
Abstract
Faeces have proved to be a suitable non-invasive DNA source for microsatellite analysis in wildlife research. For the success of such studies it is essential to obtain the highest possible PCR amplification success rate. These rates are still relatively low in most carnivorous species, especially in the otter (Lutra lutra). We therefore optimised the entire microsatellite genotyping process by combining our findings with results from previous studies to gain a high rate of reliable genotypes. We investigated the influence of otter faecal quality in relation to the quantity of slimy secretions and three levels of storage periods at −20°C on amplification success. Further, we tested the cost-effective and time-saving Chelex extraction method against the profitable QIAamp® DNA Stool Kit (Qiagen), and compared three PCR methods - a standard single-step PCR protocol, a single-locus two-step PCR procedure and a multiplex two-step PCR procedure - regarding success rate and genotyping errors. The highest amplification success rate (median: 94%; mean: 78%) was achieved using faecal samples consisting only of jelly extracted with the QIAamp® DNA Stool Mini Kit (Qiagen) immediately after collection and amplified following the time and cost efficient multiplex two-step PCR protocol. The two-step procedure, also referred to as pre-amplification approach, turned out to be the main improvement as it increases amplification success about 11% and reduces genotyping errors about 53%, most notably allelic dropouts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microsatellite genotyping of non-invasive DNA sources like faeces is a novel and increasingly applied approach to analyse the genetic structure of species. So far it is the only available technique to study population structure, population size, genetic diversity, and relatedness of elusive animals (Amos and Pemberton 1992; Bruford and Wayne 1993; Queller et al. 1993; Kohn and Wayne 1997; Reed et al. 1997) such as otters (Lutra lutra).
However, faecal samples typically contain low quantities of target DNA in a bacterial-enriched environment that includes PCR-inhibitors (Sidransky et al. 1992; Tschirch 1995; Murphy et al. 2000) and is likely exposed to hydrolytic, oxidative, and enzymatic degradation (Kohn et al. 1995; Frantzen et al. 1998; Idaghdour et al. 2003). Thus, the success of a microsatellite analysis is significantly influenced by the age of the scat (Jansman et al. 2001; Dallas et al. 2003) and the exposure to weather conditions (Farrell et al. 2000; Murphy et al. 2000). It has been demonstrated that diet also affects the amplification success rate strongly being high for herbivorous species (Flagstad et al. 1999; Banks et al. 2002), intermediate for omnivorous species (Gerloff et al. 1995; Goossens et al. 2000; Frantz et al. 2003), and usually rather low in studies with carnivores (Reed et al. 1997; Kohn et al. 1999; Piggott and Taylor 2003). The first microsatellite studies analysing otter faeces from wild populations obtained amplification success rates of only 20% (Coxon et al. 1999; Dallas et al. 2003), which is close to the lower end even for carnivorous species. Such a low amplification success reduces considerably the suitability of faecal samples for genetic studies in this species. Therefore, it is of paramount importance to optimise the genetic techniques to obtain an adequate amplification success rate. The first attempt to optimise microsatellite analysis for the otter was presented recently (Hájková et al. 2006). Hájková et al. (2006) investigated the impact of collection temperature and sample type on amplification success, tested three buffers and ethanol for their efficiency in preservation and compared the extraction results of two similar stool kits (Qiagen, Invitek). In our approach we tried to systemise the optimisation by breaking down the whole procedure of a microsatellite analysis into its relevant parts. Four successive steps, each depending on the previous one, are crucial for microsatellite analyses: (1) the collection of samples, (2) the subsequent storage method, (3) the extraction of DNA, and (4) the amplification of DNA using PCR (polymerase chain reaction). In this study, we concentrated our optimisation effort on each of the four steps.
Hájková et al. (2006) found a pronounced effect of sample quality. However, even within the same species sample quality can vary depending on study area, diet, time of year, and also on the microsatellite loci and amplification protocol used. Therefore, we tested the influence of faecal quality on the amplification success as well. Hájková et al. (2006) reported that storage time had no effect on DNA amplification success. This is in stark contrast to a number of studies (Frantzen et al. 1998; Murphy et al. 2002) and needs further investigation. Hence, we studied the impact of storage periods on the success rate using three different levels of storage time at −20°C (one day/one week/two weeks). Although it has been shown that for the extraction of faecal DNA stool kits (e.g. Qiagen) produce high success rates (e.g. Goossens et al. 2000; Roeder et al. 2004; Hájková et al. 2006), they are also the most expensive. We examined whether the cheap and time-saving Chelex method can achieve comparable results and could be used instead. Finally, to include recent advances in PCR techniques, we optimised the PCR conditions of single-locus and multiplex PCR and investigated whether a two-step amplification approach (similar to Piggott et al. 2004; Hedmark and Ellegren 2006) yielded higher amplification success rates and lower genotyping errors than standard PCR.
Materials and methods
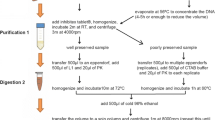
Sample collection
Spraint and anal jelly samples were collected from a wild otter population in Upper Lusatia, Saxony, Eastern Germany. Collections were made mainly during the morning hours on days without rain or frost. In all trials only freshly deposited faeces from the previous night were used. The external layer of the spraint which contains sloughed gut cells was wiped off with a commercially available cotton swab. Each cotton swab sample was stored in a separate sterile 500 ml tube. A pilot study demonstrated that this technique decreases the risk of sampling prey hard parts, such as bones or fish scales, while increasing the proportion of sloughed gut cells sampled. It is also manageable in the field, and has a reduced risk of contamination while maintaining a sufficient amplification success rate relative to other methods.
