated from the incoming water samples (abalone effluent), and it was observed that Ulva reduces their numbers within the system (Fig. 2). This is not surprising as several species of Ulva have been known to display anti-Vibrio activity (Lu et al. 2008) or harbour bacteria, as mentioned above, that can produce compounds to restrict their growth (Ismail et al. 2018).
Bacterial community profiling by 16S rDNA sequencing
Bacterial diversity in an IMTA and non-IMTA system
The bacterial diversity remained stable within the samples collected from effluent water, fertilised seawater and Ulva as no significant differences (p > 0.05) were observed for both the Shannon and the Simpson diversity indexes, with F-statistics of 1.13 and 1.52, respectively (Fig. 3). Interestingly, the richness estimator Chao1 was significantly higher for the water samples across both systems, when compared to Ulva samples (ANOVA, F = 7.11, p = 0.001), indicating a high degree of unique ASVs in the water column. This could be explained by the interchangeable nature of the water with the environment, whereby fresh seawater is constantly pumped in from the ocean and through the abalone raceway systems (in the case of the IMTA), after which it is gravity fed into the Ulva raceways. Alternatively, the microbiome modulating effect of Ulva and its associated microbiome could be reducing the presence of specific bacteria on the Ulva sampled in this study.
Ulva and water bacterial community structure
Several molecular studies have found that bacterial communities may differ between a host and its environment, and that limited core communities (~15%) specific to the host at lower taxonomic ranks are present (Burke et al. 2011; Bengtsson et al. 2010; 2012). Cluster analyses comparing the microbial communities of seawater and U. australis and U. rigida, respectively (Burke et al. 2011; Califano et al. 2020) further support the clear separation observed between the Ulva and the seawater observed in the current study. The separate clustering of inlet and outlet water samples from the non-IMTA vs IMTA systems observed in the current study is expected as anthropogenic perturbations, such as the nutrient increases associated with high density aquaculture and recirculating water within aquaculture systems, are known to influence bacterial communities (Schneider et al. 2007). The three distinct microbial community clusters observed for the inlet, outlet and Ulva samples within the effluent water systems further highlight how different systems and species combinations can impact microbial diversity, and in this case indicates that Ulva and/or its associated microbiome are capable of altering the microbial community structure within the water column.
The distinct differences in the microbiome observed between the Ulva and water samples within a system and between the two systems suggests that Ulva can improve biosecurity of aquaculture systems, as it has its own unique microbiota that can aid in modulating the microbiota of the water column, particularly in nutrient rich water (as seen for the abalone effluent in this study). Given the role of microbes in aquatic animal health and the functionality/welfare of ecosystems, understanding the complexities underlying the differences in microbial community structure between Ulva and the surrounding seawater and between IMTA and non-IMTA systems is essential.
Water column bacteria in an IMTA vs non-IMTA system
Differences within bacterial communities are known to occur from variations within system designs, such as methods of filtration, physical parameters of the system as well as cleaning and disinfection techniques, which are known to impact free-living bacterial communities (Schneider et al. 2006; Wietz et al. 2009; Schreier et al. 2010).
In the current study the bacterial communities associated with the seawater samples from both systems (Fig. 5) showed similarities to seawater samples collected from two different rock pools containing Ulva at Bare Island, La Perouse (Burke et al. 2011). Bacteria belonging to Gammaproteobacteria, Flavobacteriaceae, Alphaproteobacteria and Rhodobacteriaceae were identified in the current study, with communities belonging to the genera Pseudoalteromonas, Vibrio, Shewanella, along with 99 other genera, exhibited higher abundance in water samples, when compared with Ulva (Fig. 6). Water samples from both the non-IMTA and IMTA systems in this study had a high abundance of ASVs belonging to the heterotrophic bacterial genus Pseudoalteromonas, which was not reported by Burke et al. (2011) but has been found in association with wild-collected Ulva from Portugal (Grueneberg et al. 2016). This genus has previously been associated with healthy recirculating aquaculture systems (Schreier et al. 2010). Previous studies have suggested that Pseudoalteromonas acts as an inducer for larval settlement for various marine invertebrates (Hadfield 2011). Therefore, bacterial communities associated with the water may not only influence the Ulva growing in the raceways but could also positively impact abalone larval settlement.
The seawater entering an abalone farm could be a vector for both opportunistic bacterial pathogens, as well as naturally occurring bacteria that may be neutral or probiotic. Overall, the bacteria associated with the non-IMTA seawater samples belonged predominantly to genera common in the marine environment, such as Pseudoalteromonas, Vibrio, Sulfitobacter, Tenacibaculum and Psychrobium (Fig. 6). This was also observed for the inlet effluent water samples of the IMTA systems that contained the bacteria Psychromonas, Psychrilyobacter, Colwellia and Psychrosphaera. The presence of the genus Vibrio in both the IMTA and non-IMTA systems could be a cause for concern, as some members of this genus are known potential pathogens (Rodrick 1991).
Vibrio are ubiquitous in the marine environment and the potential risk they may pose for disease in the integrated systems appears to be mitigated by the modulatory effect of Ulva on the microbiome, as they have a statistically significantly lower (FDR-corrected p < 0.05) abundance in samples collected from Ulva (Fig. 6) and the abundance of Vibrio spp. decreases in effluent water following bioremediation with Ulva. Previous studies have found that certain probiotic bacteria, such as Lactobacillus and Phaeobacter, have the ability to reduce the abundance of Vibrio in brine shrimp (Artemia) and turbot (Psette maxima and Scophthalmus maximus) aquaculture systems (Prol-Garcia and Pintado 2013; Ofelio et al. 2021; Pintado et al. 2023), where Phaeobacter has been used to colonise U. ohnoi in IMTA systems to improve biofiltration capabilities. However, neither of these taxa were found in the dataset generated in the current study. Given the reduction in Vibrio following bioremediation with Ulva, this study provides additional evidence for the microbial gardening capabilities of Ulva as described in an IMTA-based algal production system with seabream (Califano et al. 2020), without the need to supplement with probiotic bacteria.
This idea is further supported by the differential abundance analysis (Fig. 6), where it could be observed that the majority of bacteria that had a significantly (p < 0.05) higher abundance in the water samples, had a very low abundance on Ulva samples. In contrast, the bacteria that had a higher abundance on Ulva samples (Fig. 7) were still present in the water column, suggesting that bacteria in the water column do not influence those of Ulva as much as Ulva modulates the microbiota of the water column. It has also been suggested that moderate levels of fertilization, either chemical or via the addition of agricultural waste, can act as a feed for microbial communities, promoting a balanced ecosystem in nutrient deprived recirculating aquaculture systems, and therefore, preventing shifts from neutral bacterial consortia to potentially pathogenic communities (Bentzon-Tilia et al. 2016). Of the differentially abundant bacteria in the IMTA and non-IMTA systems, many genera have previously been detected in marine environments and do not appear to be pathogenic. For example, Lutimonas, which was in higher abundance in the water samples of both systems (Fig. 6), are members of the family Flavobacteriaceae and have been isolated from the abalone Haliotis tuberculata, polychaetes and tidal flat sediments. They are believed to exclusively inhabit marine invertebrates and seawater and have not been associated with algae (Yang et al. 2007; Kim et al. 2011; 2016a; 2016b; Gobet et al. 2018). The genus Psychrilyobacter, which also had a higher abundance in seawater, are common marine bacteria that have been isolated from seawater, sediment and crustacea (Romanenko et al. 2004; 2009) and have been studied for their bioremediatory potential (Abd-Elnaby et al. 2016) and roles in abalone digestive tracts (Gobet et al. 2018).
