
Abstract
Lontra longicaudis is a top predator of freshwater systems currently categorized as ‘data deficient’ by the IUCN. Thus far, no genetic studies have been carried out in Argentina. Using 8 microsatellite loci, we analysed the genetic diversity and individual relatedness in a population located at the Lower Delta of Paraná River, Argentina. Our data, together with available genetic information for other populations and habitat suitability data obtained from species distribution models previously published, were used to test whether genetic diversity, inbreeding, and parentage coefficients are related to habitat suitability both at landscape and at regional scale. Our results show that the levels of heterozygosity found in the Lower Delta are lowest reported to date. A low overall degree of relatedness between otter dyads was found. However, almost 20% showed relatedness values higher than 0.25, suggesting local reproduction. Finally, we found a positive association between habitat quality and levels of heterozygosity, both at landscape and regional scale, suggesting that in highly suitable habitats, the otters are genetically more variable. Our study constitutes the first one on the genetics of L. longicaudis in Argentina and provides baseline information for the development of conservation plans for the Lower Paraná River Delta population.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Genetic diversity is one of the most important attributes of a population, since those species that exhibit greater variability can adapt to environmental changes better and are less susceptible to extinction (Frankham et al., 2010; Allendorf et al., 2013). Reduction in population size can cause loss of genetic variability within populations and the emergence of negative genetic effects. Small, isolated populations may suffer effects of inbreeding, leading to a decrease in reproductive success and an increase in the probability of extinction (Luck et al., 2003; Freeland, 2005). This phenomenon has been repeatedly mentioned as an impediment to the population growth of mammals found at low densities (Mills & Smouse, 1994; Lacy, 1997; Avise, 2004; O’Grady et al., 2006). Early detection of potentially negative genetic changes maximizes our ability to implement a management approach to limit or reverse the negative effects before they become substantial or irreversible.
The Lower Delta of the Paraná River is one of the most important wetlands in Argentina (Kalesnik & Quintana, 2006). At the same time, this wetland with high levels of biodiversity is one of the most endangered in the country (Dinerstein et al., 1995). Its proximity to several of the biggest Argentinean cities is one of the main change promoters since the wetland is exposed to considerable pollution sources, advance of urban frontiers, degradation and fragmentation of habitat resulting from construction of polders for use in forestry and fruit cultivation, grooving of waterways, and/or construction for tourism enterprises (Bó, 2005). This deterioration in vegetation and micro-environment structure complexity of the coast could affect population abundances of mammalian species, as has been previously described in other wetlands (Fahrig, 2003; Alho, 2008).
Otters are considered to be a bioindicator of the conservation status of the wetlands they inhabit. Because each individual occupies relatively large tracts of land, they are sensitive to the effects of habitat fragmentation (Parera, 1996; Kruuk, 2006). The Neotropical river otter, Lontra longicaudis (Olfers 1818), is a top predator in many South and Central American freshwater systems. It is distributed from northern Mexico (32°N) to Argentina (32°S) and is adapted to a wide variety of aquatic habitats, including seashores and elevations up to 3000 m (Blascher, 1987; Emmons & Feer, 1997). Its main habitat requirements include wide riparian vegetation, the availability of potential sites to construct their burrows, and clean and fast rivers and streams (Larivière, 1999). Some authors consider that the species can tolerate the modification of their environment and, in consequence, can be found in areas with intense human activity (MacDonald & Mason, 1992; Pardini & Trajano, 1999; Gómez et al., 2014). Its diet is based on fish, but it can include aquatic invertebrates such as crabs, crayfish, and mussels, and terrestrial prey including mammals, birds, and insects (Pardini & Trajano, 1999; Quadros & Monteiro-Filho, 2001; Chemes et al., 2010). Gallo-Reynoso (1989) found that the linear home range of the species in Mexican populations is 2–4 km during the dry season and 7–9 km during the rainy season. For Brazilian populations, Trinca et al. (2013) found mean linear home ranges of 3 km for females and of 11.5 km for males. The species is thought to be solitary, but many individuals may settle and feed in the same area (Kruuk, 2006). Reproductive pairs can be found during the breeding season (Larivière, 1999), while familiar groups composed by mothers and their offspring can be occasionally observed (Parera, 1993). Some evidence of female philopatric behaviour has been found (Trinca et al., 2013). Many threats affect population numbers and genetic diversity of the species, such as habitat degradation, declining prey populations, human persecution, water pollution, and other anthropogenic impacts (Waldemarin & Álvarez, 2008, Trinca et al., 2013). Nevertheless, the Neotropical river otter is a poorly known species for which local levels of genetic diversity and demographic parameters are virtually absent (Trinca et al., 2013). The species is currently categorized as ‘data deficient’ by the IUCN red list (Waldemarin & Álvarez, 2008), which makes it imperative to collect biological information on the species to assess its conservation status. It is also found in Appendix I of CITES (Larivière, 1999), and it is considered an endangered species at national level, according to the latest survey conducted by the Sociedad Argentina para el Estudio de los Mamíferos (Ojeda et al., 2012). Some recent studies examined the genetic diversity and population structure of Neotropical river otters (Trinca et al., 2007; Weber et al., 2009; Ortega et al., 2012; Trinca, 2012; Trinca et al., 2012, 2013). However, these studies were mainly carried out in Brazil, and none of them included Argentinean populations.
Species distribution models (SDMs) are associative models relating occurrence or abundance data at known locations of individual species (distribution data) to information on the environmental characteristics of those locations (Elith & Leathwick, 2009). Several publications have reviewed the available SDMs, their advantages, and limitations (Guisan & Thuiller, 2005; Heikkinen et al., 2006; Elith & Leathwick, 2009; Cassini, 2011, 2013). An implicit assumption of the SDMs is that the predicted ecological niche of a species should reflect its fitness or adaptive landscape (Nagaraju et al., 2013). Thus, individuals inhabiting high-quality habitats (i.e. habitats with better resources for survival and reproduction) are expected to have higher fitness than those inhabiting poorly suitable habitats. High values of fitness have been related with high genetic diversity and, consequently, low inbreeding and genetic relatedness among individuals (Foerster et al., 2003; Frankham et al., 2010; Hedrick, 2011). Therefore, it is expected that a high habitat suitability index would be positively associated with high genetic diversity and negatively associated with inbreeding and parentage coefficients.
