
Abstract
A concise synthetic strategy has been developed for the synthesis of the pentasaccharide repeating unit of the cell wall O-antigen of Escherichia coli O43 strain involving stereoselective β-D-mannosylation and α-L-fucosylation using corresponding trichloroacetimidate intermediates and perchloric acid supported over silica (HClO4-SiO2) as glycosylation promoter. The yield and stereoselectivity of the glycosylations were very good.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diarrhoeal outbreaks associated with gastrointestinal infections are serious health concern in the developing countries due to the lack of adequate sanitation [1]. In the recent past, it became severe health hazard in the developed countries also due to the intake of contaminated food and water [2]. Most frequently found bacteria causing enteric infections are Shigella [3], Salmonella [4], Vibrio Cholerae [5], enteropathogenic Escherichia coli (E. coli) [6], Campylobacter jejuni [7] etc. Among several enteropathogenic microbes causing diarrhoeal infections, pathogenic Escherichia coli (E. coli) strains are important. E. coli is a Gram-negative, facultative anaerobic pathogen predominantly found in the gastrointestinal tract of humans [8]. Despite their harmless existence in the gut flora and beneficial contribution, a certain species of E. coli acquired virulence factors and causes severe intestinal and urinary infections in humans and animals [9, 10]. They are associated with gastrointestinal infections, particularly “traveller’s diarrhoea” [11] and classified in several sub-types such as, enteropathogenic E. coli (EPEC), enterohemorrhagic E. coli (EHEC), enterotoxigenix E. coli (ETEC), enteroinvasive E. coli (EIEC), enteroaggregative E. coli (EAEC), diffusely adherent E. coli (DAEC) etc. [12]. Among different sub-classes, shiga-toxin (ST) producing EHEC strains are associated with diarrhoeal diseases with life threatening complications such as haemorrhagic colitis [13]. Shiga-toxin producing E. coli O43 strain belongs to EHEC and has been responsible for several diarrhoeal outbreaks in the past [14]. Because of the important role of the cell wall polysaccharides of the pathogenic bacterial strains during the initial stage of infection to the host, a large number of polysaccharides from several bacterial strains have been characterized. Recently, the structure of the repeating unit of the O-antigenic polysaccharide of E. coli O43 has been reported by Perepelov et al [15]. It is a pentasaccharide composed of one β-N-acetyl-D-galactosamine, two α-L-fucose, one α-D-mannose and one β-D-mannose moieties. Bacterial cell wall polysaccharides have been used in the preparation of glycoconjugate vaccines because of their notable antigenic properties [16,17,18,19,20]. Development of alternative approaches for the control of bacterial infections is the thrust area in medicinal chemistry to overcome the failure of the antibiotic activities on the multi-drug-resistant (MDR) bacterial strains [21]. Isolation of oligosaccharides from the natural bacterial culture is tedious as well as suffers from several shortcomings such as difficulties associated with the removal of biological impurities, handling of live bacterial strains, heterogeneity in the isolated oligosaccharide fragments, lack of sufficient quantity etc. Alternatively, significant quantities of pure oligosaccharide fragments with conserved structure and free from biological impurities can be obtained by efficient chemical synthesis. Significant quantities of oligosaccharides are required for their detailed biological studies. In this context, a straightforward synthesis of the pentasaccharide repeating unit of the O-antigenic polysaccharide of E. coli O43 is presented herein.
Results and discussion
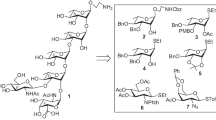
The target pentasaccharide 1 contains two L-fucose moieties having α-linkages, which require stereoselective 1,2-cis-glycosylations for their incorporations. There is a β-linked D-mannose moiety also present in the molecule whose incorporation is considered as a difficult task even after development of a variety of β-D-mannosylation techniques till date. Initially, It was decided to adopt a block glycosylation strategy to carry out stereoselective glycosylation between a disaccharide acceptor with a trisaccharide thioglycoside donor minimizing the number of reaction steps. For this purpose a number of suitably protected monosaccharide intermediates 2 [22], 3 [23], 4, 5 [24], 6 and 7 [25] have been selected, which either were known in the literature or prepared following earlier literature reports (Fig. 1). In order to achieve better yield and stereoselectivity in the glycosylation steps, it was decided to use glycosyl trichloroacetimidate derivatives [26] as glycosyl donors in most of the cases and perchloric acid supported over silica (HClO4-SiO2) [27] as a solid acid glycosylation promoter for the activation of glycosyl trichloroacetimidate derivatives [28].

Structure of the synthesized pentasaccharide repeating unit and its synthetic intermediates
Ethyl 2-O-benzyl-4,6-O-benzylidene-1-thio-α-D-mannopyranoside (8) [29], prepared from D-mannose following earlier reported reaction conditions was treated with levulinic acid in the presence of diisopropyl dicarbodimide (DIC) [30] and DMAP to furnish levulinic acid ester (9) in 74% yield. Regioselective reductive ring opening of the benzylidene acetal in compound 9 using a combination of triethylsilane and trifluoroacetic acid (TFA) [31] followed by acetylation of the free hydroxyl group furnished ethyl 4-O-acetyl-2,6-di-O-benzyl-3-O-levulinyl-1-thio-α-D-mannopyranoside (6) in 68% over all yield (Scheme 1).

Reagents and conditions: (a) levulinic acid, DIC, DMAP, CH2Cl2, r t, 5 h, 74%; (b) Et3SiH, TFA, CH2Cl2, 0 °C, 3 h; (c) Ac2O, pyridine, r t, 2 h, 68%
Stereoselective 1,2-cis-glycosylation of p-methoxyphenyl 2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (2) with 3-O-acetyl-2,4-di-O-benzyl-α-L-fucopyranosyl trichloroacetimidate (3) in the presence of perchloric acid supported over silica (HClO4-SiO2) [27, 28] as the glycosylation activator in a mixed solvent, CH2Cl2–Et2O (2:3; v/v) to enhance the 1,2-cis glycosylation [32], furnished the disaccharide derivative, which was subsequently de-O-acetylated using sodium methoxide to give the disaccharide acceptor 10 in 67% over all yield. A minor quantity (~5%) of 1,2-trans glycosylation product was also formed, which was separated by column chromatography. The NMR spectroscopic analysis of compound 10 confirmed its formation [signals at δ 5.46 (s, PhCH), 5.06 (d, J = 3.5 Hz, H-1B), 4.72 (d, J = 8.0 Hz, H-1A) in 1H NMR and at δ 102.3 (C-1A), 101.1 (PhCH), 99.9 (C-1B) in 13C NMR spectra] (Scheme 2)

Reagents and conditions: (a) HClO4-SiO2, CH2Cl2 –Et2O (2:3, v/v), −10 °C, 2 h; (b) 0.1 M CH3ONa, CH3OH, r, t, 2 h, 67%.