At the studied site, the abalone effluent water originates from the nearby shallow coastal environment, passes through the abalone rearing tanks and then flows into the Ulva raceways. This means that the bacterial communities present in the inlet water of the abalone effluent Ulva raceways are derived from the incoming seawater but are also influenced by the abalone and their associated microbiota (Schreier et al. 2010), as well as by the feed used in the system. Moreover, the presence of other organisms in abalone rearing tanks, such as polychaetes (Ruck and Cook 1998) may further shift the microbial community structure. The primary nutrients entering the abalone effluent raceways are in the form of particulate and dissolved organic matter that originates from digested feed and faeces/dissolved excreta from the abalone, as well as any uneaten feed that may remain in the wastewater. Bacteria responsible for the degradation of organic matter are known as heterotrophs and in RAS, heterotrophs are the dominant water column bacteria and aid in maintaining water quality (Itoi et al. 2006; Attramadal et al. 2012). Heterotrophic bacterial communities in aquaculture systems have been found to be dominated by Alphaproteobacteria and Gammaproteobacteria (Sugita et al. 2005; Wietz et al. 2009), which were also detected in the current study as dominant phyla. Opportunistic bacteria, which could include heterotrophs, are capable of proliferating rapidly (r-strategists) by using available resources, and are gradually outcompeted by the slower growing specialists, such as the nitrifying bacteria (k-strategists) (Salvesen et al. 1999; Defoirdt 2016).
Common nitrifying bacteria in aquaculture systems include species of the genera Nitrosomonas, Nitrosococcus, Nitrobacter, Nitrosolobus, Nitrosovibrio and Nitrococcus (Belser 1979; Teske et al. 1994). The low abundance of nitrifying bacteria in the abalone wastewater raceways in the current study is noteworthy as nitrifying bacteria have been frequently associated with effluent water (Harms et al. 2003; Lydmark et al. 2007; Paungfoo et al. 2007). The particulate organic matter released by the abalone in the effluent water could be enhancing the growth of heterotrophic bacteria which, in turn, outcompete the slower growing nitrifying bacteria. It is however known that, due to their competitive nature, excessive levels of heterotrophic bacteria in integrated aquaculture systems may become problematic if nitrification rates are restricted when oxygen levels are low (Michaud et al. 2006). However, on the studied farm, the bubbler systems in the seawater non-IMTA system, the paddlewheel systems used in the abalone effluent IMTA raceways, and the aeration in the abalone tanks, aid in oxygenating the water column, in addition to the oxygen produced by Ulva from photosynthesis during the day. Macroalgae also release superoxide ions and hydrogen peroxides as a defence mechanism against bacteria, and the production of these reactive oxygen species (ROS) could reduce the number of associated bacteria on the surface of the algae (Weinberger 2007; Egan et al. 2013; Zhao et al. 2021). Alternatively, the low abundance of nitrifying bacteria may also be attributed to the systems design (i.e., land-based pump-ashore systems with high flow/water replacement rates, typically around 0.25 to 0.5 tanks volumes per hour) and the biofiltration capacity of Ulva, which has previously been reported to outperform traditional bacterial biofilms (Bolton et al. 2016). Ulva has in fact been shown to consistently remove more that 70% of available nitrogen, mostly ammonia (the main excretory product of abalone), when grown in integrated system, possibly outcompeting bacteria for this nutrient source.
Benefits of the bacterial communities associated with Ulva in an IMTA and non-IMTA system
The differences in abundance of several genera on the Ulva in both the IMTA and non-IMTA system in the current study concur with studies showing that the microbiome of Ulva is distinct to that of the surrounding water (Pintado Valverde et al. 2018; Califano et al. 2020). This is evident in the current study considering that only 25 ASVs were in higher abundance in Ulva samples (Fig. 7), as opposed to the 99 identified as having a higher abundance in water samples (Fig. 6). In general, cultivating Ulva changes the diversity and abundance of ASVs in laboratory and aquaculture settings when compared to newly obtained specimens (Ghaderiardakani et al. 2022), consequently harbouring beneficial bacteria (Califano et al. 2020).
Both Ulva libraries (IMTA and non-IMTA) were characterised by ASVs from common marine environmental bacterial genera, such as Leucothrix, Polaribacter, Hellea, Glaciecola, Granulosicoccus, Rubidimonas, as well as members of the families Saprospiraceae and Rhodobacteraceae. Of these, Leucothrix and Granulosicoccus have been found multiple times worldwide in other land-based Ulva systems in Belgium, Portugal and Israel (Califano et al. 2020; Nguyen et al. 2023; van der Loos et al. 2024). Granulosicoccus is a gammaproteobacterium that comprises two species that have been documented exclusively in marine environments, where they have been found in surface seawater, brown algae and on leaves of seagrass (Kurilenko et al. 2010; Baek et al. 2014; Park et al. 2014), as well as with other Ulva species (van der Loos et al. 2024). The presence of Rubidimonas is interesting given that previous studies have found this genus in high abundance in association with Ulva towards the end of an experiment (after 32 weeks) in a land-based laboratory facility, but almost absent at the start of the experiment (van der Loos et al. 2024). This suggests the Ulva sampled in the current study displayed a mature microbial profile. In the current study, a higher abundance of the genera Portibacter, Sulfitobacter, Rubidimonas, Octadecabacter, Hellea and Granulosicoccus were observed in Ulva samples (Fig. 7), across both non-IMTA and IMTA systems. Interestingly, Portibacter are members of the Saprospiraceae family and have also been isolated from RAS biofilters (Yoon et al. 2012; Li et al. 2016; Nguyen et al. 2023), and Sulfitobacter has been found in high abundance in association with Ulva samples collected from the previously mentioned land-based laboratory facility, where it was present throughout the 32 week experiment (van der Loos et al. 2024) and has been identified in association with Ulva in other aquaculture environments (Ghaderiardakani et al. 2019; van der Loos et al. 2021), where they are likely promoting Ulva growth (Amin et al. 2015; Califano et al. 2020).
Genera belonging to the family Arenciellaceae and class Flavobacteriales, as well as the genera Reinchenbachiella and Maribacter, exhibited an increased abundance in the effluent water systems from the inlet to outlet samples and were prevalent in high abundance on the Ulva (Fig. 7). Notably, Maribacter strains have previously been identified as important inducers for Ulva morphogenesis (Weiss et al. 2017), releasing high amounts of the morphogen thallusin in algal culture (Ulrich et al. 2022), particularly when present in combination with Roseovarius strains (Spoerner et al. 2012). This genus could also play a role in Ulva morphogenesis in the studied aquaculture environment, as Roseovarius was also identified, albeit in relatively low abundance (denoted as “Others” in Fig. 5). In the current study, the addition of inorganic phosphorus and nitrogen to the non-IMTA system and the dissolved organic matter that the IMTA system receives likely positively impacts bacteria associated with Ulva, as fertilisation is known to promote beneficial interactions (Lara-Anguiano et al. 2013; Cooper and Smith 2015). Moreover, inorganic nitrogen and phosphorus prevents bleaching and fragmentation of the Ulva (Robertson-Andersson 2003), thus improving host health, which in turn would render it less susceptible to pathogenic microbiota.
A comparison across the dominant bacterial taxa present on the Ulva from the respective systems showed that the same bacterial taxa have a similar abundance in both systems, further highlighting that using effluent water as a means to fertilise Ulva does not introduce, nor increase the abundance of, harmful bacteria. In the current study, the bacteria present on the effluent-grown Ulva also shared similarities with those identified in RAS biofilter studies, including the genera Planctomycetes, Roseobacter, Pseudoalteromonas, Desulfovibrio and Vibrio (Schreier et al. 2010). Bacteria associated with Ulva, when utilised as a feed on an abalone farm, may also positively impact the microbiome of abalone. Ten Doeschate and Coyne (2008) noted that H. midae fed kelp cakes supplemented with Pseudoalteromonas sp. strain C4, isolated previously by Erasmus et al. (1997), grew faster than abalone fed a diet of kelp without this probiotic. The colonisation of the intestinal tract with probiotic bacteria has numerous positive effects on host function and regulation (van Baarlen et al. 2013; Bhatnagar and Lamba 2015). Moreover, Macey and Coyne (2005) indicated that abalone fed probiotic feed supplemented with Vibrio midae SY9 increased protease activity in the intestine of the abalone and improved growth, general health and disease resistance of cultured abalone. Effluent grown Ulva could be beneficial in aquaculture systems, as the Ulva may be colonised by and inoculated with probiotic bacteria from the faeces, which could in turn, re-inoculate the gut of the abalone in a microbial loop, limiting the need for continual probiotic supplementation. Probiotic bacteria within the water column have several benefits, including nutrient and niche competition (Callaway et al. 2008), aid in enzymatic digestive processes and improved growth rates (Erasmus et al. 1997; Macey and Coyne 2005), enhance immune responses and have antipathogenic properties (Nogami and Maeda 1992; Nakayama et al. 2009).