In this paper, we carried out the first genetic study of a L. longicaudis population in Argentina. Our aim was to describe and analyse the genetic diversity and individual relatedness in the Lower Delta of Paraná River. Given the high human impact in the study area, we expected a low genetic variability in the study population, accompanied by high inbreeding and genetic relatedness among individuals. In addition, data obtained in the present study, together with available genetic information for other populations and habitat suitability data obtained from species distribution models previously published, were used to test at two different ecological scales, whether genetic diversity, inbreeding, and parentage coefficients are related with habitat suitability. We expected that at lower habitat suitability, we would observe lower levels of genetic diversity, higher levels of inbreeding, and higher values of genetic relatedness among individuals.
Materials and methods
Study area
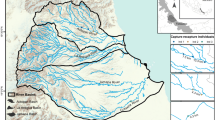
The delta of the Paraná River is a large wetland located in the east-central region of Argentina, which stretches through the final 300 km of the Paraná Basin, the second largest basin in South America (Bonetto, 1986). The Lower Delta, the last stretch of Paraná Delta, covers approximately 17,500 km2 (Malvárez, 1997) and is located approximately between 32°5′S and 58°30′W and between 34°29′S and 60°48′W. It borders to the south with a populated region of the Pampas plain with several major towns, including Buenos Aires and its suburbs, one of the 20 largest cities of the world. Our study area comprised the Buenos Aires Province portion of the Lower Delta of the Paraná River (Fig. 1).

Locations (empty circles) at the Buenos Aires portion of the Lower Delta of Paraná River (dotted line) where samples were collected for DNA genotyping in winters 2011–2013
Sample collection and DNA extraction
We conducted a latrine survey in collaboration with the Lower delta community in winters 2011–2013. Because of the extent of area sampled and logistical constraints in sampling, each sampling location was visited only once. During surveys, we collected all fresh faeces found in the study area (n = 33). Faeces were easily recognizable because they are generally found in latrines near water courses, have a characteristic smell, and contain crustacean and fish traces (Weber et al., 2009; Ortega et al., 2012; Trinca, 2012). Additionally, 7 tissue samples were obtained from road-killed animals. All samples (n = 40, Fig. 1) were preserved in plastic tubes with 96% ethanol and kept at −20°C until DNA was extracted.
Total genomic DNA was extracted from tissue samples using a standard protocol with proteinase K and phenol–chloroform (Sambrook et al., 1989). For faeces, we used two commercial kits, the QIAamp DNA Stool Mini Kit (Qiagen) or the ZR Fecal DNA MiniPrep (Zymo Research). Quality of DNA was assessed by electrophoresis on 1% agarose gels in combination with molecular size standards. Concentration of extracted DNA was not determined since DNA obtained from faeces is a mixture of the predator’s genome with those of prey items, gut microbes, and environmental contaminants (Bradley & Vigilant, 2002). The sampling location of each sample was recorded using a GPS (GPSmap 60C, Garmin).
Microsatellite amplification and sequencing
Total genomic DNA was used to amplify 11 microsatellite loci (LUT453, LUT733, LUT782, LUT701, 04OT02, 04OT04, 04OT05, 04OT17, 04OT22, RIO11, RIO7), previously developed for the European otter, Lutra lutra (Linnaeus 1758), and the North American river otter, Lontra canadensis (Schreber 1777) (Dallas & Piertney, 1998; Beheler et al., 2003, 2005; Huang et al., 2005). We first optimized the PCR with non-fluorescent primers and tested which loci correctly amplified for our population. Afterwards, loci were amplified with a replaced forward primer labelled with a fluorochrome (FAM or HEX). Amplifications were performed in 10 μl final volume in tubes containing 3 μl of empirically diluted template DNA, 1× PCR buffer (Promega), 1.5 mM to 4 mM MgCl2, 200 μM dNTPs, 0.2 μM of each primer, and 0.5 U Taq DNA Polymerase (Promega). We added 10% trehalose for DNA samples obtained from faeces in order to improve the PCR conditions (Trinca, 2012). Reactions were performed in a MyCycler system (BIO-RAD) with the following conditions: 94°C for 30 s, 40 cycles of 94°C for 45 s, 57–66°C for 45 s, and 72°C for 90 s, and a final extension of 72°C for 20 min. All samples were examined by gel electrophoresis to determine amplification. For samples lacking the expected PCR products, we performed at least two new DNA extractions. Faecal samples were discarded if less than 5 loci were amplified successfully after six non-simultaneous repetitions of the PCR. Fragment analyses were performed in an ABI3130xl sequencer at the Genomic Unit of the Instituto Nacional de Tecnología Agropecuaria (INTA, Argentina). Computer-generated results were analysed using GeneMarker, version 2.6.0 (Softgenetics).
Habitat suitability estimation
Two different sources with information on habitat suitability were used, depending on the geographical scale. At landscape level, we used previously published data of our group (Gómez et al., 2014). The measurements of habitat quality in the Lower Delta of the Parana River were obtained by applying a logit regression model to presence/absence data of L. longicaudis in a grid of 114 cells of 0.05 × 0.05° in which 15 environmental predictors were measured (for details see Gómez et al., 2014).
At regional level, we employed maps that were obtained by applying a MaxEnt model to 565 occurrence data in a 17.056.351 km2 grid that covered 14 countries encompassing the Neotropical otter historical distribution (Lopes-Rheingantz et al., 2014, maps available at: http://goo.gl/G6BaqG). The grid was divided into cells of 0.2 × 0.2° in which 11 environmental predictors were measured (for details see Lopes-Rheingantz et al., 2014). The map provides suitability values using a coloured reference. We used the GPS location of populations in which genetic studies were carried out (described later) to obtain these values from the map.