After having the disaccharide acceptor (10), attempts were made to obtain the trisaccharide thioglycoside derivative (13). Following a recently reported reaction condition for functional group influenced stereoselective β-mannosylation [24], an orthogonal glycosylation [33] of the D-mannosyl thioglycoside acceptor (4) with 3-O-p-methoxybenzyl (PMB) protected D-mannosyl trichloroacetimidate donor (5) [24] in the presence of HClO4-SiO2 furnished β-mannosidic linkage containing disaccharide thioglycoside derivative 11 in 65% yield. The anomeric thioglycosidic group in the acceptor remained unaffected under the reaction condition. The stereochemistry of the newly formed β-mannosidic linkage was characterized by its NMR spectroscopic analysis [signals at δ 5.54 (s, PhCH), 5.47 (br s, H-1A), 4.01 (br s, H-1B) in 1H NMR and at δ 101.4 (PhCH), 98.0 (JH1,C1 = 158.5 Hz, C-1B, β), 80.8 (JH1,C1 = 174.5 Hz, C-1A, α) in 13C NMR spectra]. The incorporation of the β-mannosidic linkage in compound 11 was unambiguously confirmed from its JH1,C1 value (158.5 Hz) in the 1H coupled 13C NMR spectrum. The JH1,C1 value in 1H coupled 13C NMR spectrum appears in the region of <160 Hz for β-glycosidic linkage whereas, JH1,C1 value appears in the region of >170 Hz for α-glycosidic linkage [34, 35]. Oxidative removal of the PMB group from compound 11 using DDQ [36] furnished the disaccharide acceptor 12 in 73% yield, which was allowed to couple with a L-fucosyl trichloroacetimidate derivative (3) in the presence of HClO4-SiO2 in a mixed solvent CH2Cl2–Et2O (2:3; v/v) [32] under orthogonal 1,2-cis-glycosylation approach [33] without affecting the thioglycoside to furnish trisaccharide thioglycoside derivative 13 in 68% yield together with a minor quantity (~7%) of 1,2-trans glycosylation product, which was separated by column chromatography. Spectroscopic analysis of compound 13 confirmed its formation [signals at δ 5.48 (s, PhCH), 5.47 (br s, H-1A), 4.93 (d, J = 3.5 Hz, H-1C), 4.00 (br s, H-1B) in 1HNMR and at δ 101.7 (PhCH), 97.6 (JH1,C1 = 159.0 Hz, C-1B, β), 96.0 (JH1,C1 = 172.0 Hz, C-1C, α), 80.7 (JH1,C1 = 174.0 Hz, C-1A, α) in 13C NMR spectra]. Next the trisaccharide derivative 13 was allowed to react with the disaccharide acceptor 10 in the presence of a combination of NIS and HClO4-SiO2 [37] to provide desired pentasaccharide derivative following a block glycosylation strategy. Unfortunately, the expected pentasaccharide derivative was not formed and the decomposition of the trisaccharide donor 13 was observed leaving the disaccharide acceptor 10 unaffected. Application of other thiophilic glycosylation activators such as DMTST [38], NIS-TfOH [39], methyl triflate [40] also did not lead to the formation of the desired product except decomposition of the glycosyl donor 13. Such unexpected outcome of the block glycosylation strategy forced to identify a different approach for the synthesis of the desired pentasaccharide 1 (Scheme 3).

Reagents and conditions: (a) HClO4-SiO2, CH2Cl2, −40 °C, 1 h, 65%; (b) DDQ, CH2Cl2-H2O, r t, 5 h, 73%; (c) HClO4-SiO2, CH2Cl2 –Et2O (2:3, v/v), −10 °C, 1 h, 68%; (d) NIS, HClO4-SiO2, MS 4 Å, CH2Cl2, −10 °C, 2 h, 0%
In this endeavor, a sequential glycosylation strategy has been adopted for the synthesis of the pentasaccharide 1. Stereoselective glycosylation of the disaccharide acceptor 10 with D-mannosyl thioglycoside donor 6 in the presence of a combination [37] of NIS and HClO4-SiO2 furnished a trisaccharide derivative, which on subsequent treatment with hydrazine acetate [41] resulted the trisaccharide acceptor 14 in 72% over all yield. NMR spectroscopic analysis of compound 14 supported its formation [signals at δ 5.41 (s, PhCH), 5.30 (br s, H-1C), 5.07 (d, J = 3.5 Hz, H-1B), 4.68 (d, J = 8.0 Hz, H-1A) in 1H NMR and at δ 102.0 (C-1A), 101.2 (PhCH), 99.9 (C-1B), 98.8 (C-1C) in 13C NMR spectra]. Following the recently reported reaction condition [24] on the functional group induced β-mannosidic glycosylation as well as removal of PMB group in one-pot [42] by tuning the reaction condition, treatment of trisaccharide acceptor 14 with D-mannosyl trichloroacetimidate donor (5) in the presence of HClO4-SiO2 furnished tetrasaccharide derivative 15 in 65% yield. The NMR spectroscopic analysis unambiguously established the formation of the newly formed β-mannosidic linkage together with other glycosidic linkages [signals at δ 5.41 (s, PhCH), 5.40 (br s, H-1C), 5.11 (d, J = 3.5 Hz, H-1B), 4.69 (d, J = 8.0 Hz, H-1A), 4.15 (br s, H-1D) in 1H NMR and at δ 102.1 (JH1,C1 = 156.0 Hz, C-1A, β), 101.9 (PhCH), 101.2 (PhCH), 99.7 (JH1,C1 = 171.0 Hz, C-1B, α), 98.9 (JH1,C1 = 159.0 Hz, C-1D, β), 98.6 (JH1,C1 = 173.5 Hz, C-1C, α) in 13C NMR spectra]. The incorporation of the β-mannosidic linkage in compound 15 was unambiguously confirmed from its JH1,C1 value (159.0 Hz) in the 1H coupled 13C NMR spectrum [34, 35]. Compound 15 was then allowed to couple with L-fucosyl trichloroacetimidate derivative 7 under 1,2-cis glycosylation condition using HClO4-SiO2 as glycosylation activator in a mixed solvent CH2Cl2-Et2O (2:3, v/v) [32] to furnish pentasaccharide derivative 16 in 70% yield together with a minor quantity (~5%) of 1,2-trans glycosylation product, which was separated using column chromatography. The NMR spectroscopic analysis of compound 16 confirmed the formation of a new α-glycosidic linkage together with existing glycosidic bonds [signals at δ 5.48 (br s, H-1C), 5.05 (d, J = 3.5 Hz, H-1B), 4.88 (d, J = 3.5 Hz, H-1E), 4.73 (d, J = 8.0 Hz, H-1A), 3.91 (br s, H-1D) in 1H NMR and at δ 101.9 (PhCH), 101.4 (C-1A), 100.3 (C-1B), 98.5 (C-1C), 97.5 (C-1D), 96.6 (C-1E) in 13C NMR spectra]. Finally, compound 16 was subjected to a sequence of reactions comprising (a) transformation of azido group in acetamide using thioacetic acid [43]; (b) removal of acetyl group using sodium methoxide and (c) hydrogenolysis over Pearlman’s catalyst [44] to furnish compound 1 in 60% over all yield. NMR spectroscopic analysis confirmed the formation of compound 1 [signals at δ 5.24 (d, J = 1.5 Hz, H-1C), 5.06 (d, J = 4.0 Hz, H-1E), 4.97 (d, J = 8.5 Hz, H-1A), 4.94 (d, J = 4.0 Hz, H-1B), 4.78 (br s, H-1D) in 1H NMR and at δ 101.4 (JH1,C1 = 174.0 Hz, C-1C, α), 101.3 (JH1,C1 = 171.0 Hz, C-1B, α), 100.8 (JH1,C1 = 157.0 Hz, C-1A, β), 96.7 (JH1,C1 = 158.5 Hz, C-1D, β), 95.9 (JH1,C1 = 171.0 Hz, C-1E, α) in 13C NMR spectra]. The anomeric configuration of the glycosidic bonds were unambiguously confirmed from the JH1,C1 values in the 1H coupled 13C NMR spectrum, particularly in case of the β-mannosidic linkage (JH1,C1 = 158.5 Hz). In addition, appearance of C-5 carbons at δ 77.1 (C-5D) of the β-mannosidic moiety and at δ 73.3 (C-5C) of the α-mannosidic moiety in the deprotected pentasaccharide 1 also confirmed the presence of β-mannose in the molecule (Scheme 4).