To test for the effect of the sample quality on the PCR amplification, 20 samples were classified into three types of faeces according to the quantity of slimy secretions: spraint (consisting of prey remains and almost no mucus), spraint plus mucus (consisting of prey remains and a layer of mucus), and jelly (gelatinous secretion of anal scent glands without prey remains). Supplementary data, such as weather conditions and collection time, were recorded to test these factors for correlation with DNA amplification success. Faecal samples were extracted with the most reliable extraction method (see section "Comparison of extractions"), PCR amplified at six loci using several PCR protocols, and separated in an ABI PRISM® 3100 Genetic Analyser (see section "PCR amplification").
Storage time
The effect of storage time on genotyping success was tested using 15 faecal samples. Three swabs were taken from each faecal sample at three different positions of the surface using a separate cotton swab each time. All swabs were frozen at −20°C in a 500 ml tube within 10 h of sampling. Amplification success could thus be tested for each scat for each of the three storage times: (1) one day, (2) one week, and (3) two weeks. To avoid any methodological bias the order of the subsamples was randomised before proceeding with the following steps. DNA from all 45 subsamples was extracted with the most reliable extraction method (see section "Comparison of extractions"). Eight microsatellite loci with fragment length 127–211 bp (Lut 435, 457, 604, 615, 701, 717, 733, 832; Dallas and Piertney 1998; Dallas et al. 2002) were amplified following the single-locus two-step PCR protocol (see section "DNA amplification"; annealing temperature for Lut 717: 57°C; Lut 832: 55°C), and separated by electrophoresis on 1.75% agarose gels in TBE buffer (68.5 mM Tris HCl, 89 mM boric acid, 2.5 mM EDTA). DNA was ethidium bromide stained and visualised using BIO-RAD Gel Doc 1000.
DNA extraction
DNA extractions were carried out in a separate laboratory that was free of concentrated otter DNA or PCR products. Aerosol resistant pipette tips were used in all working steps. Negative controls were included in each extraction to monitor contamination.
Two different extraction methods were tested: The Chelex® 100 method is a very fast, simple, and cost-effective technique that has been used in previous studies to isolate DNA from hair (Walsh et al. 1991; Vigilant 1999; Frantz et al. 2004) and faeces (Paxinos et al. 1997; Reed et al. 1997; Palomares et al. 2002; Berry and Sarre, unpublished). In contrast, the QIAamp® DNA Stool Mini Kit (Qiagen) is more time-consuming and costly but produces relatively high-quality template DNA. The Qiagen kit is based on the GuSCN/silica method (Frantz et al. 2003) and has been applied to faecal DNA extractions several times (Goossens et al. 2000; Morin et al. 2001; Frantz et al. 2003; Nsubuga et al. 2004; Roeder et al. 2004).
The Chelex extraction protocol involved an initial wash with 0.75 ml PBS puffer (pH 7.4), which was added to the cotton swab in the 500 ml tube and homogenised by vortexing. 500 μl of the supernatant was transferred to a fresh tube containing 500 μl 10% H2O-Chelex-solution and 4 μl of proteinase K (10 mg/ml) was added. Samples were then vortexed before incubation over night at 55°C with rotation. The following day samples were briefly vortexed, then boiled for 20 min followed by a 5 min centrifugation at 16,000g. The supernatant was removed into a new tube and centrifuged again for 5 min at 16,000g before the supernatant was transferred into a fresh tube and stored at −20°C.
The Qiagen kit extraction was carried out according to the manufacturer’s instructions except for the initial steps. Here the cotton swab was suspended in 1.7 ml of ASL buffer (warmed to 70°C) in the 500 ml tube and vortexed for 20 s. After 2 min of incubation at room temperature the extraction was performed as from step 4 of the manufacturer’s instructions.
DNA was extracted on the day of collection from 47 faecal samples, which were each wiped at two different positions with separate cotton swabs to allow a comparison of both extraction methods for the same scat. Only samples for which at least three microsatellites could be successfully amplified were included in the final comparison, this was achieved for 24 samples. Amplification was carried out with six microsatellites (Lut 435, 457, 604, 615, 701, 733; Dallas and Piertney 1998; Dallas et al. 2002) following the single-locus two-step PCR protocol (see section "DNA amplification"). PCR products were initially screened by agarose gel electrophoresis and only successfully amplified samples were genotyped on ABI PRISM® 3100 Genetic Analyser (Applied Biosystems).
PCR amplification
PCRs were prepared using aerosol resistant pipette tips in a DNA UV-cleaner box and all reactions included a PCR negative control.
In a pilot study 12 microsatellites designed by Dallas and Piertney (1998) and Dallas et al. (2002) (Lut 435, 457, 604, 615, 701, 715, 717, 733, 782, 818, 832, 833) were tested for their amplification success rate, allelic richness and heterozygosity in otter faecal DNA originating from Saxony. Three of the 12 markers had a very low amplification success rate (Lut 782: 14,9%; Lut 818: 12,8%; Lut 833: 10,6%). Moreover, Lut 782 turned out to be monomorphic.