Data analysis
We estimated the probability of identity (P ID) (i.e. the probability of randomly drawn individuals sharing an identical genotype; Mills et al., 2000; Waits et al., 2001) and the probability of identity between siblings (P ID-SIB), using GenAlEx 6.501 (Peakall & Smouse, 2012). The presence of null alleles was estimated using ML-Relate (Kalinowski et al., 2006). The mean number of alleles per locus (A), observed (H o) and expected (H e) heterozygosity, and polymorphic information content (PIC) were estimated using the Microsatellite Toolkit (Park, 2001). PIC refers to the value of a marker for detecting polymorphism within a population, depending on the number of detectable alleles and the distribution of their frequency; thus, it provides an estimate of the discriminating power of the marker (Nagy et al., 2012). Departures from Hardy–Weinberg equilibrium and linkage disequilibrium were calculated with Genepop v4.2.2 (Raymond & Rousset, 1995; Rousset, 2008). Allelic richness (AR) and inbreeding coefficient (F IS) were estimated with Fstat v2.9.3.2 (Goudet et al., 1996).
In order to compare our results to those previously published for other Neotropical river otter populations (Weber et al., 2009; Trinca et al., 2013), two coefficients of relatedness (r) were used: Queller & Goodnight (1989) estimator was calculated using GenAlEx v6.501 software (Peakall & Smouse, 2012), while a coefficient based on maximum likelihood estimations was calculated using ML-Relate software (Kalinowski et al., 2006). For both coefficients, individuals that do not share any allele (i.e. are not related) should theoretically have an r = 0 and second-degree relatives (half-siblings) should have an r = 0.25. In the case of first-degree relatives, full-siblings should have a mean r = 0.5, while parent–offspring should have an r = 0.5 (Queller & Goodnight, 1989; Kalinowski et al., 2006).
The presence of a population structure in our dataset was tested using Structure v2.3.4 (Pritchard et al., 2000). This approach uses a Bayesian clustering analysis to assign individuals to clusters (K) without prior knowledge of their population assignment. Given the geographical distribution of samples (Fig. 1), the distance between them, and the linear home range of Neotropical river otters, Structure simulations were performed with a number of presumed clusters from K = 1 to 3. We performed 20 runs per K value. For each run, the initial burn-in period was set to 200,000 followed by 1,000,000 Markov Chain Monte Carlo (MCMC) iterations. Longer burn-in or MCMC did not change the results indicating the convergence of data. The most probable number of clusters was determined by plotting the estimated likelihood values [LnP(D)] as a function of K (Falush et al., 2003). We chose a proportion of membership threshold value of q ≥ 0.9 to assign individuals to clusters. This value provides a statistical cut-off within the range of suggested values in the literature (Manel et al., 2002) and indicates that ≥90% of ancestry can be attributed to the respective subpopulation.
The relationship between genetic traits and habitat suitability estimated by SDMs was tested using simple linear regression models in which genetic variables were defined as dependent (response) variables and habitat suitability as the independent (explanatory) variable. In the Lower Delta of Paraná River, the 29 individuals that were genetically sampled (see results section) were found to occupy 8 of the 114 cells used in a previous study (Gómez et al., 2014). For each of these 8 cells the following variables were measured: H o, H e, A, AR, F IS, and r (both Queller & Goodnight, 1989; Kalinowski et al., 2006 coefficients). A similar procedure was used to test the relationship between genetic diversity and habitat suitability at a regional scale. In this case, we analysed data obtained from 7 populations where genetic diversity indexes were available: southern Mexico (Ortega et al., 2012), French Guiana, Northeastern Brazil, Brazilian Amazon, Southern Brazil, Southern South America (Trinca, 2012; Trinca et al., 2013) and Lower Delta (this study). One study could not be included (Weber et al., 2009) because only 5 loci were used and only 37% of individuals were genotyped (10 from 27) at more than three loci. We used H o, H e, A, and AR as genetic diversity variables. Habitat suitability values were obtained from the literature (Lopes-Rheingantz et al., 2014). The relation of habitat suitability with the inbreeding and relatedness coefficients could not be tested at regional scale since inbreeding and relatedness data are not available for most of the populations.
Results
Microsatellite genotyping and probability of identity
We were able to obtain DNA from 36 out of 40 samples analysed. Thirty-one of them were faeces obtained from latrines, while the other 5 were obtained from road-killed individuals. Twenty-nine (24 faeces and 5 tissues) out of these 36 samples (80.5%) were successfully genotyped at 5 or more loci. If only faecal samples were considered (n = 31), genotyping success decreased to 24 samples (77.4%), and this value decreased to 17 samples (54.8%) for those that amplified at least at 8 loci. Three of the 11 microsatellite loci used in genotyping had to be discarded from the analyses due to unspecific amplification or low amplification success, and hence, the final dataset used in this study was composed of 29 individuals of which 22 were genotyped at 8 loci, 6 at 7 loci, and 1 at 5 loci.
Calculations of the probabilities of identity indicated that the 8 selected microsatellites were powerful enough to distinguish between individuals, even in the case of siblings. Theoretical P ID and P ID-SIB values across all loci were 1.3 × 10−5 and 8.5 × 10−3, respectively. All samples showed different multilocus genotypes, suggesting some degree of gene flow in our study area. However, the existence of resident otters cannot be discarded since some individuals in the population were highly related (see below).