Putative functional capabilities of bacterial communities in water and Ulva samples
Microbiota are known to strongly influence carbon, nitrogen and phosphorus cycling in aquatic environments due to metabolic interactions with particulate- (POM) and dissolved (DOM) organic matter (Pomeroy et al. 2007). The proteins in abalone feed and faeces are commonly decomposed by heterotrophic bacteria to ammonia, after which nitrifying bacteria convert the ammonia to nitrate (Bender et al. 2004; Itoi et al. 2006). The latter also applies to the dissolved ammonia directly excreted by then abalone. The co-existence of bacterial genera, with different functional roles, is often desirable, as can be observed in the symbiotic relationship between heterotrophic bacteria and nitrifiers, allowing for simultaneous carbon and ammonia oxidation (Kindaichi et al. 2004; Dong and Sun 2007). Similar to a study conducted by Schreier et al. (2010), bacteria with the potential for nitrification, denitrification and nitrate producers were also identified in the current study, including Pseudomonas, and various members belonging to the classes Bacteroidetes, Proteobacteria and Firmicutes. The differential presence of the bacterial genus Desulfotalea in the current study, which are sulfate-reducing bacteria, also indicates that there is potential for organic substrates to be oxidised to produce acetate and CO2 (Widdel and Bak 1992; Inagaki et al. 2002).
In the current study the inlet seawater is more susceptible to change as a result of the variation introduced by the water being pumped on land from the adjacent ocean, as this seawater interacts with various other biotic factors, such as the nearby kelp forest for example. Putative functional capability profiles showed that the bacteria associated with Ulva across both systems had greater metabolic potential across majority of the KEGG metabolic pathways identified in this study (Fig. 8). Though limited inferences can be made given that this study was based on 16S data alone, an upregulated potential capability for the metabolism of lipids, carbohydrates, energy, terpenes, amino acids, cofactors and vitamins, as well as the biosynthesis of secondary metabolites was observed for Ulva-associated taxa (Fig. 8), which could influence the health of the Ulva by improving nutrient availability. Additionally, these bacteria showed potential capabilities for xenobiotic degradation and metabolism, whereby bacteria make use of substances that would otherwise be toxic to them by breaking them down and using them as carbon, phosphorus, sulphur or nitrogen sources (Datta et al. 2020). This potential of the bacterial communities to degrade potentially toxic compounds and their metabolites could aid in the biofiltration and bioremediation action of the Ulva in these IMTA systems.
It is possible that the use of Ulva as a biofilter maintains a microbial balance in the water column of aquaculture systems through microbial antibiotic biosynthesis, however, a shotgun metagenomics approach would need to be implemented to assess this further and confirm the functional roles of the microbial communities in these systems. The bacteria inhabiting the Ulva could protect the Ulva holobiont, as well as the abalone by inhibiting bacterial species that could be pathogenic, such as Vibrio, without the need to administer antibiotics. This is beneficial considering that administering antibiotics in high dosages could negatively affect beneficial microflora and has the potential to result in the development of antibiotic resistance and the potential spread of these resistance genes to human and animal pathogens (Handlinger et al. 2006).
Implications for farm management
A common concern with biofilters in integrated aquaculture systems, such as the Ulva used in the current study, is their potential to harbour bacterial pathogens that may either infect the co-produced organisms within the system, or consumers after processing. In the current study, evidence was provided to support that the presence of Ulva in the raceways does not act as a sink for harmful bacteria and instead is capable of positively impacting the microbiome through antimicrobial action and therefore, would not act as a biosecurity risk when used as a means of bioremediation in IMTA systems or when used as an abalone feed. For example, members of the genus Vibrio, displayed a reduction in abundance from the inlets to the outlets of an Ulva raceway, following bioremediation by Ulva. It was further demonstrated that the modulatory effect of Ulva and/or its associated microbiota is increased with increasing nutrient levels, as the aforementioned effect was more pronounced in the IMTA systems.
Seawater is thought to have a lower proportion of opportunistic microorganisms when sufficient time is provided for it to mature (k-selection) and reach a stable microbial community (Blancheton et al. 2013). It is likely that IMTA systems are capable of promoting similar microbial maturation benefits, whereby bacterial communities may establish a stable carrying capacity within the system and be composed of dominant organisms that remain balanced under the given environmental conditions (Attramadal et al. 2012).
Similarly, Ulva that has remained in a system for sufficient time to enable microbial maturation may also serve as a stable source of host-specific microbes that positively interact with the water column, the Ulva growing in a system, as well as the abalone co-produced within a system when provided as a feed. Ulva also improves farm productivity through its interaction with beneficial microorganisms (Wichard 2015; 2023; Alsufyani et al. 2020) and the continual vegetative growth of Ulva in effluent water raceways will facilitate the microbial maturation process. If this is the case, microbially matured Ulva should exhibit less of a biosecurity concern because as a stable carrying capacity on the host (Ulva) is reached, there will be less niche availability for colonisation and growth of opportunistic pathogens. Therefore, there is potential for microbial manipulation in IMTA systems by incorporating Ulva, but an improved understanding of these mechanisms in response to different environments is required (Ghaderiardakani et al. 2020; van der Loos et al. 2024). Beneficial microorganisms have the potential to improve future farming practices (Li et al. 2023) and can support growth even under environmental stress (Hmani et al. 2023).
The ability of Ulva, as well as its associated microbiota, to act as a natural disinfectant and antifouling agent (Egan, et al. 2000; Ravikumar, et al. 2016) further reduces the risk for proliferation of opportunistic bacteria and the spread of disease should effluent grown Ulva be used as a feed. Nonetheless, the extent of Ulva’s ability to act as a bioremediation unit under varying physiochemical conditions, including seasonal assessments and microbiome characterisation across increasing recirculation rates in abalone-Ulva IMTA systems, requires further investigation, particularly given that the bacteria present on biological filters, such as Ulva, rely on interactions with the environment as a function of nutrient input.
Data availability
All next-generation sequencing data are publicly available on the NCBI SRA database (accession nr: PRJNA1075670). All colony forming unit (CFU) data are available from the corresponding author upon request.
Code availability
All code used in this manuscript is described in the manuscript. Any additional information is available from the corresponding author upon request.