Six of the remaining nine microsatellites had the same optimal annealing temperature (58°C) and were hence suitable for multiplex PCR. Conditions were optimised for single and multiplex PCR of these six loci (combinations: Lut 435, 604, 701; Lut 457, 615, 733) based on the original PCR conditions described in Dallas et al. (1999). The major difference between the original standard PCR protocol and the optimised single-locus and multiplex protocols described here is that two consecutive PCR reactions were carried out, with PCR product from the first amplification being used as the template for a second PCR reaction. During the single-locus two-step procedure only one locus was amplified per reaction, whereas the multiplex two-step approach contained the primers for three markers in both (first and second) PCR steps. Furthermore, compared to the original PCR protocol the optimised protocols included higher (Taq DNA Polymerase, primers) and lower (MgCl) concentrations of PCR reagents, longer reaction times during amplification (30 s vs. 15 s), the use of locus-specific annealing temperatures and a greater extent of cycles (first PCR: 45, second PCR: 40 vs. 35). To test the effect of performing two consecutive PCRs, the product from the first amplification was also genotyped for all samples identified as positive using the single-locus PCR protocol.
All three protocols (original, single-locus, multiplex) were performed in 25 μl volumes containing 3 μl of DNA extract (6 μl of PCR product for the second PCR). The final reaction concentrations for both single-locus and multiplex PCRs consisted of 1 × reaction buffer (Taq PCR Core Kit, Qiagen) with 1.5 mM MgCl2, 200 μM of each dNTP, 0.6 μM of each primer (0.4 μM for the second PCR) and 0.5 units Taq Polymerase (Taq PCR Core Kit). Whereas the original protocol uses a touch-down profile, we amplified at the locus-specific annealing temperature. The PCR profile was: initial 2 min 15 s at 90°C, and then cycles of 30 s at 90°C, 30 s at 58°C and 30 s at 72°C. The first PCR was replicated for 45 cycles and the second PCR for 40 cycles. Amplification ended with a final extension at 72°C for 1 min. Reactions were carried out in a BIOMETRA T3 Thermocycler. Forward primers were end-labelled at the 5′-end with a fluorescent dye and the pigtail ‘GTTGCTT’ was added to the 5′-end of reverse primers to enhance the 3′ adenosine overhang. This avoids typing error due to variability in non-templated nucleotide addition at the 3′-end of PCR products (Brownstein et al. 1996).
DNA from 20 faecal samples was extracted immediately after collection using the most reliable extraction method (see section "Comparison of extractions") and stored at −20°C. For each of these 20 samples, three PCRs were performed with the six loci, giving a total of 60 PCRs per amplification protocol tested. PCR products were separated and visualised in an ABI PRISM® 3100 Genetic Analyser (Applied Biosystems) and analysed using ABI Prism® GeneMapper\({\texttrademark}\) Software V.3.0.
In all comparisons PCR amplification success rate was used as an indicator for the quality of the particular method and was estimated as the median percentage of successfully amplified products either over all samples per locus (see sections "Effect of storage time", "Comparison of extraction", "Comparison of amplification protocols") or over all loci per sample (see sections "Sample collection", "Comparison of amplification protocols"). Due to sample sizes between 15 and 24, non-parametric tests (Kruskal–Wallis test, multiple comparisons and Wilcoxon test) were used to test for significant differences between methods (P = 0.05 after Bonferroni adjustments for multiple testing, thus in three tests (see sections "Sample collection", "Effect of storage time", "Comparison of amplification protocols") the level of significance is P = 0.0167).
In addition, the three PCR protocols (original, single-locus, multiplex) were evaluated by comparing the rate of false alleles and allelic dropout. Due to three replications of each sample with each PCR protocol (nine PCRs per locus and sample in total) the criteria established by Frantz et al. (2003) could be used to obtain reliable genotypes for each successfully amplified sample. Therefore, genotyping errors were ascertained by comparing scored genotypes with the reliable genotype. False alleles can occur in all positive samples, whereas allelic dropout can only be detected in positive heterozygous samples (Creel et al. 2003; Broquet and Petit 2004). Hence, both rates were estimated considering only such genotypes.
Results
Sample collection
There was no correlation between collection time (within the first 20 h after defecation) and amplification success rate in any of the three trials (storage time, extraction, amplification) (Kruskal–Wallis test: H (2, n = 59) = 2.82, P = 0.24). Due to constant weather conditions during all sample collections, predictions about weather effects on PCR amplification could not be made. Rather, the quantity of slimy secretions influenced amplification success significantly in all three PCR protocols (Kruskal–Wallis test: original: H (2, n = 20) = 9.92, P = 0.0070; single-locus: H (2, n = 20) = 9.42, P = 0.0090; multiplex: H (2, n = 20) = 11.13, P = 0.0038). PCR amplification success was low for spraint and spraint plus mucus samples but high for samples that consisted only of jelly (Table 1). Pairwise comparisons using multiple comparisons showed a significant increase in amplification success rate for jelly samples compared with spraint samples. Compared with spraint plus mucus samples only the multiplex protocol achieved a significantly increased success rate for jelly samples (Table 1).