Reagents and conditions: (a) NIS, HClO4-SiO2, MS 4 Å, CH2Cl2, −15 °C, 2 h; (b) AcOH, NH2NH2·H2O, CH3OH-CH2Cl2 (1:1), 0 °C-r t, 2 h, 72%; (c) HClO4-SiO2, CH2Cl2, −40 °C, 2 h, then at 20 °C for 30 min, 65%; (d) HClO4-SiO2, CH2Cl2-Et2O (2:3, v/v), −20 °C, 1 h, 70%; (e) CH3COSH, pyridine, r t, 12 h; (f) 0.1 M CH3ONa, CH3OH, r, t, 3 h; (g) H2, 20% Pd(OH)2-C, CH3OH, r t, 24 h, 62% in three steps
Conclusion
In summary, a straightforward synthetic strategy has been developed for the synthesis of the pentasaccharide repeating unit of the cell wall O-antigen of Escherichia coli O43 using sequential glycosylation strategy after a failed attempt to achieve the target pentasaccharide using a block synthetic scheme. Satisfactory yield with appropriate stereochemistry has been achieved applying PMB group influenced β-D-mannosylation reaction using D-mannosyl trichloroacetimidate as glycosyl donor. 1,2-Cis glycosylation of L-fucose moieties have also been achieved in very good yield. The yields of the glycosylation steps were very good with satisfactory stereochemistry at the newly formed glycosidic linkages.
Experimental
General methods
All reactions were monitored by thin layer chromatography over silica gel coated TLC plates. The spots on TLC were visualized by warming ceric sulphate (2% Ce(SO4)2 in 2 N H2SO4) sprayed plates on hot plate. Silica gel 230–400 mesh was used for column chromatography. NMR spectra were recorded on Brucker Avance 500 MHz using CDCl3 as solvent and TMS as internal reference unless stated otherwise. Chemical shift value is expressed in δ ppm. ESI-MS were recorded on a Thermo Scientific Orbitrap Velos Pro TM mass spectrometer. Optical rotations were recorded in a Jasco P-2000 spectrometer. Commercially available grades of organic solvents of adequate purity are used in all reactions. HClO4-SiO2 was prepared following the report of Chakraborti et al. [27]
Ethyl 4-O-acetyl-2,6-di-O-benzyl-3-O-levulinyl-1-thio-α-D-mannopyranoside (6)
To a solution of compound 9 (3 g, 5.99 mmol) in dry CH2Cl2 (25 mL) were added triethylsilane (4.8 mL, 30 mmol) at 0 °C followed by the addition of trifluoroacetic acid (TFA) (1.75 mL, 22.8 mmol) and the reaction mixture was stirred at the same temperature for 3 h. The reaction mixture was diluted with CH2Cl2 (50 mL), washed with saturated NaHCO3 solution (2 × 50 mL), dried (Na2SO4), and concentrated. To the solution of the crude product in pyridine (10 mL) was added Ac2O (10 mL) and the reaction mixture was stirred at room temperature for 2 h. The solvents were removed and co-evaporated with toluene (2 × 25 mL) under reduced pressure. The crude product was purified over SiO2 using hexane-EtOAc (6:1) as eluant to give pure compound 6 (2.2 g, 68%). Yellow oil; [α]D + 11 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.34–7.22 (m, 10 H, Ar-H), 5.38 (t, J = 10.0 Hz, 1 H, H-4), 5.29 (br s, 1 H, H-1), 5.10 (dd, J = 9.5 Hz, 3.0 Hz, 1 H, H-3), 4.66 (d, J = 11.5 Hz, 1 H, PhCH), 4.56 (d, J = 11.5 Hz, 1 H, PhCH), 4.54 (d, J = 11.5 Hz, 1 H, PhCH), 4.47 (d, J = 11.5 Hz, 1 H, PhCH), 4.28–4.20 (m, 1 H, H-5), 3.87 (br s, 1 H, H-2), 3.59–3.50 (m, 2 H, H-6a,b), 2.64–2.60 (m, 4 H, 2 CH2), 2.57–2.41 (m, 2 H, SCH2CH3), 2.11 (s, 3 H, CH3CO), 1.93 (s, 3 H, CH3COO), 1.26 (t, J = 7.4 Hz, 3 H, SCH2CH3); 13C NMR (125 MHz, CDCl3): δ 205.5 (CO), 171.5, 169.6 (2 CO), 138.0–127.7 (Ar-C), 81.6 (C-1), 77.0 (C-2), 73.4 (PhCH2), 72.6 (PhCH2), 71.9 (C-5), 70.1 (C-4), 69.2 (C-6), 67.2 (C-3), 37.6 (CH2), 29.6 (CH3CO), 27.9 (CH2), 25.0 (SCH2CH3), 20.7 (COCH3), 14.8 (SCH2CH3); HRMS (ESI) for C29H36O8S [M + H]+: Calcd. 545.2209; found, 545.2200.
Ethyl 2-O-benzyl-4,6-O-benzylidene-3-O-levulinyl-1-thio-α-D-mannopyranoside (9)
To a solution of compound 8 (4 g, 9.94 mmol) in anhydrous CH2Cl2 (30 mL) were added levulinic acid (1.3 mL, 12.76 mmol), DIC (1.7 mL, 10.85 mmol) and DMAP (1.2 g, 9.82 mmol) and the reaction mixture was allowed to stir at room temperature for 5 h. The reaction mixture was diluted with H2O (50 mL) and extracted with CH2Cl2 (100 mL). The organic layer was washed with 2 M HCl (50 mL), H2O (100 mL), dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (5:1) as eluant to give pure compound 9 (3.7 g, 74%); Yellow oil; [α]D + 6 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.39–7.12 (m, 10 H, Ar-H), 5.49 (s, 1 H, PhCH), 5.20 (br s, 1 H, H-1), 5.12 (dd, J = 9.5 Hz, 3.0 Hz, 1 H, H-3), 4.61 (br s, 2 H, PhCH2), 4.20–4.10 (m, 3 H, H-5, H-6a,b), 3.99 (d, J = 1.2 Hz, 1 H, H-2), 3.80 (t, J = 10.0 Hz, 1 H, H-4), 2.67–2.47 (m, 6 H, 2 CH2, SCH2CH3), 2.07 (s, 3 H, CH3CO), 1.22 (t, J = 7.4 Hz, 3 H, SCH2CH3); 13C NMR (125 MHz, CDCl3): δ 205.9 (CO), 171.9 (CO), 137.7–126.3 (Ar-C), 101.8 (PhCH), 83.1 (C-1), 77.9 (C-2), 76.3 (C-5), 73.4 (PhCH2), 71.0 (C-4), 68.6 (C-6), 64.5 (C-3), 37.8 (CH2), 29.8 (CH3CO), 27.9 (CH2), 25.3 (SCH2CH3), 14.9 (SCH2CH3); HRMS (ESI) for C27H32O7S [M + H]+: Calcd. 501.1947; found, 501.1940.