References
Abd-Elnaby HM, Abou-Elela GM, Ghozlan HA, Hussein H, Sabry SA (2016) Characterization and bioremediation potential of marine Psychrobacter species. Egypt J Aquat Res 42:193–203
Alsufyani T, Califano G, Deicke M, Grueneberg J, Weiss A, Engelen AH et al (2020) Macroalgal-bacterial interactions: Identification and role of thallus in in morphogenesis of the seaweed Ulva (Chlorophyta). J Exp Bot 71:3340–3349
Altschul SF, Gish W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Amin SA, Hmelo LR, van Tol HM, Durham BP, Carlson LT, Heal KR, Morales RL, Berthiaume CT, Parker MS, Djunaedi B, Ingalls AE, Parsek MR, Moran MA, Armbrust EV (2015) Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522:98–101
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46
Anderson MJ, Ellingsen KE, Mcardle BH (2006) Multivariate dispersion as a measure of beta diversity. Ecol Lett 9:683–693
Anguiano-Beltrán C, Searcy-Bernal R, Leonardo Lizárraga-Partida M (1998) Pathogenic effects of Vibrio alginolyticus on larvae and postlarvae of the red abalone Haliotis rufescens. Dis Aquat Organ 33:119–122
Aníbal J, Madeira HT, Carvalho LF, Esteves E, Veiga-Pires C, Rocha C (2014) Macroalgae mitigation potential for fish aquaculture effluents: an approach coupling nitrogen uptake and metabolic pathways using Ulva rigida and Enteromorpha clathrata. Environ Sci Pollut Res Int 21:13324–13334
Arahal DR, Castillo AM, Ludwig W, Schleifer KH, Ventosa A (2002) Proposal of Cobetia marina gen. nov., comb. nov., within the family Halomonadaceae, to include the species Halomonas marina. Syst Appl Microbiol 25:207–211
Aranda CP, Valenzuela C, Barrientos J, Paredes J, Leal P, Maldonado M, Godoy F, Osorio CG (2012) Bacteriostatic anti-Vibrio parahaemolyticus activity of Pseudoalteromonas sp. strains. World J Microbiol Biotechnol 28:2365–2374
Armstrong E, Yan L, Boyd KG, Wright PC, Burgess JG (2001) The symbiotic role of marine microbes on living surfaces. Hydrobiologia 461:37–40
Aßhauer KP, Wemhauer B, Daniel R, Meinicke P (2015) Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31:2882–2884
Attramadal KJK, Salvesen I, Xue R, Øie G, Størseth TR, Vadstein O, Olsen Y (2012) Recirculation as a possible microbial control strategy in the production of marine larvae. Aquac Eng 46:27–39
Bachoo T, Bolton JJ, Macey BM, Kandjengo L, Reddy MM (2023) Resolving the identity of commercially cultivated Ulva (Ulvaceae, Chlorophyta) in integrated seaweed-abalone aquaculture farms in South Africa. J Phycol 59:1272–1283
Baek K, Choi A, Kang I, Im M, Cho JC (2014) Granulosicoccus marinus sp. nov., isolated from Antarctic seawater, and emended description of the genus Granulosicoccus. Int J Syst Evol Microbiol 64:4103–4108
Balcázar JL, Gallo-Bueno A, Planas M, Pintado J (2010) Isolation of Vibrio alginolyticus and Vibrio splendidus from captive-bred seahorses with disease symptoms. Antonie Van Leeuwenhoek 97:207–210
Bansemer MS, Qin JG, Harris JO, Duong DN, Hai T, Howarth GS, Stone D (2016) Growth and feed utilisation of greenlip abalone (Haliotis laevigata) fed nutrient enriched macroalgae. Aquaculture 452:62–68
Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C (2014) Venn: an interactive Venn diagram viewer. BMC Bioinf 15:293
Belser L (1979) Population ecology of nitrifying bacteria. Ann Rev Microbiol 33:309–333
Bender J, Lee R, Sheppard M, Brinkley K, Phillips P, Yeboah Y, Wah RC (2004) A waste effluent treatment system based on microbial mats for black sea bass Centropristis striata recycled-water mariculture. Aquac Eng 31:73–82
Bengtsson MM, Sjøtun K, Øvreås L (2010) Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea. Aquat Microb Ecol 60:71083
Bengtsson MM, Sjøtun K, Lanzén A, Øvreås L (2012) Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J 6:2188–2198
Ben-Haim Y, Rosenberg E (2002) A novel Vibrio sp. pathogen of the coral Pocillopora damicornis. Mar Biol 141:47–55
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Bentzon-Tilia M, Sonnenschein EC, Gram L (2016) Monitoring and managing microbes in aquaculture - towards a sustainable industry. Microb Biotechnol 9:576–584
Bhatnagar A, Lamba R (2015) Antimicrobial ability and growth promoting effects of feed supplemented with probiotic bacterium isolated from gut microflora of Cirrhinus mrigala. J Integr Agric 14:583–592
Blancheton JP, Attramadal KJK, Michaud L, d’Orbcastel ER, Vadstein O (2013) Insight into bacterial population in aquaculture systems and its implication. Aquac Eng 53:30–39
Bolinches J, Lemos ML, Barja JL (1988) Population dynamics of heterotrophic bacterial communities associated with Fucus vesiculosus and Ulva rigida in an estuary. Microb Ecol 15:345–357
Bolton JJ, Robertson-Andersson DV, Shuuluka D, Kandjengo L (2009) Growing Ulva (Chlorophyta) in integrated systems as a commercial crop for abalone feed in South Africa: a SWOT analysis. J Appl Phycol 21:575–583
Bolton JJ, Cyrus MD, Brand MJ, Joubert M, Macey BM (2016) Why grow Ulva? Its potential role in the future of aquaculture. Perspect Phycol 3:113–120
Brink M, Rhode C, Macey BM, Christison KW, Roodt-Wilding R (2019) Metagenomic assessment of body surface bacterial communities of the sea urchin, Tripneustes gratilla. Mar Genom 47:100675
Brink-Hull M, Cyrus MD, Macey BM, Rhode C, Hull KL, Roodt-Wilding R (2022) Dietary effects on the reproductive performance of the sea urchin Tripneustes gratilla l: Implications for broodstock conditioning. Aquaculture 552:738035
Bryers J, Characklis W (1982) Processes governing primary biofilm formation. Biotechnol Bioeng 24:2451–2476
Buchan A, González J, Moran M (2005) Overview of the marine Roseobacter lineage. Appl Environ Microbiol 71:5665–5677
Buchholz C, Krause G, Buck B (2012) Seaweed and man. In: Wiencke C, Bischof K (eds) Seaweed Biology. Springer, Berlin, pp 417–493
Burke C, Thomas T, Lewis M, Steinberg P, Kjelleberg S (2011) Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J 5:590–600
Califano S, Kwantes M, Abreu MH, Costa R, Wichard T (2020) Cultivating the macroalgal holobiont: effects of integrated multi-trophic aquaculture on the microbiome of Ulva rigida (Chlorophyta). Front Mar Sci 7:52
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Meth 13:581–583
Callahan BJ, McMurdie PJ, Holmes SP (2017) Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J 11:2639–2643
Callaway TR, Edrington TS, Anderson RC, Harvey RB, Genovese KJ, Kennedy CN, Venn DW, Nisbet DJ (2008) Probiotics, prebiotics and competitive exclusion for prophylaxis against bacterial disease. Anim Health Res Rev 9:217–225
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7:335–336
Chao A (1984) Nonparametric estimation of the number of classes in a population. Scan J Stat 11:265–270
Chesson P, Warner R (1981) Environmental variability promotes coexistence in lottery competitive systems. Am Nat 117:923–943
Chopin T, Buschmann AH, Halling C, Troell M, Kautsky N, Neori A, Kraemer GP, Zertuche-González JA, Yarish C, Neefus C (2001) Integrating seaweeds into marine aquaculture systems: a key toward sustainability. J Phycol 37:975–986
Chung IK, Kang YH, Yarish C, Kraemer GP, Lee JA (2002) Application of seaweed cultivation to the bioremediation of nutrient-rich effluent. Algae 17:187–194
Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Austral Ecol 18:117–143
Cooper MB, Smith AG (2015) Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr Opin Plant Biol 26:147–153
Cotton A (1910) On the growth of Ulva latissima, L. in water polluted by sewage. Bull Misc Inform Kew 1910:15–19
Cripps S, Bergheim A (2000) Solids management and removal for intensive land-based aquaculture production systems. Aquac Eng 22:33–56
Crisp D, Ryland J (1960) Influence of filming and of surface texture on the settlement of marine organisms. Nature 185:119