Effect of storage time
On average, DNA was obtained from 80% of the samples (n = 15) extracted after one day, while only 63% and 60% of the same faecal samples (n = 15 each) that were extracted after one or two weeks amplified successfully (represented as bands in agarose gel, Fig. 1). Thus, by increasing the storage time we observed a significant decrease in amplification success rate, whereby the highest decline was detected after one week (Wilcoxon tests: 1 day–1 week: Z = 2.366, P = 0.018; 1 day–2 weeks: Z = 2.521, P = 0.012; 1 week–2 weeks: Z = 1.400, P = 0.161).

Comparison of amplification success rate of three different storage times at −20°C. Results are from 45 subsamples of 15 faecal samples that were extracted using the Qiagen kit one day, one week, or two weeks after storing at −20°C. Each sample from each treatment was amplified once with eight microsatellite loci. The percentage of successful amplification (detected as bands in an agarose gel) for each locus in each storage time was calculated by pooling the data across the 15 samples. Boxes represent the interquartile range with the median square, while error bars signify outliers
Comparison of extractions
As shown in Fig. 2, the two extraction methods differ significantly in amplification success rate (Wilcoxon test: Z = 2.201, P = 0.028). Also, differences between extraction methods in successful PCRs were highly significant for each microsatellite loci (Fisher test: P < 0.001, df = 1). All six loci amplified in 17 out of 24 (70.8%) kit-extracted samples, whereas none of the Chelex-extracted samples yielded positive amplifications at all six loci simultaneously. Although positive extracts purified with the Chelex method often showed higher signal intensity as the same kit-extracted sample, some negative Chelex-extracted faeces did not even display primer dimers, which indicate the presence of PCR inhibitors (Kohn et al. 1995; Reed et al. 1997; Vigilant 1999; Palomares et al. 2002). Potential PCR inhibition, caused by components in faecal extracts, were tested in supplementary assays by amplifying tissue DNA (already successfully amplified in former PCRs) in which kit- or Chelex-extracted faecal DNA were added. Negative amplifications were only noted from mixtures of tissue and Chelex-extracted faecal DNA, whereas all mixtures containing tissue and kit-extracted faecal DNA resulted in positive PCRs.

Median of the amplification success rate of two extraction methods tested with 24 subsamples respectively. One amplification with six microsatellite loci was carried out for each sample and each extraction method. The amplification success rate (represented as bands in an agarose gel and in an ABI PRISM® 3100 Genetic Analyser) was obtained by averaging the positive samples over six loci. Variance across loci is illustrated by a box plot chart. Boxes represent the interquartile range with the median square, while error bars signify outliers
Comparison of amplification protocols
Positive amplification products occurred in 27% of faecal samples amplified using the original protocol compared with 51% using the optimised single-locus protocol, and 47% using the multiplex protocol (Fig. 3). Thus, the optimised single-locus two-step PCR protocol showed a near significant improvement in amplification success compared to the original protocol (Wilcoxon tests: Single-locus–Original: Z = 2.201, P = 0.028; Multiplex–Original: Z = 2.201, P = 0.028). Between the single-locus and the multiplex protocol no significant difference was found (Wilcoxon test: Z = 0.944, P = 0.345). Additionally, the percentage of samples, in which a genotype could be obtained at least once for all six loci simultaneously, declined from 40% using the multiplex protocol to 35% using the single-locus protocol to finally 25% using the original protocol, thus highlighting the improved PCR conditions (Fig. 4). A similar pattern was noted when the number of positive replicates over all samples and microsatellite loci was considered (Fig. 4).

Amplification success rate of three PCR protocols obtained by amplifying 20 faecal samples in triple replication with six microsatellite loci. The percentages of successful amplification per treatment were calculated as the median of all positive samples across each loci. Boxes represent the interquartile range with the median square, while error bars signify outliers

Amplification success rate of three PCR protocols (original, single-locus, multiplex) regarding several criteria: (1) samples with at least one positive amplification in all six loci, (2) at least one positively amplified replicate over all samples and loci, (3) at least two positively amplified replicates over all samples and loci, (4) three positively amplified replicates over all samples and loci. Data obtained by amplifying 20 faecal samples in triplicate with six microsatellite loci
When considering genotyping errors, false alleles differed between 0% following the original or the optimised single-locus protocol and 0.02% using the multiplex protocol. Allelic dropout varied from 13 to 56% in the original protocol (median 27%), from 16 to 48% in the single-locus protocol (median 38%), and from 24 to 42% in the multiplex protocol (median 29%). In all positive amplification products amplified according to the original protocol, the signal intensity was conspicuously reduced compared to those of the optimised protocol, making precise allele detection much more complicated. Furthermore, separation and visualisation by the less sensitive electrophoresis in agarose gels frequently failed due to low peak size (often < 50 units).
Of each successfully amplified sample using the single-locus two-step protocol the first PCR product was also genotyped. Compared to the first PCR, the second amplification showed an about 11% increase in success rate that ranged from 6 to 28% across samples. Moreover, the appearance of genotyping errors (mainly allelic dropouts, 46 of 47) decreased about 53% after performing the second amplification ranging from 14% to 100% between loci. From all samples that generated a genotyping error during the first PCR only 47% repeated this error after the second amplification. Only once a sample showed artefacts after performing the second amplification, although the first PCR amplified the reliable genotype.