p-Methoxyphenyl (2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (10)
A solution of compound 2 (1 g, 2.50 mmol) and compound 3 (1.6 g, 3.01 mmol) in anhydrous CH2Cl2-Et2O (15 mL; 2:3 v/v) was cooled to - 10 °C under argon. To the cooled reaction mixture was added HClO4-SiO2 (50 mg) and it was allowed to stir at same temperature for 2 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (25 mL). The organic layer was successively washed with satd. NaHCO3 (25 mL) and H2O (25 mL), dried (Na2SO4) and concentrated. A solution of the product in 0.1 M CH3ONa in CH3OH (15 mL) was stirred at room temperature for 2 h and neutralized with Amberlite IR-120 (H+) resin. The reaction mixture was filtered and concentrated under reduced pressure to give the crude product, which was purified over SiO2 using hexane-EtOAc (3:1) as eluant to give pure compound 10 (1.2 g, 67%). Yellow oil; [α]D - 8 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.31–7.17 (m, 15 H, Ar-H), 7.07 (d, J = 9.0 Hz, 2 H, Ar-H), 6.79 (d, J = 9.0 Hz, 2 H, Ar-H), 5.46 (s, 1 H, PhCH), 5.06 (d, J = 3.5 Hz, 1 H, H-1B), 4.90 (d, J = 11.5 Hz, 1 H, PhCH), 4.80 (d, J = 11.5 Hz, 1 H, PhCH), 4.72 (d, J = 8.0 Hz, 1 H, H-1A), 4.62 (d, J = 11.5 Hz, 1 H, PhCH), 4.59 (d, J = 11.5 Hz, 1 H, PhCH), 4.33 (d, J = 12.5 Hz, 1 H, H-6aA), 4.23 (d, J = 3.0 Hz, 1 H, H-4A), 4.12–4.09 (m, 4 H, H-2A, H-3B, H-5B, H-6bA), 3.83 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-2B), 3.77 (s, 3 H, OCH3), 3.58 (d, J = 2.0 Hz, 1 H, H-4B), 3.42 (br s, 1 H, H-5A), 3.36 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-3A), 1.08 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 155.8–114.5 (Ar-C), 102.3 (C-1A), 101.1 (PhCH), 99.9 (C-1B), 80.2 (C-3A), 78.6 (C-4B), 76.6 (C-2B), 75.2 (PhCH2), 74.8 (C-4A), 72.0 (PhCH2), 70.5 (C-3B), 68.9 (C-6A), 67.1 (C-5B), 66.5 (C-5A), 61.5 (C-2A), 55.5 (OCH3), 16.9 (CCH3); HRMS (ESI) for C40H43N3O10 [M + H]+: Calcd. 726.3026; found, 726.3017.
Ethyl [2-O-benzyl-4,6-O-benzylidene-3-O-(p-methoxybenzyl)-β-D-mannopyranosyl]-(1 → 3)-4-O-acetyl-2,6-di-O-benzyl-1-thio-α-D-mannopyranoside (11)
A solution of compound 4 (1.2 g, 2.69 mmol) and compound 5 (2 g, 3.21 mmol) in anhydrous CH2Cl2 (25 mL) was cooled to - 40 °C under argon. To the cooled reaction mixture was added HClO4-SiO2 (80 mg) and it was allowed to stir at same temperature for 1 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (50 mL). The organic layer was successively washed with satd. NaHCO3 (50 mL) and H2O (50 mL), dried (Na2SO4) and concentrated under reduced pressure to give the crude product, which was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 11 (1.6 g, 65%). Yellow oil; [α]D - 13 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.47–6.80 (m, 22 H, Ar-H), 6.80 (d, J = 9.0 Hz, 2 H, Ar-H), 5.54 (s, 1 H, PhCH), 5.47 (br s, 1 H, H-1A), 5.40 (t, J = 10.0 Hz, 1 H, H-4A), 4.86 (d, J = 11.5 Hz, 1 H, PhCH), 4.76 (d, J = 11.5 Hz, 1 H, PhCH), 4.68 (d, J = 11.5 Hz, 1 H, PhCH), 4.59–4.43 (m, 5 H, 5 PhCH), 4.26–4.20 (m, 2 H, H-5A, H-6aA), 4.08 (dd, J = 9.5 Hz, 3.5 Hz, 1 H, H-3A), 4.05 (t, J = 10.0 Hz, 1 H, H-4B), 4.01 (br s, 1 H, H-1B), 3.83–3.79 (m, 1 H, H-6bA), 3.78 (s, 3 H, OCH3), 3.77 (br s, 1 H, H-2A), 3.64–3.60 (m, H-2B, H-6aB), 3.56 (dd, J = 12.5 Hz, 2.5 Hz, 1 H, H-6bB), 3.26 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-3B), 3.03–2.99 (m, 1 H, H-5B), 2.76–2.58 (m, 2 H, SCH2CH3), 1.87 (s, 3 H, COCH3), 1.32 (t, J = 7.4 Hz, 3 H, SCH2CH3); 13C NMR (125 MHz, CDCl3): δ 169.7 (CO), 159.2–113.7 (Ar-C), 101.4 (PhCH), 98.0 (JH1,C1 = 158.5 Hz, C-1B), 80.8 (JH1,C1 = 174.5 Hz, C-1A), 78. 6 (C-4B), 76.9 (C-3B), 75.7 (C-2B), 74.3 (PhCH2), 74.0 (C-2A), 73.8 (C-3A), 72.3 (PhCH2), 71.6 (PhCH2), 71.2 (PhCH2), 70.4 (C-5A), 69.4 (C-6B), 68.5 (C-6A), 67.7 (C-5B), 67.6 (C-4A), 55.1 (OCH3), 25.4 (SCHCH3), 20.9 (COCH3), 14.9 (SCH2CH3); HRMS (ESI) for C52H58O12S [M + H]+: Calcd. 907.3727; found, 907.3720.