Genetic diversity and inference of population structure
Across the eight loci, we observed 39 alleles in 29 otters. Low to high levels of genetic variability were observed in our dataset, with loci exhibiting between 2 and 11 alleles (mean value = 4.88 ± 2.75 alleles per locus), a mean allelic richness of 4.573, and mean H o and H e of 0.563 and 0.526, respectively (Table 1). No pair of loci exhibited linkage disequilibrium after Bonferroni correction (P < 0.002), suggesting that the markers were independent. A significant deviation from HWE (P < 0.001) was observed for two loci (RIO07 and RIO11; Table 1). However, when two homozygotes for rare alleles were excluded from the analysis, all loci were in HWE. Statistical indication for the presence of null alleles was found for 7 of the 8 loci, with frequencies varying from 0.017 for LUT733 to 0.159 for 04OT02 (Table 1). The polymorphic information content of each locus ranged from 0.093 for 04OT04 to 0.808 for RIO11 (Table 1). F IS values across all loci were not significantly different from zero after Bonferroni correction [the indicative adjusted nominal level (5%) for multiple comparisons was P = 0.006], indicating a random mating in the population (Table 1). Structure runs were performed for K = 1–3 based on the distribution of the 39 alleles at the 8 loci. The highest posterior probability of the data was obtained at K = 1 [LnP(D) = −443.3], suggesting the existence of only one genetic unit in the Lower Delta of Parana River. All individuals were assigned to that unit with a probability ≥0.90.
Relatedness
The overall degree of relatedness (r) between river otter dyads (i.e. pairs of individuals) estimated using ML-Relate software (Kalinowski et al., 2006) ranged from zero to 0.80, with a mean of 0.102 (SD = 0.163). For the Queller & Goodnight (1989) estimator, values ranged from −1.000 to 0.806, with a mean of −0.044 (SD = 0.324) (Table 2). However, 20 dyads (4.93%) showed r values based on maximum likelihood estimations higher than 0.5, and 64 dyads (15.76%) showed values higher than 0.25 (Table 2). For Queller & Goodnight (1989) relatedness coefficient, 17 dyads (4.19%) showed values higher than 0.5, and 71 dyads (17.49%) showed values higher than 0.25 (Table 2), suggesting that some individuals in the population were highly related. In addition, maximum likelihood estimations showed that 26 of the 29 individuals had at least one parent–offspring or full-sibling relationship with another member of the population, and 28 of the 29 showed some degree of relatedness with another individual.
Genetic traits and habitat suitability
At Lower Delta, we found significant positive relationships between H o, H e and habitat suitability (Fig. 2a; Table 3). Habitats that were predicted to be highly suitable had significantly greater heterozygosity than sites that were predicted to be poorly suitable. No other significant relationships were found at landscape scale (Table 3). At regional scale, we also found a significant positive relationship between H o and habitat suitability (Fig. 2b; Table 3), while other relationships were non-significant. Neither at landscape scale nor at regional scale, we found significant relationships between observed and expected heterozygosity and sample size (R < 0.52, P > 0.37), indicating that our findings are not the result of an artefact in the number of individuals analysed in each cell/population.

Simple linear regressions between observed heterozygosity and habitat suitability at landscape (a) and regional (b) scales
Discussion
Amplification success and probability of identity
The use of faeces as a non-invasive method is widespread for studying elusive or endangered species (Long et al., 2008) and has been extensively used to obtain genetic information of otters in the last decade (Hájkova et al., 2006; Prigioni et al., 2006; Centrón et al., 2008; Weber et al., 2009; Mowry et al., 2011; Guertin et al., 2012; Ortega et al., 2012; Pickles et al., 2012; Trinca et al., 2013). However, extraction and posterior amplification success of DNA depend on different factors (Hoffman & Amos, 2005; Kalz et al., 2006; Caudron et al., 2007; Nardelli et al., 2011). In our study, the amplification success was high in comparison with other studies, even considering the samples which could be amplified for 8 loci (54.8%). Previous studies in the European otter Lutra lutra have reported amplification rates between 20 and 40% (Dallas et al., 2003; Prigioni et al., 2006), while for Lontra longicaudis, the success values range between 24.4% (Ortega et al., 2012) and 41% (Trinca et al., 2013). Differences found between this work and previous work could be explained by different factors. First, sampling campaigns in our study were carried out during winter. Lampa et al. (2008) suggest that climate can affect amplification success, since fungal and bacterial proliferation is one of the main causes of contamination and degradation of DNA. Second, only those samples that were considered fresh were collected and analysed in the present study. A recent review (Nardelli et al., 2011) shows that the increase in post-defecation time is related with a decreasing probability of marker amplification.
Genetic diversity
L. longicaudis in the Lower Delta of Paraná River showed values in mean number of alleles and mean allelic richness similar to or slightly lower than reported previously for the species (Weber et al., 2009; Ortega et al., 2012; Trinca, 2012; Trinca et al., 2013). However, a comparison of allelic richness between these studies should be taken with caution since the number of individuals genotyped at all loci is not the same. In addition, the levels of heterozygosity found in our study (H o = 0.563, H e = 0.526, Table 1) are in accordance with those reported for mustelid populations that suffer some kind of demographic impact as the endangered giant otter, Pteronura brasiliensis (Gmelin 1788) (H o = 0.56–0.57; Pickles et al., 2012), the near threatened European otter, Lutra lutra (H o = 0.50, H e = 0.58; Mucci et al., 2010), and the endangered sea otter, Enhydra lutris (Linnaeus 1758), whose levels of mean expected heterozygosity decreased from 0.77 to 0.52 after hunting impact (Larson et al., 2002). A comparison of our results with other reports on the species (Weber et al., 2009; Ortega et al., 2012; Trinca, 2012; Trinca et al., 2013) shows that the Lower Delta population is less variable, except for the individuals studied by Weber et al. (2009) where a very low observed heterozygosity was found (range between 0.229 and 0.355). However, the reported values in that study were obtained analysing only 5 loci.
The low genetic diversity found in the Lower Delta population could be due to the fact that microsatellites used in our study were developed for other otter species. However, genetic diversity found using the same markers was high in other L. Longicaudis populations. Some other ideas can be raised. First, the moderate heterozygosity found can be explained as an effect of poaching, a common activity in our study area (Gómez et al., 2014). Several studies have reported reduced levels of genetic diversity in a variety of exploited species of mammals, including otters (McCullough et al., 1996; Whitehouse & Harley, 2001; Larson et al., 2002; Martínez et al., 2002; Nabata et al., 2004; Nyström et al., 2006). Another explanation would emerge from the central–marginal hypothesis (Carson, 1959; Eckert et al., 2008), which predicts the lowest levels of genetic diversity at the edge of a species geographic distribution, a situation experienced by the Lower Delta otter population.