Discussion
In order to maximise the amplification success of DNA from otter faeces, we investigated the effect of four factors: faecal quality, storage period at −20°C, extraction methods, and PCR conditions. All four factors had a strong influence on the amplification success rate.
The first crucial step of using faeces for microsatellite analysis is the sample collection which is related to faecal quality. The freshness of faeces is a determining factor for PCR amplification success (Jansman et al. 2001; Dallas et al. 2003), hence only freshly deposited scats from the previous night (up to 20 h after defecation) were collected. However, within these approximately 20 h after defecation, we could not detect a decline in amplification success rate. Comparable results were obtained by Coxon et al. (1999) and Hájková et al. (2006), who also failed to detect significant differences within the first hours after defecation. In addition, the quantity of slimy secretions is an important influencing factor. We found that samples consisting only of jelly showed a very high PCR success rate, whereas PCR success rate was low for samples of spraints with or without mucus. This result confirms previous studies that deal with the amplification success of faecal DNA from otters (Coxon et al. 1999; Hájková et al. 2006) and seals (Reed et al. 1997). Anal jelly may contain less PCR inhibitors, bacteria, and enzymes than spraint that is composed mainly of prey remains. One reason, why spraint plus mucus samples achieved comparable low amplification success rates as spraint samples, may be that the mucilage layer on spraint cannot be equated with the secretion of anal scent glands called jelly. In the large intestine mucus, secreted from goblet cells, is used as a lubricant for faeces which must pass over membranes (Liebich 1999; Welsch 2006). Whereas the secretion of anal scent sacs either act as visual and olfactory stimuli used in the social organisation of the population (Gorman et al. 1978; Macdonald and Mason 1987) or is of gastric origin being produced when otters have not eaten for 18–24 h (Conroy and French 1991; Carss and Parkinson 1996). Hájková et al. (2006) chose the classification (i) spraint, (ii) spraint with jelly, (iii) jelly and equated consequently the thin layer of mucus on spraint with the anal jelly. In contrast, we distinguish in our study between these two types of slimy secretion, which is supported by the high difference in amplification success between spraint plus mucus and jelly samples. Also, climatic conditions can have a high impact on the amplification rate of faeces (Farrell et al. 2000; Murphy et al. 2000). Humidity, for instance, might provide a better microclimate for bacteria and enzymes, while longer periods of rainy weather might wash away cells from the surface of the scat. Reduced amplification success rate in wet periods were detected for faeces from carnivores in western Venezuela (Farrell et al. 2000). During our pilot study we were able to observe the same findings. Hence, we only sampled faeces during periods of dry diurnal weather (i.e. without rain or frost). Finally, the collection method itself might interfere with the amplification success. It has been demonstrated that homogenisation of faecal samples yields reduced PCR success rates compared to surface wash or homogenisation of surface scrape (Piggott and Taylor 2003). In light of this and the results of our pilot study we used commercially available cotton swabs for the sample collection.
Storing faecal samples in a freezer at −20°C is a frequently practised method that should provide protection against further degradation (Tikel et al. 1996; Reed et al. 1997; Frantzen et al. 1998; Wasser et al. 1997; Ernest et al. 2000; Frantz et al. 2003; Piggott and Taylor 2003). Therefore, we considered this storage method to be suitable in combination with our collection technique as reagents or buffer solutions may remove cells from the cotton swab. However, we could show that amplification success rate decreased drastically with increased storage time (i.e. 20% within two weeks). As a result, we recommend that DNA extraction should be performed immediately after collection. Degradation during the thawing process is an unlikely reason for this effect since all subsamples were treated in the same manner. In contrast to our study, Hájková et al. (2006) recently reported that PCR success rate did not decline in otter faecal samples that were preserved up to 234 days in a freezer at −20°C, stored in 96% ethanol or buffers of kits. Our results demonstrated that the decline of amplification success rate was highest after one week of storage. Afterwards the decline was rather low. A possible explanation for the results of Hájková et al. (2006) may be that they extracted most of the samples after the first week of storing. However, contrasting results may also have occurred because samples were stored in a buffer solution which may preserve faecal samples over an intermediate period of time.
In addition to collection and preservation, the extraction of samples is the third factor strongly influencing quality and quantity of template DNA (Wasser et al. 1997; Flagstad et al. 1999; Banks et al. 2002). The efficiency of DNA purification can vary greatly among species and even among individuals (Taberlet and Luitkart 1999; Gossens et al. 2000; Piggott and Taylor 2003). Due to low DNA concentration in faeces (Gerloff et al. 1995; Murphy et al. 2000) and a high proportion of PCR inhibitors, nucleases, bacteria, and enzymes (Deuter et al. 1995; Kohn et al. 1995; Reed et al. 1997; Frantzen et al. 1998) a rapid and easy to handle (Reed et al. 1997) species specific protocol should be used to isolate a maximum of DNA while removing PCR inhibitors simultaneously. Therefore, two extraction methods were tested. The crucial difference between the quick, cheap, and simple Chelex method and the more time-consuming QIAamp® DNA Stool Mini Kit (Qiagen) is the thorough purification of extracts with the Qiagen kit, whereas Chelex, as an alkaline chelating resin, removes only polyvalent metal ions (Walsh et al. 1991; Reed et al. 1997). In consideration of the 69% higher amplification success rate obtained by extracting samples with the Qiagen kit, we suspect that the washing and purification steps of the Qiagen kit remove PCR inhibitors to the greatest possible extent. The higher amplification success justifies the time-consuming washing steps. In our study the Chelex extractions must still have contained a large number of PCR inhibitors, since some Chelex-extracted samples not only failed to amplify, but also showed no sign of primer dimers, which indicate the presence of PCR inhibitors (Kohn et al. 1995; Reed et al. 1997; Vigilant 1999; Palomares et al. 2002). Moreover, control amplifications that contained tissue extracts and Chelex-purified faecal samples also failed to amplify. This indicates that the addition of Chelex extracts to successfully amplifying tissue samples can inhibit their amplification. Other studies on otter faeces compared the Qiagen kit with the similar Invitek kit whereby the Invitek kit yielded a higher amplification success rate (Hájková et al. 2006).