Cyrus MD, Bolton JJ, Macey BM, de Wet L (2014) The use of a formulated feed containing cultivated seaweed (Ulva, Chlorophyta) to promote rapid growth and enhanced production of high quality roe in the sea urchin Tripneustes gratilla. Aquacult Res 45:159–176
Cyrus MD, Bolton JJ, Macey BM (2015) The role of the green seaweed Ulva as a dietary supplement for full life cycle grow-out of Tripneustes gratilla. Aquaculture 446:187–197
Cyrus MD, Bolton JJ, Sholtz R, Macey BM (2015) The advantages of Ulva (Chlorophyta) as an additive in sea urchin formulated feeds: Effects on palatability, consumption and digestibility. Aquac Nutr 21:578–591
Dale C, Moran N (2006) Molecular interactions between bacterial symbionts and their hosts. Cell 126:453–465
Datta S, Singh S, Kumar V, Singh Dhanjal D, Sindu GK, Amin DS, Kumar S, Singh J, Singh J (2020) Endophytic bacteria in xenobiotic degradation. In: Kumar A, Singh V (eds) Microbial Endophytes: Prospects for Sustainable Agriculture. Woodhead Publishing, Duxford, pp 125–156
De Prisco J (2020) An investigation of some key physio-chemical water quality parameters of an Integrated Multi-Trophic Aquaculture (IMTA) system operating recirculation methodology in the Western Cape of South Africa. M.Sc. thesis, University of Cape Town
Defoirdt T (2016) Implications of ecological niche differentiation in marine bacteria for microbial management in aquaculture to prevent bacterial disease. PLoS Pathogens 12:e1005843
Dhariwal A, Chong J, Habib S, King IL, Agellon LB, Xia J (2017) MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acid Res 45:180–188
Dixon P (2003) VEGAN, a package of R functions for community ecology. J Veg Sci 14:927–930
Dong Z, Sun T (2007) A potential new process for improving nitrogen removal in constructed wetlands-Promoting coexistence of partial-nitrification and ANAMMOX. J Ecol Eng 31:69–78
Du ZJ, Lv GQ, Rooney AP, Miao TT, Xu QQ, Chen GJ (2011) Agarivorans gilvus sp. nov. isolated from seaweed. Int J Syst Evol Microbiol 61:493–496
Egan S, Thomas T, Holmström C, Kjelleberg S (2000) Phylogenetic relationship and antifouling activity of bacterial epiphytes from the marine alga Ulva lactuca: brief report. Environ Microbiol 2:343–347
Egan S, Harder T, Burke C, Steinberg P, Kjelleberg S, Thomas T (2013) The seaweed holobiont: understanding seaweed-bacteria interactions. FEMS Microbiol Rev 37:462–476
Erasmus JH, Cook PA, Coyne VE (1997) The role of bacteria in the digestion of seaweed by the abalone Haliotis midae. Aquaculture 155:377–386
Farasat M, Khavari-Nejad R, Nabavi S, Namjooyan F (2014) Antioxidant activity, total phenolics and flavonoid contents of some edible green seaweeds from northern coasts of the Persian Gulf. Iran J Pharm Res 13:163
Gao M, Du D, Bo Z, Sui L (2019) Poly-β-hydroxybutyrate (PHB)-accumulating Halomonas improves the survival, growth, robustness and modifies the gut microbial composition of Litopenaeus vannamei postlarvae. Aquaculture 500:607–612
Garrett T, Bachoo M, Zhang Z (2008) Bacterial adhesion and biofilms on surfaces. Prog Nat Sci 18:1049–1056
Gauger E, Smolowitz R, Uhlinger K, Casey J, Gómez-Chiarri M (2006) Vibrio harveyi and other bacterial pathogens in cultured summer flounder, Paralichthys dentatus. Aquaculture 260:10–20
Gauthier M (1976) Alteromonas rubra sp. nov., a new marine antibiotic-producing bacterium. Int J Syst Evol Microbiol 260:459–566
Gauthier M, Flatau G (1976) Antibacterial activity of marine violet-pigmented Alteromonas with special reference to the production of brominated compounds. Can J Microbiol 22:1612–1619
Gauthier M, Breittmayer V (1979) A new antibiotic-producing bacterium from seawater. Int J Syst Evol Microbiol 29:366–372
Geldart M (2022) The potential for increasing the recirculation rate in a commercial integrated abalone/Ulva aquaculture system. Honour’s thesis, University of Cape Town, South Africa
Ghaderiardakani F, Califano G, Mohr J, Abreu M, Coates J, Wichard T (2019) Analysis of algal growth- and morphogenesis- promoting factors in an integrated multi-trophic aquaculture system for farming Ulva spp. Aquac Environ Interact 11:375–391
Ghaderiardakani F, Quartino ML, Wichard T (2020) Microbiome-dependent adaptation of seaweeds under environmental stresses: a perspective. Front Mar Sci 7:575288
Ghaderiardakani F, Langhans L, Kurbel VB, Fenizia S, Wichard T (2022) Metabolite profiling reveals insights into the species-dependent cold stress response of the green seaweed holobiont Ulva (Chlorophyta). Environ Exp Bot 200:104913
Giovannoni S (2000) Evolution, diversity and molecular ecology of marine prokaryotes. In: Kirchmann DL (ed) Microbial Ecology of Oceans. Liss/Wiley, New York, pp 47–84
Glöckner FO, Yilmaz P, Quast C, Gerken J, Beccati A, Ciuprina A, Bruns G, Yarza P, Peplies J, Westram R, Wal Ludwig (2017) 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol 261:169–176
Gobet A, Mest L, Perennou M, Dittami SM, Caralp C, Coulombet C, Huchette S, Roussel S, Michel G, Leblanc C (2018) Seasonal and algal diet-drive patterns of the digestive microbiota of the European abalone Haliotis tuberculata, a generalist marine herbivore. Microbiome 6:60
Godinho VM, Furbino LE, Santiago IF, Pellizzari FM, Yokoya NS, Pupo D, Alves TM, Junior PA, Romanha AJ, Zani CL, Cantrell CL, Rosa CA, Rosa LH (2013) Diversity and bioprospecting of fungal communities associated with endemic and cold-adapted macroalgae in Antarctica. ISME J 7:1434–1451
Goecke F, Labes A, Wiese J, Imhoff J (2010) Chemical interactions between marine macroalgae and bacteria. Mar Ecol Prog Ser 409:267–299
Grueneberg J, Engelen AH, Costa R, Wichard T (2016) Macroalgal morphogenesis induced by waterborne compounds and bacteria in coastal seawater. PLoS ONE 11:e0146307
Hadfield MG (2011) Biofilms and marine invertebrate larvae: what bacteria produce that larvae use to choose settlement sites. Ann Rev Mar Sci 3:453–470
Handlinger J, Harris J, Carson J, Taylor D (2006) Abalone aquaculture subprogram: the potential for antibiotic use in abalone for disease control. Fisheries Research and Development Corporation (FRDC), Canberra
Hardegen J, Amend G, Wichard T (2023) Lifecycle-dependent toxicity and removal of micropollutants in algal cultures of the green seaweed Ulva (Chlorophyta). J Appl Phycol 35:2031–2048
Harms G, Layton AC, Dionisi HM, Gregory IR, Garrett VM, Hawkins SA, RobinsonSayler KGGSal (2003) Real-time PCR quantification of nitrifying bacteria in a municipal wastewater treatment plant. Environ Sci Technol 37:343–351
Hastuti YP, Syarifuddin NR, Tridesianti S, Fatma YS, Supriyono E (2020) Application of Halomonas sp. HIB-F to Litopenaeus vannamei aquaculture system. AACL Bioflux 13:2116–2126
Henschel J, Cook P (1990) The development of a marine fouling community in relation to the primary film of microorganisms. Biofouling 2:1–11
Hmani I, Ghaderiardakani F, Ktari L, El Bour M, Wichard T (2023) High-temperature stress induces bacteria-specific adverse and reversible effects on Ulva (Chlorophyta) growth and its chemosphere in a reductionist model system. Bot Mar 67:131–138
Holmström C, Kjelleberg S (1999) Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol Ecol 30:285–293
Hong D, Hien H, Son P (2007) Seaweeds from Vietnam used for functional food, medicine and biofertilizer. J Appl Phycol 19:817–826
Inagaki F, Sakihama Y, Inoue A, Kato C, Horikoshi K (2002) Molecular phylogenetic analyses of reverse-transcribed bacterial rRNA obtained from deep-sea cold seep sediments. Environ Microbiol 4:277–286
Infante-Villamil S, Huerlimann R, Jerry DR (2021) Microbiome diversity and dysbiosis in aquaculture. Rev Aquac 13:1077–1096
Ismail A, Ktari L, Ben Redjem Romdhane Y, Aoun B, Sadok S, Boudabous A, El Bour M (2018) Antimicrobial fatty acids from green alga Ulva rigida (Chlorophyta). Biomed Res Int 2018:3065959