Ethyl (3-O-acetyl-2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-(2-O-benzyl-4,6-O-benzylidene-β-D-mannopyranosyl)-(1 → 3)-4-O-acetyl-2,6-di-O-benzyl-1-thio-α-D-mannopyranoside (13)
To a solution of compound 11 (1.5 g, 1.65 mmol) in CH2Cl2 (20 mL) was added a solution of DDQ (680 mg, 2.99 mmol) in H2O (5 mL) and the reaction mixture was allowed to stir at room temperature for 5 h. The reaction mixture was diluted with CH2Cl2 (50 mL), washed with satd. NaHCO3 (25 mL), dried (Na2SO4) and concentrated to give the crude product, which was purified over SiO2 using hexane-EtOAc (3:1) as eluant to give pure product 12 (950 mg, 73%). A solution of compound 12 (900 mg, 1.14 mmol) and compound 3 (730 mg, 1.37 mmol) in CH2Cl2–Et2O (15 mL; 2:3, v/v) was cooled to - 10 °C under argon. To the cooled reaction mixture was added HClO4-SiO2 (50 mg) and it was allowed to stir at same temperature for 1 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (50 mL). The organic layer was successively washed with satd. NaHCO3 (50 mL) and H2O (50 mL), dried (Na2SO4) and concentrated under reduced pressure to give the crude product, which was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 13 (900 mg, 68%). Yellow oil; [α]D + 5 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.43–7.17 (m, 30 H, Ar-H), 5.48 (s, 1 H, PhCH), 5.47 (br s, 1 H, H-1A), 5.35 (t, J = 10.0 Hz, 1 H, H-4A), 5.33 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-3C), 4.93 (d, J = 3.5 Hz, 1 H, H-1C), 4.78–4.43 (m, 10 H, 10 PhCH), 4.23–4.18 (m, 3 H, H-3A, H-5A, H-6aA), 4.07–4.01 (m, 2 H, H-2C, H-5C), 4.00 (br s, 1 H, H-1B), 3.96 (t, J = 10.0 Hz, 1 H, H-4B), 3.79 (br s, 1 H, H-2A), 3.78–3.75 (m, 1 H, H-6bA), 3.63 (d, J = 2.0 Hz, 1 H, H-4C), 3,62–3.50 (m, 4 H, H-2B, H-3B, H-6a,bB), 3.05–2.98 (m, 1 H, H-5B), 2.70–2.63 (m, 2 H, SCH2CH3), 1.90 (s, 3 H, COCH3), 1.74 (s, 3 H, COCH3), 1.28 (t, J = 7.4 Hz, 3 H, SCH2CH3), 0.80 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.1, 170.0 (2 CO), 138.2–126.2 (Ar-C), 101.7 (PhCH), 97.6 (JH1,C1 = 159.0 Hz, C-1B), 96.0 (JH1,C1 = 172.0 Hz, C-1C), 80.7 (JH1,C1 = 174.0 Hz, C-1A), 78.9 (C-4C), 76.8 (C-4B), 76.5 (C-2B), 75.9 (C-3B), 75.6 (PhCH2), 75.2 (PhCH2), 74.1 (C-2A), 73.9 (2 C, C-4A, C-5C), 73.6 (C-2C), 73.4 (PhCH2), 73.3 (PhCH2), 71.2 (PhCH2), 70.5 (C-3A), 69.4 (C-6B), 68.5 (C-6A), 67.9 (C-3C), 67.5 (C-5B), 65.9 (C-5A), 25.3 (SCH2CH3), 20.7, 20.6 (2 COCH3), 15.9 (CCH3), 14.9 (CCH3); HRMS (ESI) for C66H74O16S [M + H]+: Calcd. 1155.4776; found, 1155.4766.
p-Methoxyphenyl (4-O-acetyl-2,6-di-O-benzyl-α-D-mannopyranosyl)-(1 → 3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (14)
To a solution of compound 10 (1 g, 1.38 mmol) and compound 6 (900 mg, 1.65 mmol) in anhydrous CH2Cl2 (15 mL) was added MS 4 Å (1 g) and it was cooled to - 15 °C under argon. To the cooled reaction mixture were successively added NIS (370 mg, 1.64 mmol) and HClO4-SiO2 (10 mg) and it was allowed to stir at same temperature for 2 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (25 mL). The organic layer was successively washed with 5% Na2S2O3 (25 mL), satd. NaHCO3 (25 mL) and H2O (25 mL), dried (Na2SO4) and concentrated under reduced pressure to give the crude product. To a solution of the crude product in CH3OH-CH2Cl2 (15 ml; 1:1 v/v) were added AcOH (300 μL, 5.25 mmol) and NH2NH2·H2O (255 μL, 5.25 mmol) and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with CH2Cl2 (25 mL), successively washed with 2 N HCl (25 mL), H2O (25 mL), dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 14 (1.1 g, 72% in two steps). Yellow oil; [α]D + 9 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.47–7.06 (m, 27 H, Ar-H), 6.80 (d, J = 9.0 Hz, 2 H, Ar-H), 5.41 (s, 1 H, PhCH), 5.30 (br s, 1 H, H-1C), 5.07 (d, J = 3.5 Hz, 1 H, H-1B), 4.98 (t, J = 10.0 Hz, 1 H, H-4C), 4.85 (d, J = 11.5 Hz, 1 H, PhCH), 4.77 (d, J = 11.5 Hz, 1 H, PhCH), 4.68 (d, J = 8.0 Hz, 1 H, H-1A), 4.60 (d, J = 11.5 Hz, 1 H, PhCH), 4.50 (d, J = 11.5 Hz, 1 H, PhCH), 4.46 (d, J = 11.5 Hz, 1 H, PhCH), 4.38 (d, J = 11.5 Hz, 1 H, PhCH), 4.35–4.23 (m, 2 H, H-6aA, PhCH), 4.22 (d, J = 3.0 Hz, 1 H, H-4A), 4.20 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-3A), 4.15–4.08 (m, 2 H, H-2A, H-5B), 4.00 (d, J = 12.5 Hz, 1 H, H-6bA), 3.98 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-2B), 3.94 (d, J = 11.5 Hz, 1 H, PhCH), 3.93–3.90 (m, 1 H, H-5C), 3.81–3.77 (m, 1 H, H-3C), 3.76 (s, 3 H, OCH3), 3.71–3.70 (m, 1 H, H-2C), 3.57 (d, J = 1.5 Hz, 1 H, H-4B), 3.52 (dd, J = 12.5 Hz, 6.0 Hz, 1 H, H-6aC), 3.47 (dd, J = 12.5 Hz, 2.5 Hz, 1 H, H-6bC), 3.41 (br s, 1 H, H-5A), 3.39 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-3B), 1.97 (s, 3 H, COCH3), 0.99 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.3 (COCH3), 155.7–114.4 (Ar-C), 102.0 (C-1A), 101.2 (PhCH), 99.9 (C-1B), 98.8 (C-1C), 80.6 (C-3B), 79.9 (C-4B), 77.9 (C-2C), 77.1 (C-2B), 76.7 (C-3A), 75.1 (PhCH2), 74.6 (C-4A), 73.3 (PhCH2), 72.3 (PhCH2), 71.9 (PhCH2), 70.5 (C-4C), 70.4 (C-5C), 69.6 (C-6A), 69.5 (C-3C), 68.9 (C-6C), 67.6 (C-5B), 66.5 (C-5A), 61.3 (C-2A), 55.5 (OCH3), 20.97 (COCH3), 16.8 (CCH3); HRMS (ESI) for C62H67N3O16 [M + H]+: Calcd. 1110.4599; found, 1110.4590.