Relatedness
The overall degree of relatedness between river otter dyads in our study area was low for Queller & Goodnight (1989) estimator (r = −0.044) and slightly higher for the maximum likelihood method (r = 0.102). Weber et al. (2009), calculating Queller & Goodnight (1989) coefficients, found low levels of parentage, ranging from r = −0.285 to r = 0.055, in two river systems of southern Brazil. Trinca et al. (2013), using maximum likelihood coefficients, found a global value of parentage of r = 0.101. They also found that otters on each of the three streams analysed tended to be more closely related among themselves than to those from other streams and that mother–daughter dyads were more often found within the same stream system than among them. Our results showed relatedness values slightly higher than previously reported ones, in accordance with lower levels of observed and expected heterozygosity, and a higher, although still low, F IS value. Even if the overall degree of relatedness between river otter dyads in the Lower Delta population is low, we found a 20% of individuals with a high degree of relatedness. The use of measures of relatedness among individuals inhabiting the same area (i.e. the spatial distribution of close relatives) to infer local reproduction has been effectively used in the past (Peery et al., 2008; Ott, 2009; Guertin et al., 2012). In our study area, almost 20% of dyads showed r values higher than 0.25, suggesting the existence of local reproduction. These high values of relatedness may be a result of low effective population size (i.e. the number of breeding otters) in the Lower Delta.
Genetic traits and habitat suitability
As expected, we found a positive association between habitat quality and levels of heterozygosity, both at landscape and regional scale, suggesting that populations living in highly suitable habitats are genetically more variable. Results of our previous work at landscape scale (Gómez et al., 2014), in which environmental factors were associated to habitat suitability in the Lower Delta of the Paraná River, showed that habitat suitability was higher in areas with polders. We proposed three explanations for this positive association: areas with polders might provide more stable environments due to protection against flooding than some natural areas of the delta; they are better protected from poaching; and/or they increase landscape connectivity. Some of these hypotheses could also explain the positive relationship between habitat quality and level of heterozygosity. Reduced poaching has been previously related to recovery of genetic diversity in the sea otter Enhydra lutris (Larson et al., 2002), while high connectivity has been related with adequate levels of genetic diversity to maintain viability in the Mexican population of L. longicaudis (Ortega et al., 2012). In this context, it is expected that otters inhabiting highly suitable areas would show higher genetic diversity, as appears to occur in the Lower Delta. At regional scale, Lopes-Rheingantz et al. (2014) found that the highest values of habitat suitability were associated with high annual temperatures and low human population density. Higher genetic diversity in places with lower human density would also be related to reduced poaching and reduced fragmentation (i.e. higher connectivity), as occurs at landscape scale.
No association was found between habitat suitability and the other measures of genetic diversity (i.e. number of alleles per locus and allelic richness) at landscape and regional scale or between habitat suitability and inbreeding and parentage at landscape scale. One possibility that could explain the first finding is the failure to detect rare alleles in cells/populations with high suitability, due to the small sampling site in each cell/population. In fact, the mean number of alleles per locus and allelic richness were similar between cells/populations with the highest and lowest suitability. The lack of association between habitat suitability and inbreeding/parentage suggest that mating between relatives is not common in low-quality cells. However, the fact that 20% of all individuals have high relatedness coefficients suggests that inbreeding is indeed present.
Even when the present work is based on a limited sample size to obtain comprehensive information on the population status of L. longicaudis in the Lower Delta of Paraná River, it constitutes the first approximation to the genetic study of the species in Argentina and complements other genetic studies carried out at the regional level. Our results showed that levels of heterozygosity found in the Lower Delta, a highly impacted wetland, are the lowest reported to date for the species. The lowest values of genetic diversity, both at landscape and regional scales, were related with low-quality habitat patches, in which connectivity and protection against poaching is reduced. Data obtained in the present study provide information for the development of conservation plans and serve as a baseline to assess future changes in the genetic diversity of L. longicaudis population in the Lower Delta.
References
Alho, C. J. R., 2008. Biodiversity of the Pantanal: response to seasonal flooding regime and to environmental degradation. Brazilian Journal of Biology 68: 957–966.
Allendorf, F. W., G. H. Luikart & S. N. Aitken, 2013. Conservation and the Genetics of Populations, 2nd ed. Willey-Blackwell, Oxford.
Avise, J. C., 2004. Molecular Markers, Natural History, and Evolution. Sinauer Associates Inc, Sunderland.
Beheler, A. S., J. A. Fike, M. L. Murfitt, O. E. Rhodes Jr & T. S. Serfass, 2003. Development of polymorphic microsatellite loci for North American river otters (Lontra canadensis) and amplification in related Mustelids. Molecular Ecology Resources 4: 56–58.
Beheler, A. S., J. A. Fike, G. Dharmarajan, O. E. Rhodes Jr & T. L. Serfass, 2005. Ten new polymorphic microsatellite loci for North American river otters (Lontra canadensis) and their utility in related mustelids. Molecular Ecology Resources 5: 602–604.
Blascher, C., 1987. Ocorrência e preservação de Lutra longicaudis (Mammalia: Mustelidae) no litoral de Santa Catarina, Vol. 22. Boletim da Fundação Brasileira para Conservação da Natureza, Rio de Janeiro: 105–117.
Bó, R., 2005. Situación ambiental en la Ecorregión Delta e Islas del Paraná. In Brown, A., U. Martínez Ortíz, M. Acerbi & J. Corcuera (eds), La situación ambiental argentina. Fundación vida silvestre argentina, Buenos Aires: 130–174.
Bonetto, A., 1986. The Paraná River system. In Davis, B. R. & K. F. Walker (eds), The Ecology of River Systems. Junk Publication, Dordrecht: 541–544.