PCR conditions need to be adjusted according to the quality and quantity of the DNA faecal extracts. For these purposes we optimised the amplification protocol designed for microsatellite loci by Dallas et al. (1999). The optimised single-locus two-step PCR protocol achieved a 24% (10% regarding positive PCRs at six loci) increase in amplification success compared to the standard PCR conditions outlined by Dallas et al. (1999). Major enhancements are the performance of two consecutive PCR reactions with the first PCR product being the template for the second PCR reaction, the use of locus-specific annealing temperatures, a greater extent of cycles, and modifications in reagent concentrations. Based on the improved protocol for single-locus PCR conditions a more time and cost efficient multiplex protocol was developed, despite previous opinions that multiplexing primers of faecal DNA is difficult to achieve (Ernest et al. 2000). By using less DNA extract the multiplex protocol allows to analyse more loci and to perform the necessary PCR repetitions. Genotyping errors (i.e. false alleles and allelic dropout) occurred at a similar rate in all three PCR protocols and can only be overcome by a large number of replicated amplifications (Taberlet et al. 1996; Kohn et al. 1999; Ernest et al. 2000; Frantz et al. 2003; Broquet and Petit 2004). Due to the low signal intensity, the detection of alleles was hampered when using the original protocol instead of the optimised protocols. As we could demonstrate the use of a two-step procedure, also referred as to pre-amplification approach, offers an increase in quality and quantity of the template DNA. This corroborates the results of recent studies (Piggott et al. 2004; Hedmark and Ellegren 2006) and highlights again the advantages of the pre-amplification approach. Piggott et al. (2004) reported an improvement in amplification success rate and genotyping error rate, whereas Hedmark and Ellegren (2006) found that allelic dropouts generated during the first PCR step were repeated to a high extent in the second amplification. Our results are in line with the ones of Piggott et al. (2004), as we observed that only 47% of allelic dropouts from the first PCR appeared also in the second amplification, while 53% of the samples generating a genotyping error during the first step (46 allelic dropouts vs. 1 false allele) amplified the reliable genotype after the second PCR. Moreover, we were able to optimise the pre-amplification approach further and obtained a still more time and cost effective protocol. Instead of performing an initial multiplex PCR with all six primers followed by a second separate amplification for each marker, we amplified three markers at once in both consecutive amplifications. Thus, the procedure outlined by Piggott et al. (2004) need seven PCR reactions per sample to amplify six loci, using our approach it demands merely four amplification steps per sample for six markers.
For comparability purposes, the amplification success rate is often calculated over all samples and loci in the literature (e.g. Hájková et al. 2006). We provide an additional amplification success rate that is based on the amplification of all six microsatellites simultaneously (Fig. 4). The restricted estimate of amplification success offers more information about the value of primers when dealing with questions such as population size estimates or parentage analyses.
In summary, DNA of otter faeces is only available in low concentrations (Gerloff et al. 1995; Murphy et al. 2000), is exposed to degradation (Frantzen et al. 1998), contains a large number of PCR inhibitors (Deuter et al. 1995; Kohn et al. 1995), and is thus vulnerable to genotyping errors (Goossens et al. 2000; Broquet and Petit 2004). This may explain why previous PCR amplification attempts have met limited success (Coxon et al. 1999; Dallas et al. 2003). However, these difficulties can be overcome with a suitable preservation technique that avoids further degradation and an extraction method that removes PCR inhibitors to a large extent. Efficient amplification conditions can compensate for low DNA concentrations while replicated PCRs can remediate genotyping errors. We could assert that the highest amplification success rate could be achieved by an extraction of jelly samples with the QIAamp® DNA Stool Mini Kit followed immediately after collection and amplified using the optimised multiplex PCR protocol. With this combination we obtained a median amplification success of 94% (mean: 78%) compared to the 20% of the original methods described by Dallas et al. (2003). To apply this method to future studies it is important to know whether the quantity of samples will be sufficient if only jelly samples can be collected. However, even if all types of faecal samples are collected and analysed the amplification success is still 47% (mean: 47%) over all samples per loci and 40% over all locus per samples that obtained a genotype at least once for all six loci. With such success rates, genetic analyses of otter faeces can provide a powerful way to study otter populations.