Itoi S, Niki A, Sugita H (2006) Changes in microbial communities associated with the conditioning of filter material in recirculating aquaculture systems of the pufferfish Takifugu rubripes. Aquaculture 256:287–295
Jaulneau V, Lafitte C, Jacquet C, Fournier S, Salamagne S, Briand X, Esquerré-Tugayé MT, Dumas B (2010) Ulvan, a sulphated polysaccharide from green algae, activates plant immunity through the Jasmonic acid signalling pathway. J Biomed Biotechnol 2010:525291
Jiravanichpaisal P, Mlyazaki T, Limsuwan C (1994) Histopathology, biochemistry, and pathogenicity of Vibrio harveyi infecting black tiger prawn Penaeus monodon. J Aquat Anim Health 6:27–35
Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acid Res 28:27–30
Kemp JOG, Britz PJ, Agüero PHT (2015) The effect of macroalgal, formulated and combination diets on growth, survival and feed utilisation in the red abalone Haliotis rufescens. Aquaculture 448:306–314
Kim KY, Park SJ, Hahm YT, Cha CJ (2011) Marinitalea sucinacia gen. nov., sp. nov., a marine bacterium of the family Flavobacteriaceae isolated from tidal flat sediment. FEMS Microbiol Lett 314:89–94
Kim HS, Kim PS, Hyun DW, Lee JY, Kang W, Shin NR, Whon TW, Bae JW (2016a) Pseudahrensia todarodis sp. nov., isolated from the gut of a Japanese flying squid, Todarodes pacificus. Int J Syst Evol Microbiol 66:1389–1393
Kim SG, Pheng S, Lee YJ, Eom MK, Shin DH (2016b) Agarivorans aestuarii sp. nov., an agar-degrading bacterium isolated from a tidal flat. Int J Syst Evol Microbiol 66:3119–3124
Kindaichi T, Ito T, Okabe S (2004) Ecophysiological interaction between nitrifying bacteria and heterotrophic bacteria in autotrophic nitrifying biofilms as determined by microautoradiography-fluorescence in situ hybridization. Appl Environ Microbiol 70:1641–1650
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acid Res 41:e1
Kraxberger-Beatty T, McGarey DJ, Grier HJ, Lim DV (1990) Vibrio harveyi, an opportunistic pathogen of common snook, Centropomus undecimalis (Bloch), held in captivity. J Fish Dis 13:557–560
Kurahashi M, Yokota A (2004) Agarivorans albus gen. nov., sp. nov., a γ-proteobacterium isolated from marine animals. Int J Syst Evol Microbiol 54:693–697
Kurilenko VV, Christen R, Zhukova NV, Kalinovskaya NI, Mikhailov VV, Crawford RJ, Ivanova EP (2010) Granulosicoccus coccoides sp. nov., isolated from leaves of seagrass (Zostera marina). Int J Syst Evol Microbiol 60:972–976
Lachnit T, Blumel M, Imhoff JF, Wahl M (2009) Specific epibacterial communities on macroalgae: phylogeny matters more than habitat. Aquat Biol 5:181–186
Lane D (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic Acids Techniques in Bacterial Systematics. John Wiley and Sons, New York, pp 115–175
Lara-Anguiano GF, Esparza-Leal HM, Sainz-Hernández JC, Ponce-Palafox JT, Valenzuela-Quiñónez W, Apun-Molina JP, Klanian MG (2013) Effects of inorganic and organic fertilization on physicochemical parameters, bacterial concentrations, and shrimp growth in Litopenaeus vannamei cultures with zero water exchange. J World Aquac Soc 44:499–510
Largo DB, Fukami K, Nishijima T (1995) Occasional pathogenic bacteria promoting ice-ice disease in the carrageenan-producing red algae Kappaphycus alvarezii and Eucheuma denticulatum (Solieriaceae, Gigartinales, Rhodophyta). J Appl Phycol 7:545–554
Lawton RJ, Mata L, de Nys R, Paul NA (2013) Algal bioremediation of waste waters from land-based aquaculture using Ulva: Selecting target species and strains. PLoS ONE 8:e0231281
Lemos M, Toranzo A, Barja J (1985) Antibiotic activity of epiphytic bacteria isolated from intertidal seaweeds. Microbial Ecol 11:149–163
Li X, Liu Y, Chen Z, Liu LZ, Liu ZP, Liu Y (2016) Membranicola marinus gen. nov., sp. nov., a new member of the family Saprospiraceae isolated from a biofilter in a recirculating aquaculture system. Int J Syst Evol Microbiol 66:1275–1280
Li J, Weinberger F, de Nys R, Thomas T, Egan S (2023) A pathway to improve seaweed aquaculture through microbiota manipulation. Trends Biotechnol 41:545–556
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Lu K, Lin W, Liu J (2008) The characteristics of nutrient removal and inhibitory effect of Ulva clathrata on Vibrio anguillarum. J Appl Phycol 20:1061
Lydmark P, Almstrand R, Samuelsson K, Mattsson A, Sörensson F, Lindgren PE, Hermansson M (2007) Effects of environmental conditions on the nitrifying population dynamics in a pilot wastewater treatment plant. Environ Microbiol 9:2220–2233
Macey BM, Coyne VE (2005) Improved growth rate and disease resistance in farmed Haliotis midae through probiotic treatment. Aquaculture 245:249–261
Macey BM, Coyne VE (2006) Colonization of the gastrointestinal tract of the farmed South African abalone Haliotis midae by the probionts Vibrio midae SY9, Cryptococcus sp. SS1, and Debaryomyces hansenii AY1. Mar Biotechnol 8:246–259
Macpherson A, Harris H (2004) Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol 4:478–485
Mahmud ZH, Neogi SB, Kassu A, Mai Huong BT, Jahid IK, Islam MS, Ota F (2008) Occurrence, seasonality and genetic diversity of Vibrio vulnificus in coastal seaweeds and water along the Kii Channel, Japan. FEMS Microbiol Ecol 64:209–218
Mao W, Zang XLY, Zhang H (2006) Sulfated polysaccharides from marine green algae Ulva conglobata and their anticoagulant activity. J Appl Phycol 18:9–14
Marshall K, Joint I, Callow M, Callow J (2006) Effect of marine bacterial isolates on growth and morphology of axenic plantlets of the green alga Ulva linza. Microbial Ecol 52:302–310
Martins P, Cleary DFR, Pires ACC, Rodrigues AM, Quintino V, Calado R, Gomes NCM (2013) Molecular analysis of bacterial communities and detection of potential pathogens in a recirculating aquaculture system for Scophthalmus maximus and Solea senegalensis. PLoS ONE 8:e80847
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217
Michaud L, Blancheton JP, Bruni V, Piedrahita R (2006) Effect of particulate organic carbon on heterotrophic bacterial populations and nitrification efficiency in biological filters. Aquac Eng 34:224–233
Moriarty D (1997) The role of microorganisms in aquaculture ponds. Aquaculture 151:333–349
Mulvaney WJ, Winberg PC, Adams L (2013) Comparison of macroalgal (Ulva and Grateloupia spp.) and formulated terrestrial feed on the growth and condition of juvenile abalone. J Appl Phycol 25:815–824
Naidoo K, Maneveldt G, Ruck K, Bolton JJ (2006) A comparison of various seaweed-based diets and formulated feed on growth rate of abalone in a land-based aquaculture system. J Appl Phycol 18:211–217
Nakanishi K, Nishimura M, Kuwano K, Saga N (1996) Bacteria that induce morphogenesis in Ulva pertusa (Chlorophyta) grown under axenic conditions. J Phycol 32:479–482
Nakayama T, Lu H, Nomura N (2009) Inhibitory effects of Bacillus probionts on growth and toxin production of Vibrio harveyi pathogens of shrimp. Lett Appl Microbiol 49:679–684
Nguyen D, Ovadia O, Guttman L (2023) Temporal force governs the microbial assembly associated with Ulva fasciata (Chlorophyta) from an integrated multi-trophic aquaculture system. Front Microbiol 14:1223204
Nicolas JL, Basuyaux O, Mazurie J, Thebault A (2002) Vibrio carchariae, a pathogen of the abalone Haliotis tubreculata. Dis Aquat Org 50:35–43
Nielsen MM, Bruhn A, Rasmussen MB, Olesen B, Larsen MM, Møller HB (2012) Cultivation of Ulva lactuca with manure for simultaneous bioremediation and biomass production. J Appl Phycol 24:449–458
Nishimori E, Hasegawa O, Numata T, Wakabayashi H (1998) Vibrio carchariae causes mass mortalities in Japanese abalone, Sulculus diversicolor supratexta. Fish Pathol 33:495–502
Nogami K, Maeda M (1992) Bacteria as biocontrol agents for rearing larvae of the crab Portunus trituberculatus. Can J Fish Aquat Sci 49:2373–2376