Bradley, B. J. & L. Vigilant, 2002. False alleles derived from microbial DNA pose a potential source of error in microsatellite genotyping of DNA from faeces. Molecular Ecology Notes 2: 602–605.
Carson, H. L., 1959. Genetic conditions which promote or retard the formation of species. Cold Spring Harbor Symposia on Quantitative Biology 24: 87–105.
Cassini, M. H., 2011. Ecological principles of species distribution models: the habitat matching rule. Journal of Biogeography 38: 2057–2065.
Cassini, M. H., 2013. Distribution ecology-from individual habitat use to species biogeographical range. Springer, New York.
Caudron, A. K., S. S. Negro, C. G. Müller, L. J. Boren & N. J. Gemmell, 2007. Hair sampling and genotyping from hair follicles: a minimally-invasive alternative for genetics studies in small, mobile pinnipeds and other mammals. Marine Mammal Science 23: 184–192.
Centrón, D., B. Ramírez, L. Fasola, D. W. MacDonald, C. Chehebar, A. Schiavini & M. H. Cassini, 2008. Diversity of mtDNA in Southern River Otter (Lontra provocax) from Argentinean Patagonia. Journal of Heredity 99: 198–201.
Chemes, S. B., A. R. Giraudo & G. Gil, 2010. Dieta de Lontra longicaudis (Carnivora, Mustelidae) en el Parque Nacional El Rey (Salta, Argentina) y su comparación con otras poblaciones de la cuenca del Paraná. Mastozoología Neotropical 17: 19–29.
Dallas, J. F. & S. B. Piertney, 1998. Microsatellite primers for the Eurasian otter. Molecular Ecology 7: 1247–1263.
Dallas, J. F., K. E. Coxon, T. Sykes, P. R. Chanin, F. Marshall, D. N. Carss, P. J. Bacon, S. B. Piertney & P. A. Racey, 2003. Similar estimates of population genetic composition and sex ratio derived from carcasses and faeces of Eurasian otter Lutra lutra. Molecular Ecology 12: 275–282.
Dinerstein, E., D. M. Olson, D. J. Graham, A. L. Webster, S. A. Primm, M. P. Bookbinder & G. Ledec, 1995. A conservation assessment of the terrestrial ecoregions of Latin America and the Caribbean. The World Bank, Washington, DC.
Eckert, C. G., K. E. Samis & S. C. Lougheed, 2008. Genetic variation across species geographical range: the central-marginal hypothesis and beyond. Molecular Ecology 17: 1170–1188.
Elith, J. & J. R. Leathwick, 2009. Species distribution models: ecological explanation and prediction across space and time. Annual Review of Ecology, Evolution, and Systematics 40: 677–697.
Emmons, L. & F. Feer, 1997. Neotropical rainforest mammals: a field guide. University of Chicago Press, Chicago.
Fahrig, L., 2003. Effects of habitat fragmentation on biodiversity. Annual Review of Ecology, Evolution, and Systematics 34: 487–515.
Falush, D., M. Stephens & J. K. Pritchard, 2003. Inference of population structure using multilocus genotype gata: linked loci and correlated allele Frequencies. Genetics 164: 1567–1587.
Frankham, R., J. D. Ballou & D. A. Briscoe, 2010. Introduction to Conservation Genetics, 2nd ed. Cambridge University Press, Cambridge.
Freeland, J. R., 2005. Molecular Ecology. Wiley, Chichester.
Foerster, K., K. Delhey, A. Johnsen, J. T. Lifjeld & B. Kempenaers, 2003. Females increase offspring heterozygosity and fitness through extra-pair matings. Nature 425: 714–717.
Gallo-Reynoso, J. P., 1989. Distribución y estado actual de la nutria o perro de agua (Lutra longicaudis annectens, Major, 1897) en la Sierra Madre del Sur, México. MSc Thesis, Facultad de Ciencias, Universidad Nacional Autónoma de México, México D.F. México.
Gómez, J. J., J. I. Túnez, N. Fracassi & M. H. Cassini, 2014. Habitat suitability and anthropogenic correlates of Neotropical river otter (Lontra longicaudis) distribution. Journal of Mammalogy 95: 824–833.
Goudet, J., M. Raymond, T. de Meeüs & F. Rousset, 1996. Testing differentiation in diploid populations. Genetics 144: 1933–1940.
Guertin, D. A., M. Ben-David, A. S. Harestad & J. E. Elliott, 2012. Fecal genotyping reveals demographic variation in river otters inhabiting a contaminated environment. The Journal of Wildlife Management 76: 1540–1550.
Guisan, A. & W. Thuiller, 2005. Predicting species distribution: offering more than simple habitat models. Ecology Letters 8: 993–1009.
Hájkova, P., B. Zemanova, J. Bryja, B. Hajek, K. Roche, E. Tkadlec & J. Zima, 2006. Factors affecting success of PCR amplification of microsatellite loci from otter faeces. Molecular Ecology Notes 6: 559–562.
Hedrick, P. W., 2011. Genetics of Populations, 4th ed. Jones & Bartlett Publishers, Sudbury, MA.
Heikkinen, R. K., M. Luoto, B. Araújo, R. Virkkala, W. Thuiller & M. T. Sykes, 2006. Methods and uncertainties in bioclimatic envelope modelling under climate change. Progress in Physical Geography 30: 751–777.
Hoffman, J. I. & W. Amos, 2005. Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion. Molecular Ecology 14: 599–612.
Huang, C. C., Y. C. Hsu, L. L. Lee & S. H. Li, 2005. Isolation and characterization of tetramicrosatellite DNA markers in the Eurasian otter (Lutra lutra). Molecular Ecology Resources 5: 314–316.
Kalesnik, F. & R. Quintana, 2006. The Paraná River Delta as a wetland mosaic. Study case: the Reserve of Biosphere MAB-UNESCO “Paraná Delta”. Revista Geociências-UnG 5: 22–37.