References
Amos B, Pemberton J (1992) DNA fingerprinting in non-human populations. Curr Opin Genet Dev 2:857–860
Banks SC, Piggott MP, Hansen BD, Robinson NA, Taylor AC (2002) Wombat coprogenetics enumerating a common wombat population by microsatellite analysis of faecal DNA. Aust J Zool 50:193–204
Broquet T, Petit E (2004) Quantifying genotyping errors in noninvasive population genetics. Mol Ecol 13:3601–3608
Brownstein MJ, Carpten JD, Smith JR (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20:1004–1006
Bruford MW, Wayne RK (1993) Microsatellites and their application to population genetic studies. Curr Opin Genet Dev 3:939–943
Carss DN, Parkinson SG (1996) Errors associated with otter Lutra lutra faecal analysis. 1. Assessing general diet from spraints. J Zool 238:301–317
Conroy JWH, French DD (1991) Seasonal patterns in the sprainting behaviour of otters (Lutra lutra L.) in Shetland. In: Reuther C, Röchert R (eds) Proceedings of 5th International Otter Colloquium, Hankensbüttel, 1989
Coxon K, Chanin P, Dallas J, Sykes T (1999) The use of DNA fingerprinting to study the population dynamics of otters (Lutra lutra) in southern Britain a feasibility study. Environment Agency R&D Project W1-025
Creel S, Spong G, Sands JL, Rotella J, Zeigle J, Joe L, Murphy KM, Smith D (2003) Population size estimation in Yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12:2003–2009
Dallas JF, Piertney SB (1998) Microsatellite primers for the Eurasian otter. Mol Ecol 7:1247–1263
Dallas JF, Bacon PJ, Carss DN, Conroy JWH, Green R, Jefferies DJ, Kruuk H, Marshall F, Piertney SB, Racey PA (1999) Genetic diversity in the Eurasian otterLutra lutra in Scotland Evidence from microsatellite polymorphism. Biol J Linn Soc 68:73–86
Dallas JF, Marshall F, Piertney SB, Bacon PJ, Racey PA (2002) Spatially restricted gene flow and reduced microsatellite polymorphism in the Eurasion otter Lutra lutra in Britain. Conserv Genet 3:15–29
Dallas JF, Coxon KE, Sykes T, Chanin PRF, Marshall F, Carss DN, Bacon PJ, Piertney SB, Racey PA (2003) Similar estimates of population genetic composition and sex ratio derived from carcasses and faeces of Eurasian otter Lutra lutra. Mol Ecol 12:275–282
Deuter R, Pietsch S, Hertel S, Müller O (1995) A method for preparation of fecal DNA suitable for PCR. Nucleic Acids Res 23:3800–3801
Ernest HB, Penedo MCT, May BP, Syvanen M, Boyce WM (2000) Molecular tracking of mountain lions in the Yosemite Valley region in California genetic analysis using microsatellites and faecal DNA. Mol Ecol 9:433–441
Farrell LE, Roman J, Sunquist ME (2000) Dietary separation of sympatric carnivores identified by molecular analysis of scats. Mol Ecol 9:1583–1590
Flagstad O, Roed K, Stacy JE, Jakobsen KS (1999) Reliable noninvasive genotyping based on excremental PCR of nuclear DNA purified with a magnetic bead protocol. Mol Ecol 8:879–883
Frantz AC, Pope LC, Carpenter PJ, Roper TJ, Wilson GJ, Delahay RJ, Burke T (2003) Reliable microsatellite genotyping of the Eurasian badger (Meles meles) using faecal DNA. Mol Ecol 12:1649–1661
Frantz AC, Schaul M, Pope LC, Fack F, Schley L, Muller CP, Roper TJ (2004) Estimating population size by genotyping remotely plucked hair the Eurasian badger. J Appl Ecol 41:985–995
Frantzen MAJ, Silk JB, Fergusen JWH, Wayne RK, Kohn MH (1998) Empirical evaluation of preservation methods for faecal DNA. Mol Ecol 7:1423–1428
Gerloff U, Schlötterer C, Rassmann K, Rambold I, Hohmann G, Fruth B, Tautz D (1995) Amplification of hypervariable simple sequence repeats (microsatellites) from excremental DNA of wild living bonobos (Pan paniscus). Mol Ecol 4:515–518
Goossens B, Chikhi L, Utami SS, de Ruiter J, Bruford MW (2000) A multi-samples multi-extracts approach for microsatellite analysis of faecal samples in an arboreal ape. Conserv Genet 1:157–162
Gorman ML, Jenkins D, Harper RJ (1978) The anal scent sacs of the otter (Lutra lutra). J Zool 186:463–474
Hájková P, Zemanová B, Bryja J, Hájek B, Roche K, Tkadlec E, Zima J (2006) Factors affecting success of PCR amplification of microsatellite loci from otter faeces. Mol Ecol Notes 6:559–562
Hedmark E, Ellegren H (2006) A test of the multiplex pre-amplification approach in microsatellite genotyping of wolverine faecal DNA. Conserv Genet 7:289–293
Idaghdour Y, Broderick D, Korrida A (2003) Faeces as a source of DNA for molecular studies in a threatened population of great bustards. Conser Genet 4:789–792
Jansman HAH, Chanin PRF, Dallas JF (2001) Monitoring otter populations by DNA typing of spraints. IUCN Otter Spec Group Bull 18:12–19
Kohn M, Knauer F, Stoffella A, Schröder W, Pääbo S (1995) Conservation genetics of the European brown bear—a study using excremental PCR of nuclear and mitochondrial sequences. Mol Ecol 4:95–103