p-Methoxyphenyl (2-O-benzyl-4,6-O-benzylidene-β-D-mannopyranosyl)-(1 → 3)-(4-O-acetyl-2,6-di-O-benzyl-α-D-mannopyranosyl)-(1 → 3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (15)
A solution of compound 14 (1 g, 0.90 mmol) and compound 5 (650 mg, 1.04 mmol) in anhydrous CH2Cl2 (10 mL) was cooled to - 40 °C under argon. To the cooled reaction mixture was added HClO4-SiO2 (40 mg) and it was allowed to stir at same temperature for 2 h. After consumption of the starting materials the reaction mixture was allowed to stir at 20 °C for 30 min. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (25 mL). The organic layer was successively washed with satd. NaHCO3 (25 mL) and H2O (25 mL), dried (Na2SO4) and concentrated under reduced pressure to give the crude product, which was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 15 (850 mg, 65%). Yellow oil; [α]D - 17 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.47–7.05 (m, 37 H, Ar-H), 6.80 (d, J = 9.0 Hz, 2 H, Ar-H), 5.41 (s, 1 H, PhCH), 5.40 (br s, 2 H, H-1C, PhCH), 5.33 (t, J = 10.0 Hz, 1 H, H-4C), 5.11 (d, J = 3.5 Hz, 1 H, H-1B), 4.94 (d, J = 11.5 Hz, 1 H, PhCH), 4.90 (d, J = 11.5 Hz, 1 H, PhCH), 4.82 (d, J = 11.5 Hz, 1 H, PhCH), 4.69 (d, J = 8.0 Hz, 1 H, H-1A), 4.65 (d, J = 11.5 Hz, 1 H, PhCH), 4.60 (d, J = 11.5 Hz, 1 H, PhCH), 4.55 (d, J = 11.5 Hz, 1 H, PhCH), 4.50–4.38 (m, 4 H, 4 PhCH), 4.30 (d, J = 12.5 Hz, 1 H, H-6aA), 4.26 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-3A), 4.23 (d, J = 2.0 Hz, 1 H, H-4A), 4.15 (br s, 1 H, H-1D), 4.13–4.07 (m, 3 H, H-2A, H-3C, H-5B), 4.04 (dd, J = 9.0 Hz, 3.5 Hz, 1 H, H-2B), 3.99 (d, J = 12.5 Hz, 1 H, H-6bA), 3.98–3.92 (m, 1 H, H-5C), 3.90–3.85 (m, 1 H, H-6aD), 3.75 (s, 3 H, OCH3), 3.72–3.71 (m, 1 H, H-2C), 3.61–3.55 (m, 4 H, H-3D, H-4D, H-6aC, H-6bD), 3.52 (d, J = 3.0 Hz, 1 H, H-4B), 3.50 (dd, J = 12.5 Hz, 2.5 Hz, 1 H, H-6bC), 3.43–3.37 (m, 3 H, H-2D, H-3B, H-5A), 2.96–2.89 (m, 1 H, H-5D), 1.85 (s, 3 H, COCH3), 1.04 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 169.9 (COCH3), 155.7–114.5 (Ar-C), 102.1 (JH1,C1 = 156.0 Hz, C-1A), 101.9 (PhCH), 101.2 (PhCH), 99.7 (JH1,C1 = 171.0 Hz, C-1B), 98.9 (JH1,C1 = 159.0 Hz, C-1D), 98.6 (JH1,C1 = 173.5 Hz, C-1C), 80.5 (C-3B), 79.4 (C-3D), 79.3 (C-4D), 78.1 (C-4B), 76.7 (C-3A), 76.1 (C-4A), 75.1 (PhCH2), 75.0 (PhCH2), 74. 7 (C-3C), 74.0 (C-2B), 73.7 (PhCH2), 73.3 (C-2C), 71.7 (PhCH2), 71.6 (PhCH2), 71.0 (C-5C), 70.5 (C-2D), 69.8 (C-6C), 68.9 (C-6A), 68.4 (C-6D), 68.1 (C-4C), 68.0 (C-5B), 67.0 (C-5D), 66.5 (C-5A), 61.3 (C-2A), 55.5 (OCH3), 20.9 (COCH3), 16.8 (CCH3); HRMS (ESI) for C82H87N3O21 [M + H]+: Calcd. 1450.5910; found, 1450.5902.
p-Methoxyphenyl (2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-(2-O-benzyl-4,6-O-benzylidene-β-D-mannopyranosyl)-(1 → 3)-(4-O-acetyl-2,6-di-O-benzyl-α-D-mannopyranosyl)-(1 → 3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (16)
A solution of compound 15 (750 mg, 0.51 mmol) and compound 7 (330 mg, 0.57 mmol) in anhydrous CH2Cl2-Et2O (10 mL; 2:3 v/v) was cooled to - 20 °C under argon. To the cooled reaction mixture was added HClO4-SiO2 (40 mg) and it was allowed to stir at same temperature for 1 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (25 mL). The organic layer was successively washed with satd. NaHCO3 (25 mL) and H2O (25 mL), dried (Na2SO4) and concentrated under reduced pressure to give the crude product, which was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 16 (665 mg, 70%). Yellow oil; [α]D - 3 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.44–7.09 (m, 52 H, Ar-H), 6.83 (d, J = 9.0 Hz, 2 H, Ar-H), 5.50 (s, 1 H, PhCH), 5.48 (br s, 2 H, H-1C, PhCH), 5.29 (t, J = 10.0 Hz, 1 H, H-4C), 5.05 (d, J = 3.5 Hz, 1 H, H-1B), 4.92 (d, J = 11.5 Hz, 1 H, PhCH), 4.88 (d, J = 3.5 Hz, 1 H, H-1E), 4.83–4.77 (m, 3 H, 3 PhCH), 4.73 (d, J = 8.0 Hz, 1 H, H-1A), 4.71–4.58 (m, 7 H, 7 PhCH), 4.55–4.48 (m, 3 H, 3 PhCH), 4.37–4.33 (m, 2 H, H-6aA, PhCH), 4.30–4.26 (m, 2 H, H-3A, H-4A), 4.23–4.16 (m, 2 H, H-5C, H-6aD), 4.12–4.00 (m, 6 H, H-2A, H-2B, H-3C, H-3D, H-5B, H-6bA), 3.98–3.93 (m, 3 H, H-4D, H-5E, PhCH), 3.91 (br s, 1 H, H-1D), 3.83–3.80 (m, 2 H, H-2C, H-2E), 3.78 (s, 3 H, OCH3), 3.77–3.74 (m, 3 H, H-4B, H-4E, H-6bD), 3.59–3.55 (m, 1 H, H-6aC), 3.53–3.44 (m, 5 H, H-2D, H-3B, H-3E, H-5A, H-6bC), 2.98–2.90 (m, 1 H, H-5D), 2.13 (s, 3 H, COCH3), 1.18 (d, J = 6.5 Hz, 3 H, CCH3), 0.75 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.6 (CO), 155.8–114.5 (Ar-C), 102.0 (PhCH), 101.9 (PhCH), 101.4 (C-1A), 100.3 (C-1B), 98.5 (C-1C), 97.5 (C-1D), 96.6 (C-1E), 80.9 (C-3B), 78.4 (C-3D), 77.8 (C-4D), 77.2 (C-4B), 76.9 (2 C, C-4A, C-4E), 76.4 (C-3A), 76.2 (C-3E), 75.2 (PhCH2), 75.1 (C-2E), 74.7 (C-2D), 74.6 (PhCH2), 74.0 (PhCH2), 73.4 (2 C, 2 PhCH2), 73.1 (C-2B), 72.7 (C-3C), 72.4 (PhCH2), 71.9 (PhCH2), 71.2 (C-2C), 71.0 (C-5E), 70.9 (PhCH2), 69.8 (C-6C), 69.0 (C-6A), 68.7 (C-6D), 67.6 (C-5D), 67.1 (2 C, C-4C, C-5C), 66.5 (C-5A), 64.7 (C-5B), 61.4 (C-2A), 55.5 (OCH3), 21.0 (COCH3), 17.4 (CCH3), 15.7 (CCH3); HRMS (ESI) for C109H115N3O25 [M + H]+: Calcd. 1866.7898; found, 1866.7890.