Kalinowski, S., A. Wagner & M. Taper, 2006. ML-Relate: a computer program for maximum likelihood estimation of relatedness and relationship. Molecular Ecology Notes 6: 576–579.
Kalz, B., K. Jewgenow & J. Fickel, 2006. Structure of an otter (Lutra lutra) population in Germany: results of DNA and hormone analyses from faecal samples. Mammalian Biology 71: 321–335.
Kruuk, H., 2006. Otters. Ecology, Behaviour and Conservation. Oxford University Press, Oxford.
Lacy, R. C., 1997. Importance of genetic variation to the viability of mammalian populations. Journal of Mammalogy 78: 320–335.
Lampa, S., B. Gruber, K. Henle & M. Hoehn, 2008. An optimisation approach to increase DNA amplification success of otter faeces. Conservation Genetics 9: 201–210.
Larivière, S., 1999. Lontra longicaudis. Mammalian Species 609: 1–5.
Larson, S., R. Jameson, J. Bodkin, M. Staedler & P. Bentzen, 2002. Microsatellite DNA and mitochondrial DNA variation in remnant and translocated sea otter (Enhydra lutris) populations. Journal of Mammalogy 83: 893–906.
Long, R., P. MacKay, W. Zielinski & J. Ray, 2008. Non-invasive Survey Methods for Carnivores. Island Press, Washington, DC.
Lopes-Rheingantz, M., J. F. Saraiva de Menezes & B. de Thoisy, 2014. Defining Neotropical otter Lontra longicaudis distribution, conservation priorities and ecological frontiers. Tropical Conservation Science 7: 214–229.
Luck, G. W., G. C. Daily & P. R. Ehrlich, 2003. Population diversity and ecosystem services. Trends in Ecology and Evolution 18: 331–336.
Macdonald, S. & C. Mason, 1992. A note on Lutra longicaudis in Costa Rica. IUCN Otter Speccialis Group Bulletin 7: 37–38.
Malvárez, A. I., 1997. Las comunidades vegetales del Delta del río Paraná. Su relación con factores ambientales y patrones de paisaje. PhD Thesis, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina.
Manel, S., P. Berthier & G. Luikart, 2002. Detecting wildlife poaching: identifying the origin of individuals with Bayesian assignment tests and multilocus genotypes. Conservation Biology 16: 650–659.
Martínez, J. G., J. Carranza, J. L. Fernandez-Garcia & C. B. Sanchez-Prieto, 2002. Genetic variation of red deer populations under hunting exploitation in southwestern Spain. Journal of Wildlife Management 66: 1273–1282.
McCullough, D. R., J. K. Fischer & J. D. Ballow, 1996. From bottleneck to metapopulation: recovery of the tule elk in California. In McCullough, D. R. (ed.), Metapopulation and Wildlife Conservation. Island Press, Washington, DC: 375–403.
Mills, L. S. & P. E. Smouse, 1994. Demographic consequences of inbreeding in remnant populations. The American Naturalist 144: 412–431.
Mills, L. S., K. L. Pilgrim, M. K. Schwartz & K. McKelvey, 2000. Identifying lynx and other North American felids based on mtDNA. Conservation Genetics 1: 285–288.
Mowry, R. A., M. E. Gompper, J. Beringer & L. S. Eggert, 2011. River otter population size estimation using noninvasive latrine surveys. Journal of Wildlife Management 75: 1625–1636.
Mucci, N., J. Arrendal, H. Ansorge, M. Bailey, M. Bodner, M. Delibes, A. Ferrando, P. Fournier, C. Fournier, J. A. Godoy, P. Hajkova, S. Hauer, T. M. Heggberget, D. Heidecke, H. Kirjavainen, H. Heinrich Krueger, K. Kvaloy, L. Lafontaine, J. Lanszki, C. Lemarchand, U. M. Liukko, V. Loeschcke, G. Ludwig, A. B. Madsen, L. Mercier, J. Ozolins, M. Paunovic, C. Pertoldi, A. Piriz, C. Prigioni, M. Santos-Reis, T. Sales Luis, T. Stjernberg, H. Schmid, F. Suchentrunk, J. Teubner, R. Tornberg, O. Zinke & E. Randi, 2010. Genetic diversity and landscape genetic structure of otter (Lutra lutra) populations in Europe. Conservation Genetics 11: 583–599.
Nabata, D., R. Masuda, O. Takahashi & J. Nagata, 2004. Bottleneck effects on the sika deer Cervus Nippon population in Hokkaido, revealed by ancient DNA analysis. Zoological Science 21: 473–481.
Nagaraju, S. K., R. Gudasalamani, N. Barve, J. Ghazoul, G. Kotiganahalli Narayanagowda & U. Shaanker Ramanan, 2013. Do ecological niche model predictions reflect the adaptive landscape of species?: a test using Myristica malabarica Lam., an endemic tree in the Western Ghats, India. PLoS One 8: e82066.
Nagy, S., P. Poczai, I. Cernák, A. M. Gorji, G. Hedegűs & J. Taller, 2012. PICcalc: an online program to calculate polymorphic information content for molecular genetic studies. Biochemical Genetics 50: 670–672.
Nardelli, M., J. I. Túnez, D. Centrón & M. H. Cassini, 2011. Técnicas de muestreo no invasivas aplicadas al estudio de los mamíferos. Interciencia 36: 404–411.
Nyström, V., A. Angerbjörn & L. Dalén, 2006. Genetic consequences of a demographic bottleneck in the Scandinavian Arctic fox. Oikos 114: 84–94.
O’Grady, J. J., B. W. Brook, D. H. Reed, J. D. Ballou, D. W. Tonkyn & R. Frankham, 2006. Realistic levels of inbreeding depression strongly affect extinction risk in wild populations. Biological Conservation 133: 42–51.
Ojeda, R. A., V. Chillo & G. Díaz Inserath, 2012. Libro Rojo: Mamíferos Amenazados de Argentina. Sociedad Argentina para el Estudio de los Mamíferos (SAREM), Buenos Aires.