Kohn MH, Wayne RK (1997) Facts from feces revisited. Trends Ecol Evol 12:223–227
Kohn MH, York EC, Kamradt DA, Haught G, Sauvajot RM, Wayne RK (1999) Estimating population size by genotyping faeces. Proc R Soc Lond B 266:657–663
Liebich HG (1999) Funktionelle Histologie der Haussäugetiere: Lehrbuch und Farbatlas für Studium und Praxis, 3rd edn. Schattauer, Stuttgart
Macdonald SM, Mason CF (1987) Seasonal marking in an otter population. Acta Theriol 32:449–462
Morin PA, Chambers KE, Boesch C, Vigilant L (2001) Quantitative polymerase chain reaction analysis of DNA from noninvasive samples for accurate microsatellite genotyping of wild chimpanzees (Pan troglodytes versus). Mol Ecol 10:1835–1844
Murphy MA, Waits LP, Kendall KC (2000) Quantitative evaluation of fecal drying methods for brown bear DNA analysis. Wildlife Soc B 28:951–957
Murphy MA, Waits LP, Kendall KC, Wasser SK, Higbee JA, Bogden R (2002) An evaluation of long-term preservation methods for brown bear (Ursus arctos) faecal DNA samples. Conserv Genet 3:435–440
Nsubuga AM, Robbins MM, Roeder AD, Morin PA, Boesch C, Vigilant L (2004) Factors affecting the amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol Ecol 13:2089–2094
Palomares F, Godoy A, Piriz A, O’Brien J, Johnson WE (2002) Faecal genetic analysis to determine the presence and distribution of elusive carnivores design and feasibility for the Iberian lynx. Mol Ecol 11:2171–2182
Paxinos E, Mcintosh C, Ralls K, Fleischer R (1997) A noninvasive method for distinguishing among canid species amplification and enzyme restriction of DNA from dung. Mol Ecol 6:483–486
Piggott MP, Taylor AC (2003) Extensive evaluation of faecal preservation and DNA extraction methods in Australian native and introduced species. Aust J Zool 51:341–355
Piggott MP, Bellemain E, Taberlet P, Taylor AC (2004) A multiplex pre-amplification method that significantly improves microsatellite amplification and error rates for faecal DNA in limiting conditions. Conserv Genet 5:417–420
Queller DC, Strassmann JE, Hughes CR (1993) Microsatellites and kinship. Trends Ecol Evol 8:285–288
Reed JZ, Tollit DJ, Thompson PM, Amos W (1997) Molecular scatology the use of molecular genetic analysis to assign species sex and individual identity to seal faeces. Mol Ecol 6:225–234
Roeder AD, Archer FI, Poinar HN, Morin PA (2004) A novel method for collection and preservation of faeces for genetic studies. Mol Ecol Notes 4:761–764
Sidransky D (1992) Identification of rare mutations in the stool of patients with curable colorectable tumors. Science 256:102–105
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194
Taberlet P, Luikart G (1999) Non-invasive genetic sampling and individual identification. Biol J Linn Soc 68:41–55
Tikel D, Blair D, Marsh HD (1996) Marine mammal faeces as a source of DNA. Mol Ecol 5:456–457
Tschirch W (1995) Neue biomedizinische Methoden für die Feldforschung - dargestellt am Beispiel des Fischotters (Lutra lutra). In: Stubbe M, Stubbe A, Heidecke D (eds) Methoden der feldökologischen Säugetierforschung (Methods in mammalian field ecology), vol 1. Martin-Luther-Universität, Halle/Saale, pp 203–210
Vigilant L (1999) An evaluation of techniques for the extraction and amplification of DNA from naturally shed hairs. Biol Chem 380:1329–1331
Walsh PS, Metzger DA, Higuchi R (1991) Chelex® 100 as a medium for simple extraction of DNA for PCR-based typing form forensic material. Biotechniques 10:506–513
Wasser SK, Houston CS, Koehler GM, Cadd GG, Fain SR (1997) Techniques for application of faecal DNA methods to field studies of ursids. Mol Ecol 6:1091–1097
Welsch U (2006) Lehrbuch Histologie, 2nd edn. Urban & Fischer, München
Acknowledgements
We would like to thank R. Klenke for helpful discussions and pertinent knowledge about the otter biology. A. Vallentin and U. Hempel are thanked for collecting faecal samples. Thanks to E. Küster and the Department of Bioanalytical Ecotoxicology and also to W. Durka and the Department of Community Ecology for giving access to the laboratories. We are grateful to the members of the Institute of Applied Ecology, University of Canberra for proof-reading the manuscript and to the anonymous reviewers who have greatly contributed to improve a first version of this paper. We kindly acknowledge private landowners for their permission to access the ponds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lampa, S., Gruber, B., Henle, K. et al. An optimisation approach to increase DNA amplification success of otter faeces. Conserv Genet 9, 201–210 (2008). https://doi.org/10.1007/s10592-007-9328-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-007-9328-9