p-Methoxyphenyl (α-L-fucopyranosyl)-(1 → 3)-(β-D-mannopyranosyl)-(1 → 3)-(α-D-mannopyranosyl)-(1 → 3)-(α-L-fucopyranosyl)-(1 → 3)-2-acetamido-2-deoxy-β-D-galactopyranoside (1)
To a solution of compound 16 (500 mg, 0.27 mmol) in pyridine (2 mL) was added CH3COSH (0.5 mL) and the reaction mixture was stirred at room temperature for 12 h. The solvents were removed and co-evaporated with toluene (3 × 20 mL) under reduced pressure and the crude product was passed through a short pad of SiO2. A solution of the N-acetylated product in 0.1 M CH3ONa in CH3OH (10 mL) was stirred at room temperature for 3 h, neutralized with Amberlite IR-120 (H+) resin, filtered and concentrated. To a solution of the de-O-acetylated product in CH3OH (5 mL) was added 20% Pd(OH)2-C (50 mg) and the reaction mixture was allowed to stir at room temperature under a positive pressure of H2 for 24 h. The reaction mixture was filtered through a Celite bed, washed with CH3OH-H2O (30 mL; 2:1 v/v) and concentrated under reduced pressure. The deprotected product was passed through a Sephadex LH-20 column using CH3OH-H2O (3:1) as eluant to give pure compound 1 (160 mg, 62%). White powder; [α]D + 11 (c 1.0, H2O); 1H NMR (500 MHz, D2O): δ 6.99 (d, J = 9.0 Hz, 2 H, Ar-H), 6.90 (d, J = 9.0 Hz, 2 H, Ar-H), 5.24 (d, J = 1.5 Hz, 1 H, H-1C), 5.06 (d, J = 4.0 Hz, 1 H, H-1E), 4.97 (d, J = 8.5 Hz, 1 H, H-1A), 4.94 (d, J = 4.0 Hz, 1 H, H-1B), 4.78 (br s, 1 H, H-1D), 4.27 (br s, 1 H, H-5E), 4.20–4.16 (m, 3 H, H-2A, H-2C, H-2D), 4.10–4.02 (m, H-3D, H-5B), 4.00–3.90 (m, 4 H, H-3A, H-3E, H-4A, H-4E), 3.88–3.84 (m, 1 H, H-6aC), 3.82–3.78 (m, 4 H, H-3C, H-6aA, H-6aD, H-6bC), 3.78 (s, 3 H, OCH3), 3.77–3.62 (m, 9 H, H-2E, H-3B, H-4B, H-4C, H-4D, H-5A, H-5C, H-6bA, H-6bD), 3.55–3.47 (m, 1 H, H-2B), 3.36–3.32 (m, 1 H, H-5D), 1.94 (s, 3 H, COCH3), 1.20 (d, J = 6.5 Hz, 3 H, CCH3), 1.02 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, D2O): δ 175.8 (COCH3), 155.4–114.8 (Ar-C), 101.4 (JH1,C1 = 174.0 Hz, C-1C), 101.3 (JH1,C1 = 171.0 Hz, C-1B), 100.8 (JH1,C1 = 157.0 Hz, C-1A), 96.7 (JH1,C1 = 158.5 Hz, C-1D), 95.9 (JH1,C1 = 171.0 Hz, C-1E), 78.8 (C-3A), 78.5 (C-3C), 78.2 (C-3B), 77.1 (C-5D), 76.3 (C-3D), 75.3 (2 C, C-4B, C-5A), 74.4 (C-4E), 73.3 (C-5C), 70.1 (C-3E), 68.2 (2 C, C-2E, C-4A), 67.8 (2 C, C-2B), 67.6 (C-5E), 67.0 (C-5B), 66.5 (C-2C), 65.7 (C-4C), 65.0 (C-4D), 64.9 (C-2D), 61.1 (C-6D), 60.7 (2 C, C-6A, C-6C), 55.7 (OCH3), 51.4 (C-2A), 22.2 (COCH3), 16.4 (CCH3), 15.0 (CCH3); HRMS (ESI) for C39H61NO25 [M + H]+: Calcd. 944.3611; found, 944.3602.
References
Mokomane, M., Kasvosve, I., de Melo, E., Pernica, J.M., Goldfarb, D.M.: The global problem of childhood diarrhoeal diseases: emerging strategies in prevention and management. Ther. Adv. Infect, Dis. 5, 29–43 (2018)
Ashbolt, N.J.: Microbial contamination of drinking water and disease outcomes in developing regions. Toxicology. 198, 229–238 (2004)
Dekker, J., Frank, K.: Salmonella, Shigella, and Yersinia. Clin. Lab. Med. 35, 225–246 (2015)
Crump, J.A., Sjölund-Karlsson, M., Gordon, M.A., Parry, C.M.: Epidemiology, clinical presentation, laboratory diagnosis, antimicrobial resistance, and antimicrobial management of invasive Salmonella Infections. Clin. Microbiol. Rev. 28, 901–937 (2015)
Morris Jr., J.G., Acheson, D.: Cholera and other types of vibriosis: a story of human pandemics and oysters on the half Shell. Clin. Infect. Dis. 37, 272–280 (2003)
Clements, A., Young, J.C., Constantinou, N., Frankel, G.: Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes. 3, 71–87 (2012)
Acheson, D., Allos, B.M.: Campylobacter jejuni infections: update on emerging issues and trends. Clin. Infect. Dis. 32, 1201–1206 (2001)
Orskov, I., Orskov, F., Jann, B., Jann, K.: Serology, chemistry, and genetics of O and K antigens of Escherichia coli. Bacterial Rev. 41, 667–710 (1977)
Kaper, J.B., Nataro, J.P., Mobley, H.L.T.: Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140 (2004)
Johnson, J.R., Russo, T.A.: Extraintestinal pathogenic Escherichia coli : “the other bad E coli ”. J. Lab. Clin. Med. 139, 155–162 (2002)
Kelly, P.: Infectious diarrhoea. Medicine. 39, 201–206 (2011)
Stenutz, R., Weintraub, A., Widmalm, G.: The structures of Escherichia coli O-polysaccharide antigens. FEMS Microbiol. Rev. 30, 382–403 (2006)
Paton, J.C., Paton, A.W.: Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 11, 450–479 (1998)
Nataro, J.P., Kaper, J.B.: Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201 (1998)
Perepelov, A.V., Guo, X., Filatov, A.V., Liu, B., Knirel, Y.A.: Structure and gene cluster of the O-antigen of Escherichia coli O43. Carbohydr. Res. 416, 32–36 (2015)
Roy, R.: New trends in carbohydrate based vaccines. Drug Discov. Today Technol. 1, 327–336 (2004)
Hölemann, A., Seeberger, P.H.: Carbohydrate diversity: synthesis of glycoconjugates and complex carbohydrates. Curr. Opin. Biotechnol. 15, 615–622 (2004)
Pozsgay, V.: Recent developments in synthetic oligosaccharide-based bacterial vaccines. Curr. Top. Med. Chem. 8, 126–140 (2008)
Morelli, L., Poletti, L., Lay, L.: Carbohydrates and immunology: synthetic oligosaccharide antigens for vaccine formulation. Eur. J. Org. Chem. 5723–5777 (2011)