Ofelio C, Planas M, Pintado J (2021) Administration of the probiotic Lactobacillus rhamnosus IMC 501 as a strategy for the control of Vibrio bacteria in the brine shrimp Artemia. Lett Appl Microbiol 73:336–342
Olafsen JA (2001) Interactions between fish larvae and bacteria in marine aquaculture. Aquaculture 200:223–247
Paillard C, Le Roux F, Borrego JJ (2004) Bacterial disease in marine bivalves, a review of recent studies: trends and evolution. Aquat Living Resour 17:477–498
Park S, Park JM, Jung YT, Yoon JH (2014) Agarivorans litoreus sp. nov., a novel gammaproteobacterium isolated from seawater and amended description of the genus Agarivorans. Antonie van Leeuwenhoek 106:1041–1047
Paungfoo C, Prasertsan P, Burrell PC, Intrsungkha N, Blackall LL (2007) Nitrifying bacterial communities in an aquaculture wastewater treatment system using fluorescence in situ hybridization (FISH), 16S rRNA gene cloning, and phylogenetic analysis. Biotechnol Bioeng 97:985–990
Penesyan A, Marshall-Jones Z, Holmstrom C, Kjelleberg S, Egan S (2009) Antimicrobial activity observed among cultured marine epiphytic bacteria reflects their potential as a source of new drugs. FEMS Microbiol Ecol 69:113–124
Pfeffer C, Oliver JD (2003) A comparison of thiosulphate-citrate-bile salts-sucrose (TCBS) agar and thiosulphate-chloride-iodide (TCI) agar for the isolation of Vibrio species from estuarine environments. Lett Appl Microbiol 36:150–151
Pintado J, Del Olmo G, Guinebert T, Ruiz P, Nappi J, Thomas T, Egan S, Masalo I, Cremades J (2023) Manipulating the Ulva holobiont: co-culturing Ulva ohnoi with Phaeobacter bacteria as a strategy for disease control in fish-macroalgae IMTA-RAS aquaculture. J Appl Phycol 35:2017–2029
Pintado Valverde J, Ruiz P, Oca Baradad J, Masalo Llora I, Jimenez de Ridder P, Cremades Ugarte J (2018) Study of the bacterial communities of the seaweed (Ulva spp.) holobiont to base management strategies for the control of harmful bacteria in IMTA-RAS. World Aquaculture Society Congress 2018. "AQUA-2018 Abstracts", p 599
Polikovsky M, Califano G, Dunger N, Wichard T, Golberg A (2020) Engineering bacteria-seaweed symbioses for modulating the photosynthate content of Ulva (Chlorophyta): significant for the feedstock of bioethanol production. Algal Res 49:101945
Pomeroy LR, le Williams PJB, Azam F, Hobbie JE (2007) The microbial loop. Oceanography 20:28–33
Primavera J (2006) Overcoming the impacts of aquaculture on the coastal zone. Ocean Coast Manag 49:531–545
Prol-García MJ, Pintado J (2013) Effectiveness of probiotic Phaeobacter bacteria grown in biofilters against Vibrio anguillarum infections in the rearing of Turbot (Psetta maxima) larvae. Mar Biotechnol 15:726–38
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acid Res 41:D590–D596
Rao D, Webb JS, Holmstro C, Case R, Low A, Steinberg P, Kjelleberg S (2007) Low densities of epiphytic bacteria from the marine alga Ulva australis inhibit settlement of fouling organisms. Environ Microbiol 73:7844–7852
Ravikumar S, Anburajan L, Meena B (2016) Antibacterial activity of Ulva reticulata from southwest coast of Kanyakumari, India. J Coast Life Med 4:246–247
Ray B, Lahaye M (1995) Cell-wall polysaccharides from the marine green alga Ulva “rigida” (Ulvales, Chlorophyta). Extraction and chemical composition. Carbohydr Res 274:251–261
Reis AM, Araújo SD Jr, Moura RL, Francini-Filho RB, Pappas G Jr, Coelho AM, Krüger RH, Thompson FL (2009) Bacterial diversity associated with the Brazilian endemic reef coral Mussismilia braziliensis. J Appl Microbiol 106:1378–1387
Rengpipat S, Tunyanun A, Fast AW, Piyatiratitivorakul S, Menasveta P (2003) Enhanced growth and resistance to Vibrio challenge in pond-reared black tiger shrimp Penaeus monodon fed a Bacillus probiotic. Dis Aquat Org 55:169–173
Robertson-Andersson DV, Potgieter M, Hansen J, Bolton JJ, Troell M, Anderson RJ, Halling C, Probyn T (2008) Integrated seaweed cultivation on an abalone farm in South Africa. J Appl Phycol 20:579–595
Robertson-Andersson D (2003) The cultivation of Ulva lactuca (Chlorophyta) in an integrated aquaculture system, for the production of abalone feed and the bioremediation of aquaculture effluent. PhD thesis, University of Cape Town, South Africa
Rodrick G (1991) Indigenous pathogens: Vibrionaceae. In: Ward DR, Hackney C (eds) Microbiology of Marine Food Products. Springer, Boston, pp 285–300
Rodrigues CJC, de Carvalho CCCR (2022) Cultivating marine bacteria under laboratory conditions: overcoming the “unculturable” dogma. Front Bioeng Biotechnol 10:964589
Rojas R, Miranda CD, Amaro AM (2009) Pathogenicity of a highly exopolysaccharide-producing Halomonas strain causing epizootics in larval cultures of the Chilean scallop Argopecten purpuratus (Lamarck, 1819). Microbial Ecol 57:129–139
Romanenko LA, Lysenko AM, Rohde M, Mikhailov VV, Stackebrandt E (2004) Psychrobacter maritimus sp. nov. and Psychrobacter arenosus sp. nov., isolated from coastal sea ice and sediments of the Sea of Japan. Int J Syst Evol Microbiol 54:1741–1745
Romanenko LA, Uchino M, Kalinovskaya NI, Mikhailov VV (2008) Isolation, phylogenetic analysis and screening of marine mollusc-associated bacteria for antimicrobial, hemolytic and surface activities. Microbiol Res 163:633–644
Romanenko LA, Tanaka N, Frolova GM, Mikhailov VV (2009) Psychrobacter fulvigenes sp. nov., isolated from a marine crustacean from the sea of Japan. Int J Syst Evol Microbiol 59:1480–1486
Ruck KR, Cook PA (1998) Sabellid infestations in the shells of South African molluscs: implications for abalone mariculture. J Shellfish Res 17:693–699
Salvesen I, Skjermo J, Vadstein O (1999) Growth of turbot (Scophthalmus maximus L.) during first feeding in relation to the proportion of r/K-strategists in the bacterial community of the rearing water. Aquaculture 175:337–350
Sangnoi Y, ChankaewO-Thong SS (2017) Indigenous Halomonas spp., the potential nitrifying bacteria for saline ammonium waste-water treatment. Pak J Biol Sci 20:52–58
Sawabe T, Makino H, Tatsumi M, Nakano K, Tajima K, Iqbal MM, Yumoto I, Ezura Y, Christen R (1998) Pseudoalteromonas bacteriolytica sp. nov., a marine bacterium that is the causative agent of red spot disease of Laminaria japonica. Int J Syst Bacteriol 48:769–774
Schneider O, Sereti V, Machiels MAM, Eding EH, Verreth JAJ (2006) The potential of producing heterotrophic bacteria biomass on aquaculture waste. Water Res 40:2684–2694
Schneider O, Chabrillon-Popelka M, Smidt H, Haenen O, Sereti V, Eding EH, Verreth JA (2007) HRT and nutrients affect bacterial communities grown on recirculation aquaculture system effluents. FEMS Microbiol Ecol 60:207–219
Schreier HJ, Mirzoyan N, Saito K (2010) Microbial diversity of biological filters in recirculating aquaculture systems. Curr Opin Biotechnol 21:318–325
Schroeder DC, Jaffer MA, Coyne VE (2003) Investigation of the role of a β(1–4) agarase produced by Pseudoalteromonas gracilis B9 in eliciting disease symptoms in the red alga Gracilaria gracilis. Microbiology 149:2919–2929
Selvam G, Sivakumar K (2013) Effect of foliar spray from seaweed liquid fertilizer of Ulva reticulata (Forsk.) on Vigna mungo L. and their elemental composition using SEM-energy dispersive spectroscopic analysis. Asian Pac J Reprod 2:119–125
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379–423
Shuuluka D (2011) Ecophysiological studies of three South African Ulva species from integrated seaweed. PhD thesis, University of Cape Town, South Africa
Simpson EH (1949) Measurement of diversity. Nature 163:688
Simpson B, Cook P (1998) Rotation diets: a method of improving growth of cultured abalone using natural diets. J Shellfish Res 17:635–640