Ortega, J., D. Navarrete & J. E. Maldonado, 2012. Non–invasive sampling of endangered neotropical river otters reveals high levels of dispersion in the Lacantun River System of Chiapas, Mexico. Animal Biodiversity and Conservation 1: 59–69.
Ott, K. E., 2009. Recolonization or local reproduction? An assessment of river otter recovery in previously-oiled areas of coastal Alaska via non-invasive genetic sampling. PhD Thesis, University of Wyoming, Laramie.
Pardini, R. & E. Trajano, 1999. Use of Shelters by the Neotropical River Otter (Lontra longicaudis) in an Atlantic Forest Stream, Southeastern Brazil. Journal of Mammalogy 80: 600–610.
Parera, A., 1993. The Neotropical River Otter Lutra longicaudis in Iberá Lagoon, Argentina. IUCN Otter Specialist Group Bulletin 8: 13–16.
Parera, A., 1996. Estimating river otter Lutra longicaudis population in Iberá Lagoon using direct sightings methodology. IUCN Otter Specialist Group Bulletin 13: 77–83.
Park, S. D. E., 2001. Trypanotolerance in west African cattle and population genetic effects of selection. PhD thesis, University of Dublin, Dublin, Ireland.
Peakall, R. & P. E. Smouse, 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28: 2537–2539.
Peery, M. Z., S. R. Beissinger, R. F. House, M. Bérubé, L. A. Hall, A. Sellas & P. J. Palsbøll, 2008. Characterizing source-sink dynamics with genetic parentage assignments. Ecology 89: 2746–2759.
Pickles, R. S. A., J. J. Groombridge, V. D. Zambrana Rojas, P. Van Damme, D. Gottelli, C. V. Ariani & W. C. Jordan, 2012. Genetic diversity and population structure in the endangered giant otter, Pteronura brasiliensis. Conservation Genetics 13: 235–245.
Prigioni, C. L., L. U. Remonti & A. L. Balestrieri, 2006. Otter Lutra lutra movements assessed by genotyped spraints in Southern Italy. Italian Journal of Mammalogy 17: 91–96.
Pritchard, J. K., M. Stephens & P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Quadros, J. & E. L. Monteiro-Filho, 2001. Diet of the neotropical otter, Lontra longicaudis, in an Atlantic forest area, Santa Catarina State, southern Brazil. Studies on Neotropical Fauna and Environment 36: 15–21.
Queller, D. C. & K. F. Goodnight, 1989. Estimating relatedness using molecular markers. Evolution 43: 258–275.
Raymond, M. & F. Rousset, 1995. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity 85: 248–249.
Rousset, F., 2008. Genepop’007: a complete reimplementation of the Genepop software for Windows and Linux. Molecular Ecology Resources 8: 103–106.
Sambrook, J., E. F. Fritsch & T. Maniatis, 1989. Molecular cloning: a laboratory manual, Vol. 3. Cold Spring Harbor Laboratory Press, New York.
Trinca, C. S., 2012. História evolutiva e ecologia molecular da Lontra Neotropical (Lontra longicaudis) (Carnivora: Mustelidae). PhD Thesis, Universidade Federal do Rio Grande do Sul, Rio Grande do Sul, Brazil.
Trinca, C. S., H. F. Waldemarin & E. Eizirik, 2007. Genetic diversity of the Neotropical otter (Lontra longicaudis Olfers, 1818) in Southern and Southeastern Brazil. Revista Brasileira de Biologia 67: 813–818.
Trinca, C. S., B. de Thoisy, F. C. W. Rosas, H. F. Waldemarin, K. P. Koepfli, J. A. Vianna & E. Eizirik, 2012. Phylogeography and demographic history of the Neotropical otter (Lontra longicaudis). Journal of Heredity 103: 479–492.
Trinca, C. S., C. F. Jaeger & E. Eizirik, 2013. Molecular ecology of the Neotropical otter (Lontra longicaudis): non-invasive sampling yields insights into local population dynamics. Biological Journal of the Linnean Society 109: 932–948.
Waits, L. P., G. Luikart & P. Taberlet, 2001. Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Molecular Ecology 10: 249–256.
Waldemarin, H. F. & R. Álvarez, 2008. Lontra longicaudis. The IUCN Red List of Threatened Species. Version 2014.3 [available on internet at www.iucnredlist.org]. Accessed on 25 Mar 2015.
Weber, L. I., C. G. Hildebrand, A. Ferreira, G. Pedarassi, J. A. Levy & E. P. Colares, 2009. Microsatellite genotyping from faeces of Lontra longicaudis from southern Brazil. Iheringia, Série Zoologia 99: 5–11.
Whitehouse, A. M. & E. H. Harley, 2001. Post-bottleneck genetic diversity of elephant populations in South Africa, revealed using microsatellite analysis. Molecular Ecology 10: 2139–2149.
Acknowledgments
This work could not have been undertaken without the help of N. Fracassi and the inhabitants of the Lower Delta. We specially thank Rubén, Clara, and Gastón and their family, for their help during field work. We also thank Andrés Pautasso, from the Museo de Ciencias Naturales “Florentino Ameghino” who kindly provided us with two tissue samples stored in the museum. We thank N.A. Pearson for his review. This work was supported by grants from the Consejo Nacional de Investigaciones Científicas y Técnicas of Argentina (PIP No. 11420100100189), the Agencia Nacional de Promoción Científica y Tecnológica of Argentina (PICT-2010-1978), and the National University of Luján (Fondos Finalidad 3.5). MHC and JIT are researchers of the CONICET.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Christian Sturmbauer
Rights and permissions
About this article

Cite this article
Trigila, A.P., Gómez, J.J., Cassini, M.H. et al. Genetic diversity in the Neotropical river otter, Lontra longicaudis (Mammalia, Mustelidae), in the Lower Delta of Parana River, Argentina and its relation with habitat suitability. Hydrobiologia 768, 287–298 (2016). https://doi.org/10.1007/s10750-015-2557-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-015-2557-x