Lee, C.-J., Lee, L.H., Frasch, C.E.: Protective immunity of pneumococcal glycoconjugates. Crit. Rev. Microbiol. 29, 333–349 (2003)
Cheesman, M.J., Ilanko, A., Blonk, B., Cock, I.E.: Developing new antimicrobial therapies: are synergistic combinations of plant extracts/compounds with conventional antibiotics the solution? Pharmacogn. Rev. 11, 57–72 (2017)
He, H., Chen, D., Li, C., Zhao, J.-H., Qin, H.-B.: Synthesis of trisaccharide repeating unit of fucosylated chondroitin sulfate. Org. Biomol. Chem. 17, 2877–2882 (2019)
Susanto, W., Kong, K.-H., Hua, K.-F., Wu, S.-H., Lam, Y.: Synthesis of the trisaccharide repeating unit of capsular polysaccharide from Klebsiella pneumonia. Tetrahedron Lett. 60, 288–291 (2019)
Gucchait, Misra, A.K.: Influence of remote functional groups towards the formation of 1,2-cis glycosides: special emphasis on β-mannosylation. Org. Biomol. Chem. 17, 4605–4610 (2019)
Lönn, H.: Synthesis of a tri- and a heptasaccharide which contain α-L-fucopyranosyl groups and are part of the complex type of carbohydrate moiety of glycoproteins. Carbohydr. Res. 139, 105–113 (1985)
Schmidt, R.R., Zhu, X.: Glycosyl Trichloroacetimidates. In: Fraser-Reid, B.O., Tatsuta, K., Thiem, J. (eds.) Glycoscience. Springer, Berlin, Heidelberg (2008). https://doi.org/10.1007/978-3-540-30429-6_11
Chakraborti, A.K., Gulhane, R.: Perchloric acid adsorbed on silica gel as a new, highly efficient, and versatile catalyst for acetylation of phenols, thiols, alcohols, and amines. Chem. Commun. 2003, (1896-1897)
Mukhopadhyay, B., Maurer, S.V., Rudolph, N., van Well, R.M., Russell, D.A., Field, R.A.: From solution phase to “on-column” chemistry: trichloroacetimidate-based glycosylation promoted by perchloric acid-silica. J. Org. Chem. 70, 9059–9062 (2005)
Si, A., Misra, A.K.: Synthesis of a pentasaccharide repeating unit corresponding to the cell wall O-antigen of Escherichia coli O59 using iterative glycosylations in one pot. Tetrahedron. 72, 4435–4441 (2016)
Love, K.R., Andrade, R.B., Seeberger, P.H.: Linear synthesis of a protected H-type II pentasaccharide using glycosyl phosphate building blocks. J. Org. Chem. 66, 8165–8176 (2001)
DeNinno, M.P., Etienne, J.B., Duplantier, K.C.: A method for the selective reduction of carbohydrate 4,6-O-benzylidene acetals. Tetrahedron Lett. 36, 669–672 (1995)
Ishiwata, A., Munemura, Y., Ito, Y.: Synergistic solvent effect in 1,2-cis-glycoside formation. Tetrahedron. 64, 92–102 (2008)
Kanie, O., Ito, Y., Ogawa, T.: Orthogonal glycosylation strategy in oligosaccharide synthesis. J. Am. Chem. Soc. 116, 12073–12074 (1994)
Kasai, R., Okihara, M., Asakawa, J., Mizutani, K., Tanaka, O.: 13C NMR study of α- and β-anomeric pairs of D-mannopyranosides and L-rhamnopyranosides. Tetrahedron Lett. 35, 1427–1432 (1979)
Bock, K., Pedersen, C.: A study of 13CH coupling constants in hexopyranoses. J. Chem. Soc. Perkin Trans. 2, 293–297 (1974)
Wuts, P.G.M.: In: Crich, D. (ed.) Handbook of Reagents for Organic Synthesis: Reagents for Glycoside, Nucleotide, and Peptide Synthesis, pp. 425–428. Wiley, Chichester (2005)
Mukherjee, C., Misra, A.K.: Glycosylation and pyranose-furanose isomerization of carbohydrates using HClO4-SiO2: synthesis of oligosaccharides containing galactofuranose. Synthesis. 683–692 (2007)
Fügedi, P., Garegg, P.J.: A novel promoter for the efficient construction of 1,2-trans linkages in glycoside synthesis, using thioglycosides as glycosyl donors. Carbohydr. Res. 149, C9–C-12 (1986)
Veeneman, G.H., van Leeuwen, S.H., van Boom, J.H.: Iodonium ion promoted reactions at the anomeric centre. II An efficient thioglycoside mediated approach toward the formation of 1,2-trans linked glycosides and glycosidic esters. Tetrahedron Lett. 31, 1331–1334 (1990)
Lönn, H.: Glycosylation using a thioglycoside and methyl trifluoromethanesulfonate. A new and efficient method for cis and trans glycoside formation. J. Carbohydr. Chem. 6, 301–306 (1986)
Zheng, X., Xu, D., Edgar, K.J.: Cellulose levulinate: a protecting group for cellulose that can be selectively removed in the presence of other ester groups. Cellulose. 22, 301–311 (2015)
Bhattacharyya, S., Magnusson, B.G., Wellmar, U., Nilsson, U.J.: The p-methoxybenzyl ether as an in situ-removable carbohydrate-protecting group: a simple one-pot synthesis of the globotetraose tetrasaccharide. J. Chem. Soc. Perkin Trans. 1, 886–890 (2001)
Shangguan, N., Katukojvala, S., Greenberg, R., Williams, L.J.: The reaction of thio acids with azides: a new mechanism and new synthetic applications. J. Am. Chem. Soc. 125, 7754–7755 (2003)
Pearlman, W.M.: Noble metal hydroxides on carbon nonpyrophoric dry catalysts. Tetrahedron Lett. 8, 1663–1664 (1967)
Acknowledgements
P. S. thanks CSIR, New Delhi for providing Senior Research Fellowship. The work is supported by SERB, India (Project No. CRG/2019/000352 dated 23.01.2020) (AKM) and Bose Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shit, P., Misra, A.K. Straightforward synthesis of the pentasaccharide repeating unit of the cell wall O-antigen of Escherichia coli O43 strain. Glycoconj J 37, 647–656 (2020). https://doi.org/10.1007/s10719-020-09933-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-020-09933-z