Singh R, Reddy C (2014) Seaweed-microbial interactions: key functions of seaweed-associated bacteria. FEMS Microbiol Ecol 88:213–230
Sison-Mangus M, Jiang S, Tran K, Kudela R (2014) Host-specific adaptation governs the interaction of the marine diatom Pseudo-nitzschia and their microbiota. ISME J 8:63
Snoeijs P (1994) Distribution of epiphytic diatom species composition, diversity and biomass on different macroalgal hosts along seasonal and salinity gradients in the Baltic Sea. Diatom Res 9:189–211
Soto D, Aguilar-Manjarrez J, Brugère C, Angel D, Bailey C, Black K et al (2008) Applying an ecosystem-based approach to aquaculture: principles, scales and some management measures. In: Building an Ecosystem Approach to Aquaculture. FAO/Universitat de Les Illes Balears Expert Workshop, 7–11 May 2007, Palma de Mallorca, Spain. FAO Fisheries and Aquaculture Proceedings No. 14, pp 15–35
Spoerner M, Wichard T, Bachhuber T, Stratmann Oertel W (2012) Growth and thallus morphogenesis of Ulva mutabilis (Chlorophyta) depends on a combination of two bacterial species excreting regulatory factors. J Phycol 48:1433–1447
Staufenberger T, Thiel V, Wiese J, Imhoff J (2008) Phylogenetic analysis of bacteria associated with Laminaria saccharina. FEMS Microbiol Ecol 64:65–77
Steinberg P, De Nys R, Kjelleberg S (2002) Chemical cues for surface colonization. J Chem Ecol 28:1935–1951
Sugita H, Nakamura H, Shimada T (2005) Microbial communities associated with filter materials in recirculating aquaculture systems of freshwater fish. Aquaculture 243:403–409
Taylor MW, Schupp PJ, Dahllöf I, Kjelleberg S, Steinberg PD (2004) Host specificity in marine sponge-associated bacteria, and potential implications for marine microbial diversity. Environ Microbiol 6:121–130
Taylor MW, Schupp PJ, De Nys R, Kjelleberg S, Steinberg PD (2005) Biogeography of bacteria associated with the marine sponge Cymbastela concentrica. Environ Microbiol 7:419–433
JASP Team (2017) JASP computer software (Version 0.12. 2)
ten Doeschate KI, Coyne VE (2008) Improved growth rate in farmed Haliotis midae through probiotic treatment. Aquaculture 284:174–179
Teske A, Alm E, Regan JM, Toze S, Rittmann BE, Stahl DA (1994) Evolutionary relationships among ammonia- and nitrite-oxidising bacteria. J Bacteriol 176:6623–6630
Thompson F, Abreu P, Wasielesky W (2002) Importance of biofilm for water quality and nourishment in intensive shrimp culture. Aquaculture 203:263–278
Troell M, Robertson-Andersson D, Anderson RJ, Bolton JJ, Maneveldt G, Halling C, Probyn T (2006) Abalone farming in South Africa: an overview with perspectives on kelp resources, abalone feed, potential for on-farm seaweed production and socio-economic importance. Aquaculture 257:266–281
Tujula NA, Crocetti GR, Burke C, Thomas T, Holmström C, Kjelleberg S (2010) Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME J 4:301–311
Ulrich JF, Grafe MS, Dhiman S, Wienecke P, Arndt H, Wichard T (2022) Thallusin quantification in marine bacteria and algae cultures. Mar Drugs 20:690
van Baarlen P, Wells JM, Kleerebezem M (2013) Regulation of intestinal homeostasis and immunity with probiotic Lactobacilli. Trends Immunol 34:208–215
van der Loos LM, D’hondt S, Willems A, De Clerck O (2021) Characterizing algal microbiomes using long-read nanopore sequencing. Algal Res 59:102456
van der Loos LM, Dhondt S, Engelen AH, Pavia H, Toth GB, Willems A, Weinberger F, De Clerck O, Steinhagen S (2022) Salinity and host drive Ulva associated bacterial communities across the Atlantic-Baltic Sea gradient. Mol Ecol 32:6260-6277
van der Loos LM, De Wilde C, Willems A, De Clerck O, Steinhagen S (2024) The cultivated sea lettuce (Ulva) microbiome: Successional and seasonal dynamics. Aquaculture 585:740692
Vandermeulen H, Gordin H (1990) Ammonium uptake using Ulva (Chlorophyta) in intensive fishpond systems: mass culture and treatment of effluent. J Appl Phycol 2:363–374
Weinberger F (2007) Pathogen-induced defense and innate immunity in macroalgae. Biol Bull 213:290–302
Weiss A, Costa R, Wichard T (2017) Morphogenesis of Ulva mutabilis (Chlorophyta) induced by Maribacter species (Bacteriodetes, Flavobacteriaceae). Bot Mar 60:197–206
Wichard T (2015) Exploring bacteria-induced growth and morphogenesis in the green macroalga order Ulvales (Chlorophyta). Front Plant Sci 6:1–19
Wichard T (2023) From model organism to application: Bacteria-induced growth and development of the green seaweed Ulva and the potential of microbe leveraging in algal aquaculture. Semin Cell Dev Biol 134:69–78
Widdel F, Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria. In: Balows A, Truper HG, Dworkin M, Harder W, Schleifer KH (eds) The Prokaryotes. Springer, New York, pp 3352–3378
Wietz M, Hall MR, Høj L (2009) Effects of seawater ozonation on biofilm development in aquaculture tanks. Syst Appl Microbiol 32:266–277
Yang SJ, Choo YJ, Cho JC (2007) Lutimonas vermicola gen. nov., sp. nov., a member of the family Flavobacteriaceae isolated from the marine polychaete Periserrula leucophryna. Int J Syst Evol Microbiol 57:1679–1684
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO (2014) The SILVA and “all-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acid Res 42:D643–D648
Yoon J, Matsuo Y, Kasai H, Yokota A (2012) Portibacter lacus gen. nov., sp. nov., a new member of the family Saprospiraceae isolated from a saline lake. J Gen Appl Microbiol 58:191–197
Zhao X, Zheng W, Qu T, Zhong Y, Xu J, Jiang Y (2021) Dual roles of reactive oxygen species in intertidal macroalgae Ulva polifera under ultraviolet-B radiation. Environ Exp Bot 189:104534
Zilber-Rosenberg I, Rosenberg E (2008) Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiology Reviews 32:723–735
Zorrilla I, Arijo S, Chabrillon M, Diaz P, Martinez-Manzanares E, Balebona MC, Moriñigo MA (2003) Vibrio species isolated from diseased farmed sole, Solea senegalensis (Kaup), and evaluation of the potential virulence role of their extracellular products. J Fish Dis 26:103–108
Acknowledgements
The authors would like to thank the following institutions for their availability of samples and the use of facilities over the course of the study: I&J Abalone Farm, Department of Forestry, Fisheries and the Environment (DFFE) (RSA), the University of Cape Town (UCT), University of the Western Cape (UWC) Next-Generation Sequencing Facility and the Stellenbosch University (SU) Central Analytical Facility (CAF). This study received funding from the European Commission as part of the EU Horizon 2020 Research & Innovation Programme ASTRAL Project under Grant Agreement No. 863034.
Funding
Open access funding provided by University of Cape Town. This study received funding from the European Commission as part of the EU Horizon 2020 Research & Innovation Programme ASTRAL Project under Grant Agreement No. 863034.
Author information
Authors and Affiliations
Contributions
The study was conceptualized by Brett M. Macey and John J. Bolton. Material preparation and data collection was performed by Kristin de Jager, and data curation, data analysis and visualisation were performed by Kristin de Jager and Marissa Brink-Hull. All authors contributed to the investigation conducted in this manuscript. The first draft of the manuscript was written by Kristin de Jager and Marissa Brink-Hull, and revisions were made by Marissa Brink-Hull and Brett M. Macey. All authors have read and approved the final manuscript and have reviewed previous versions of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Jager, K., Brink-Hull, M., Bolton, J.J. et al. Bacterial microbiome dynamics in commercial integrated aquaculture systems growing Ulva in abalone effluent water. J Appl Phycol (2024). https://doi.org/10.1007/s10811-024-03298-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10811-024-03298-8